

Abstract
With the advancement of high throughput screening, it has become easier and faster to discover hit compounds that inhibit proliferation of bacterial cells. However, development in technologies used to identify cellular targets of potent antibacterial inhibitors has lagged behind. Here we describe a novel strategy of target identification for antibacterial inhibitors using an array of Escherichia coli clones each over-expressing one essential protein. In a proof-of-concept study, eight essential genes were cloned into pLex5BA vector under the control of an inducible promoter. Over-expression of target proteins was confirmed. For two clones, one over-expressing FabI and the other over-expressing MurA enzymes, the host cells became 17-fold and 139-fold more resistant to the specific inhibitors triclosan and phosphomycin, respectively, while the susceptibility of other clones towards these inhibitors remained unchanged after induction of gene expression. Target identification via target protein over-expression was demonstrated using both mixed clone and individual clone assay formats.
Keywords: Escherichia coli, Essential genes, Drug targets, Antibacterial compounds, Over-expression, Triclosan, Phosphomycin
Introduction
Over the past 20 years, emergence and dissemination of bacterial resistance to existing antibiotics has increased at an alarming rate [1-3]. Especially troublesome is the emergence of diverse pathogens that became multi-drug resistant [4-7], posing a serious threat to global public health. During this same period, few antibiotics of New Chemical Entities (NCEs) have emerged [8], reflecting both the challenges of identifying NCEs and a reduced commitment of antibiotic drug discovery by pharmaceutical companies. One factor contributing to the dismal productivity in this therapeutic area has been the lack of a comprehensive knowledge of bacterial essential genes. Since the first sequencing of a complete microbial genome, Mycoplasma genitalium [9], a decade ago, more than 350 bacterial genomes have been completely sequenced, including Escherichia coli [10], Bacillus subtilis [11] and many important human pathogens. The rapid advances in microbial genomics could potentially lead to renewed efforts and technological breakthroughs in antibiotic drug discovery to meet the urgent medical need of new antibiotics [12].
Currently available antibiotics target approximately 20 of the estimated 200 essential gene products in bacteria [13]. The recognition of the utilities of untapped potential antibiotic targets has fostered various innovative approaches for bacterial essential gene identification, facilitating the development of a new discipline—bacterial functional genomics. Comparative genomics has been combined with conditional expression to identify essential genes in E. coli [14, 15]. Promoter replacement techniques have been developed to assess the essentiality of Staphylococcus aureus genes [16, 17]. Massive transposon mutagenesis has been applied to identify essential genes on genome-scale in M. genitalium [18], Haemophilus influenzae [19, 20], and E. coli [21]. Gene disruption methodologies have been employed to systematically identify essential genes in Streptococcus pneumoniae [22] and B. subtilis [23]. Finally, regulated antisense expression [24, 25] has been utilized to identify a comprehensive set of essential genes in S. aureus. Conjoint analysis of several computational models and experimental strategies has led to the proposition that, from a standpoint of cellular processes necessary to maintain cellular life, a minimal bacterial essential set contains 206 genes [13]. Bacterial essential genes thus far identified have been increasingly used in either cell-based or biochemical drug screens [26-28].
Historically, the most productive paradigm for antibacterial lead discovery involved a straightforward initial screen for the inhibition of bacterial growth. This was followed by laborious experimental attempts to identify drug targets and establish mechanisms of action for those discovered antibacterial compounds. Once and if the target of the hit compound was identified, antibacterial potency and selectivity were then optimized through the synthesis and testing of structural analogs of the initial hit compound. With increased exploitation of chemical space (in which distance between points approximates compound similarity), it is still simple, fast and straightforward to identify inhibitors of bacterial growth. What remains difficult is to experimentally link the inhibitor to the cellular target. There is therefore a need for development of fast and comprehensive assay systems to identify cellular targets of potent antibacterial inhibitors. Li and colleagues [29] recently reported a strategy of identifying targets of antibacterial inhibitors via suppression of inhibition by multicopy plasmid clones of an E. coli random genomic library. While this approach does not require large scale cloning efforts, it would be difficult to judge the comprehensiveness of the essential gene coverage in the clones of the genomic library without detailed validation experiments. In this report, we describe a novel strategy to identify the target of an inhibitor via a comprehensive collection (array) of E. coli clones each engineered to over-express one essential protein. This strategy is based on the concept that elevated levels of the drug target protein confer resistance because higher concentrations of a specific inhibitor are required to bind to the excess target in order to inhibit bacterial growth [30]. In this proof-of-concept report, we confirmed elevated inhibitor-specific resistance of E. coli strains over-expressing specific essential targets and demonstrated identification of cellular targets for inhibitors using either a culture of mixed clones or parallel growth of individual clones each over-expressing one particular essential gene. If this strategy is applicable to a large number of essential genes in E. coli, it would become a very useful assay tool to identify cellular targets of inhibitors of unknown mechanisms of action.
Materials and Methods
Bacterial strains, plasmid and materials
Purchased from ATCC (Manassas, VA), E. coli K-12 strain MG1655 (Genotype: F- lambda- ilvG- rfb-50 rph-1) was used as the source of chromosomal DNA with known genomic sequence [10] from which essential genes were PCR amplified and cloned. E. coli K-12 DH5α was obtained from Invitrogen (Genotype: F-Φ80ΔlacZΔM15 Δ(lacZYA-αργF) U169 recA1 endA1 hsdR17 (rk-, mk+) phoA supE44 λ-thi-1 gyrA96 relA1). Expression vector pLex5BA [31] was used to clone and express essential genes under control of the inducer isopropyl β-D-thiogalactopyranoside (IPTG). Restriction enzymes and T4 DNA ligase were purchased from Invitrogen (Carlsbad, CA). Antibiotics and inhibitors (ampicillin, phosphomycin and triclosan), IPTG and dithiothreitol (DTT) were purchased from Sigma-Aldrich (St. Louis, MO). Phenylmethylsulfonyl fluoride (PMSF) was obtained from Pierce (Rockford, IL). Difco Luria-Bertani (LB) broth powder was purchased from Fisher Scientific (Tustin, CA). E. coli cells were grown in LB broth or agar media under appropriate antibiotic selection when necessary (100 μg/ml of ampicillin).
Gene cloning
E. coli essential genes were PCR amplified using E. coli K-12 MG1655 genomic DNA as the template and cloned into the plasmid pLex5BA [31]. Essential genes were selected from the PEC (Profiling of E. coli Chromosome) database. PCR primers were designed to include the complete open reading frame (ORF) of each gene and its putative Shine-Delgarno sequence in the amplified product. Appropriate restriction endonuclease recognition sites were designed into PCR primers to facilitate the cloning process. Genes were directionally cloned downstream of an IPTG-inducible promoter present on the plasmid [31]. Recombinant plasmid clones were initially validated via restriction digest and agarose gel electrophoresis. One clone for each gene was confirmed via DNA sequencing using an ABI Big-Dye terminator Kit 3.1 and the ABI 3100 Avant Genetic Analyzer (ABI, Foster City, CA).
Over-expression of cloned essential proteins in E. coli
Based on a procedure from [32] with modifications, cell-free protein extracts were obtained from E. coli cells of various clones under appropriate inducing conditions. Briefly, an E. coli strain harboring a plasmid with an inserted essential gene was streaked on an LB agar plate with 100 μg/ml of ampicillin and grown overnight at 37 °C. An Erlenmeyer flask containing 50 ml LB medium (with 100 μg/ml ampicillin) was then inoculated with 4 colonies from the plate and grown at 37 °C until the culture reached an OD600 of 0.5. Expression of the essential gene was induced by the addition of IPTG to a final concentration of 1 mM and continued growth of the culture overnight (approximately 17 h) at 37 °C. At either 4 h after induction or after overnight induction ten milliliters of the culture were harvested and resuspended in 0.455 ml of phosphate buffered saline (PBS: 137 mM NaCl; 2.7 mM KCl; 10 mM Na2HPO4; 2 mM KH2PO4). Lysozyme was added to a final concentration of 1 mg/ml and the suspension was incubated on ice for 60 min. One milliliter of 0.2% Triton X-100 was added, the suspension was vigorously mixed, and DNase and RNase enzymes were each added to a final concentration of 5 μg/ml. The mixture was gently mixed for 20 min at 4 °C by rocking, followed by centrifugation at 3,500 × g for 30 min at 4 °C. The supernatant was collected and placed into a fresh tube and DTT and PMSF were added to a final concentration of 1 mM. Glycerol was then to a final concentration of 10%. The cell-free protein extract was either stored at -20 °C or subjected to polyacrylamide gel electrophoresis analysis using precast protein gels from Pierce (Rockford, IL). Following electrophoresis the protein gels were stained with Bio-Safe Coomassie blue from Bio-Rad (Hercules, CA).
Determination of inducer concentrations on growth of E. coli clones
A series of IPTG dilutions were combined, in wells of a Corning Costar flat-bottom 96-well microplate, with LB broth plus ampicillin inoculated with cells of E. coli expression clones (to an initial inoculation OD600 of 0.001). The plate was sealed with clear thermal sealing film (E& K Scientific, Campbell, CA) and growth of each well at 37°C was monitored kinetically by Tecan Genios plate reader equipped with an incubation chamber (Tecan, Durham, NC). Plates were read every 30 min for approximately 18 h with low speed shaking prior to every read. E. coli cell growth curves and optical density comparison charts were generated by GraphPad Prizm software (San Diego, CA).
Drug dose response and resistance testing
Individual E. coli clones carrying plasmids containing essential genes were grown in LB medium with 100 μg/ml ampicillin for maintenance of the plasmid, in the presence of serial dilutions of specific inhibitors available, either with or without the inducer IPTG (at final concentration of 1 mM). The growth experiments were performed as described above using Corning Costar flat-bottom 96-well microplates and Tecan Genios plate reader. Inhibitor dose response curves were generated by GraphPad Prizm software.
Target identification using mixed clone assay format
Eight E. coli over-expressing clones were mixed and grown in concentrations of either triclosan or phosphomycin at levels that inhibit the parental strain under inducing or noninducing conditions. Cells grown from two treatment conditions (8 μg/ml triclosan and 2 μg/ml phosphomycin) in the presence of 1 mM IPTG were diluted and plated on selectable media to obtain isolated colonies. Plasmid DNAs were isolated and restriction digest was performed to determine the identities of insert DNAs. Sequence confirmation of the plasmid inserts was carried out as described above.
Target identification using individual clone assay format
To test if cellular targets of antibacterial inhibitors can also be identified using individually grown clones, over-expressing clones were arrayed and grown in separate microplate rows under various conditions. Some wells of the microplate contained triclosan (4 μg/ml), others contained phosphomycin (2 μg/ml). Some wells contained no antibacterial inhibitors to serve as innoculum control. Clones were grown either with or without IPTG. Cell growth in the microplate was monitored in Tecan Genios plate reader as describe above.
Results
Gene cloning
PCR-amplified DNA fragments for eight E. coli essential genes (Table 1) were cloned into pLex5BA and transformed into E. coli DH5α host cells. The length and identity of insert DNA from recombinant plasmids were confirmed via restriction digest analysis (Fig. 1) and DNA sequencing (data not shown). These eight essential genes were chosen to demonstrate a proof of concept for our strategy of target identification via target protein over-expression because they encode a variety of essential target proteins involved in several distinct cellular functions or pathways. MurA and MurG are enzymes involved in bacterial peptidoglycan (cell wall) biosynthesis and are critical for maintaining cellular integrity. FabD and FabI are essential enzymes involved in fatty acid biosynthesis. Tryptophanyl tRNA synthetase (TrpS), peptide chain release factor 2 (PrfB), 30S ribosomal protein S12 and 50S ribosomal protein L10 are involved in different facets of protein synthesis. Conveniently, triclosan and phosphomycin are known antibacterial inhibitors specific for FabI and MurA enzymes, respectively, and are available commercially to test our strategy.
Table 1.
E. coli essential genes cloned.
| Gene Name | Gene Product (Target) | Gene Size (base pairs) | E. coli Clone Name |
|---|---|---|---|
| fabD | Malonyl CoA-ACP transacylase | 930 | FabD clone |
| fabI | Enol-acyl carrier protein reductase | 789 | FabI clone |
| murA | UDP-N-acetylglucosamine enolpyruvyl transferase | 1260 | MurA clone |
| murG | UDP-N-acetylglucosamine--N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase | 1068 | MurG clone |
| prfB | Peptide chain release factor 2 | 1023 | PrfB clone |
| rplJ | 50S ribosomal protein L10 | 498 | RplJ clone |
| rpsL | 30S ribosomal protein S12 | 375 | RpsL clone |
| trpS | Tryptophanyl tRNA synthetase | 1005 | TrpS clone |
Figure 1.

Agarose gel stained with ethidium bromide after electrophoresis. Lane 1, DNA ladder; Lanes 2-10, various plasmids digested with EcoR I and BamH I: lane 2, vector pLex5BA; lane 3, pLex5BA-fabD; lane 4, pLex5BA-fabI; lane 5, pLex5BA-murA; lane 6, pLex5BA-murG; lane 7, pLex5BA-prfB; lane 8, pLex5BA-rplJ; lane 9, pLex5BA-rpsL; lane 10, pLex5BA-trpS. Note, due to restriction sites and Shine-Delgarno sequences designed into PCR primers, the sizes of inserts shown in the gel are slightly bigger than the ORF sizes listed in Table 1.
Protein over-expression
To confirm over-expression of cloned essential proteins, clones were grown to stationary phase and induction was initiated by adding IPTG to a final concentration of 1 mM. Cell culture samples were taken at either 4 h or 17 h (overnight) after induction. Examples of protein over-expression are shown in Figure 2. Results indicated that for FabI, MurA and TrpS clones, there was significant over-expression of essential proteins at 4 h after induction and that continued induction beyond 4 h did not appear to further increase the protein expression. Specifically, there were protein bands of similar intensity present in both 4 hr and overnight post-induction samples of a MurA clone that match the calculated molecular weight (44.8 kD) of MurA protein (Fig. 2). Similarly, cells of a TrpS clone produced an obviously over-expressed polypeptide band that corresponds to TrpS enzyme’s calculated molecular weight (37.4kD) (Fig. 2). In contrast, over-expression of a FabI clone resulted in a protein band corresponding to a molecular weight of around 33 kDalton (Fig. 2), which is inconsistent with its calculated molecular weight of 28 kD. This type of discrepancies between calculated molecular weight of a polypeptide and electrophoretic outcome is not unusual. Interestingly, Bergler and coworkers over-expressed the same protein (FabI, used to be called EnvM) from E. coli and had obtained a similar result [33]. This led us to believe that the observed over-exppressed protein from FabI clone belonged to FabI enzyme. Of eight essential gene clones tested, all except PrfB clone demonstrated detectable over-expression by protein gel electrophoresis of cell-free extracts (data not shown).
Figure 2.
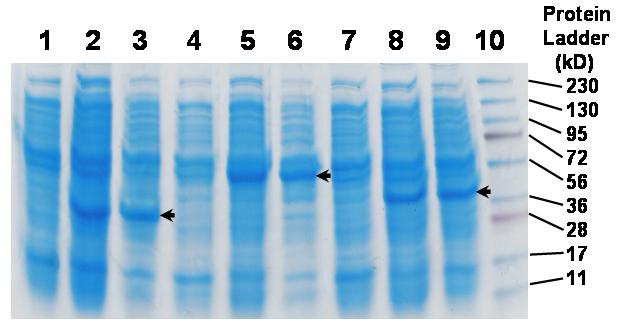
Acrylamid gel electrophoresis separation of protein extracts after induction of essential protein expression by IPTG. Protein extracts from cell cultures of FabI clone (lanes 1-3), MurA clone (4-6) and TrpS clone (7-9) before adding inducer (1 mM IPTG) (lanes 1, 4, 7), 4 h after induction (lanes 2, 5, 8) and 17 h after induction (lanes 3, 6, 9). Lane 10, protein ladder.
Effects of induction on cell growth
To determine whether or not the IPTG inducer of the over-expression of these essential proteins caused adverse effects on growth of these clones (since robust growth from each clone was critical for target identification assays), individual clones were grown in the presence of a series of IPTG concentrations between 0.01 mM to 10 mM. Our results indicated that inducer concentrations as high as 10 mM (ten times as high as used in our experiments) did not cause significant growth defects on TrpS, RplJ, FabI and MurA clones (Fig. 3). Similar results were obtained with FabD, RpsL, PrfB, MurG and vector clones (data not shown)
Figure 3.

Effect of inducer concentrations on the growth of four E. coli clones (TrpS, RplJ, FabI and MurA). Cell growth in the wells of a microplate was monitored kinetically using a plate reader. For each concentration of inducer for every clone, three replicate wells were used. Late log-phase (10 h) plate reading values were used to generate the bar graph.
Drug dose response and resistance testing
To confirm the concept that target over-expression increases resistance to specific inhibitors, a FabI clone was grown in various concentrations of triclosan (the specific inhibitor of the FabI enzyme) in the LB medium with ampicillin in the presence and absence of 1 mM IPTG. Triclosan dose response curves for the FabI clone exhibited a significant shift in the presence of the inducer as compared to that in the absence of an inducer (Fig. 4). The IC50 values increased from 584 ng/ml to 9,927 ng/ml, a 17-fold increase in resistance, when over-expression of FabI was induced. In contrast, as expected since the vector contains no insert, the IC50 values for the vector clone did not change at all (Fig. 4). When a MurA clone was grown in various concentrations of phosphomycin, the specific inhibitor of the MurA enzyme, the increase in resistance was even more dramatic (a 139-fold change in IC50 values as shown in Fig. 5). Interestingly, the elevated resistance conferred by induction of the target expression did not require the presence of ampicillin in the medium (Fig. 6). Without ampicillin in the LB medium, cells of MurA clone grew fairly well in the presence of 8 μg/ml of phosphomycin under the condition of induction (Fig. 6). In the absence of IPTG, cells failed to grow in 8 μg/ml of phosphomycin. Similar growth profiles were observed for the FabI clone in the absence of ampicillin (data not shown).
Figure 4.

Increased resistance to triclosan via over-expression of inhibitor-specific essential gene product, FabI. The FabI clone was grown in presence of serial dilutions of triclosan either with or without 1 mM IPTG. A vector control clone containing vector alone served as a control. For each condition (in respect to inhibitor and inducer), three replicate wells were used. Late log-phase (10 h) plate read values were used to generate the dose response graph.
Figure 5.

Increased resistance to phosphomycin via over-expression of inhibitor-specific essential gene product MurA. The MurA clone was grown in presence of serial dilutions of phosphomycin either with or without 1 mM IPTG. For each condition (in respect to inhibitor and inducer), three replicate wells were used. Late log-phase (10 h) plate read values were used to generate the dose response graph.
Figure 6.

Growth profiles for cells of MurA clone in the absence of ampicillin. Cells were grown in LB media without ampicillin in wells of a microplate in the presence or absence of IPTG and/or phosphomycin. Growth was monitored kinetically in a plate reader at 37 °C for 17.5 h, with optical density reading every 0.5 h.
To determine that the increased resistance was specific for target and inhibitor pairs involved, IC50 values were obtained from dose response curves for each E. coli clone treated with triclosan and phosphomycin both in the presence and absence of an inducer. The ratio of IC50 induced vs IC50 non-induced was used as a measure of changes in a clone’s susceptibility toward a particular inhibitor. Results indicated that elevated resistance to triclosan was observed only in the FabI clone, but not in any of the other seven over-expression clones (Fig. 7). Similarly, elevated phosphomycin resistance was only evident in the MurA clone but not in the other seven clones (Fig. 7). Even over-expression of enzymes (FabD and MurG) in the same respective pathways did not cause increases in resistance to triclosan and phosphomycin, respectively (Fig. 7). These results indicate that the inhibitor resistance as a result of target protein over-expression was highly specific.
Figure 7.

Specificity of elevated resistance conferred by target protein over-expression. Each of the eight clones plus the vector clone was treated with a series of concentrations of either triclosan or phosphomycin in the presence or absence of the inducer (1 mM IPTG). Dose response curves for each clone were plotted for either induced or noninduced conditions. IC50 values were obtained. The ratio of IC50 values (IC50 induced vs IC50 noninduced) was used as a measure of fold increase in resistance.
Target identification using mixed clone assay
The specificity nature of the enhanced resistance due to target over-expression became the scientific basis for our strategy of identifying cellular targets using a collection of E. coli clones each over-expressing a particular target protein. When eight over-expressing clones were mixed and subjected to treatment by inhibitory (to the parental strain) concentrations of triclosan and phosphomycin, respectively, cell growth was observed in the presence of 1 mM IPTG in both treatments over a range of inhibitor concentrations (data not shown). We suspected that the cell growth observed was due to the enhanced resistance of the one clone that was over-expressing a specific target protein. To test this, cultures with growth from the wells containing 2 μg/ml of phosphomycin and 8 μg/ml of triclosan were serially diluted and plated out on LB agar plates with 100 μg/ml of ampicillin. Isolated colonies from each inhibitor treatment were grown and plasmids were isolated to perform restriction digest analysis. Restriction analysis results indicated that 100% of the colonies from phosphomycin treatment contained murA gene inserts and 100% of the colonies from triclosan treatment contained fabI gene inserts (Fig. 8a and 8b). Subsequent plasmid DNA sequencing further confirmed that the growth was indeed the result of the clones over-producing the specific target proteins to which the specific inhibitors binds (data not shown).
Figure 8.
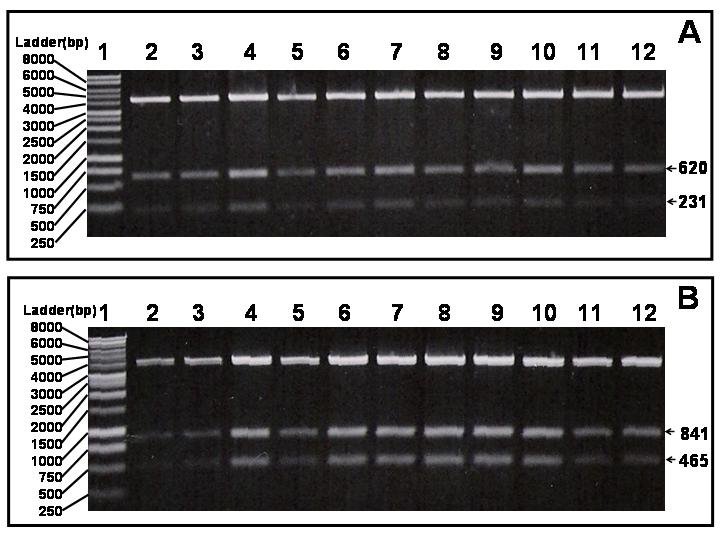
Restriction digest analysis of isolated plasmids from colonies obtained from mixed strain treatment with either triclosan (4 μg/ml) (A) or phosphomycin (2 μg/ml) (B). A, fabI gene contains an internal Pst I site. EcoR I/Pst I double digest of pLex5BA (which also contains a Pst I site in the polylinker) containing cloned fabI gene would produce two small fragments (620 and 231 bps) in additional to the vector band. B, murA gene contains an internal Hind III site. EcoR I/Hind III double digest of pLex5BA (which also contains a Hind III site in the polylinker) containing cloned murA gene would produce two fragments (841 and 465 bps) in addition to the vector band.
Target identification using individual clone assay
To test whether or not treatment by an inhibitor of individually grown over-expression clones also could identify the target of the inhibitor, eight over-expression clones were individually grown in the presence or absence of inhibitors triclosan (4 μg/ml) and phosphomycin (2 μg/ml), with or without 1 mM IPTG (Fig. 9). As can be seen from Figure 9, for those wells without inhibitors, cells grew normally with or without inducer as expected (used as inoculum control). Microplate wells G5 and G6, the only two wells containing the FabI clone with both triclosan and IPTG present, produced robust growth similar to the wells of the inoculum control (Fig. 9). For wells treated with phosphomycin, only two wells (F1 and F2) from the MurA clone row containing both phosphomycin and the inducer grew normally.
Figure 9.
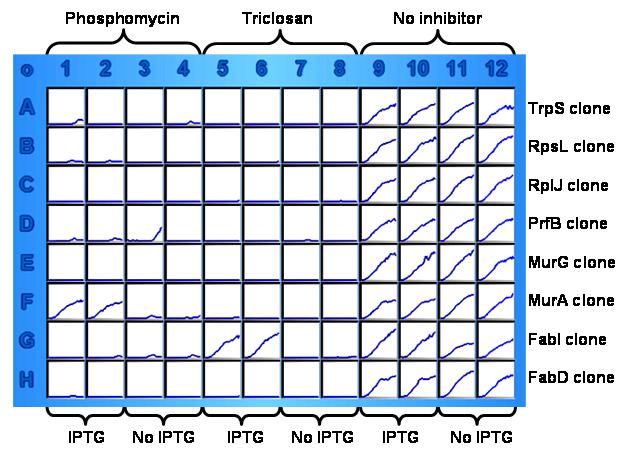
A kinetic growth image of a microplate-based target identification assay. The wells of each row contained one individual clone (marked on the right). Columns 1 to 4 contained 2 μg/ml of phosphomycin. Columns 5 to 8 contained 8 μg/ml of triclosan. Columns 9 to 12 contained no inhibitor to serve as cell growth control. For each inhibitor treatment, half of the wells also contained 1 mM IPTG while the other half did not (shown at the bottom).
Discussion
Recognition of the mechanism of antibiotic resistance due to drug target over-expression was first reported decades ago [34]. This line of research has received little attention over the years, probably because the earlier examples were laboratory phenomena with no clinical relevance [34, 35]. More recently, however, it has been reported that target over-expression probably accounts for acquired resistance to isoniazid and ethambutol in clinical isolates of mycobacteria [36, 37]. In these studies, over-expression of targets through promoter mutations appeared to be responsible for resistance in drug resistant clinical isolates. In a laboratory setting, bacterial target proteins have been over-expressed either to study the function of the enzymes or to examine the interaction of inhibitors and target proteins. Over-expression of prolipoprotein signal peptidase (gene product of lsp) was shown to confer its host cells a high level of resistance to globomycin, an inhibitor of lipoprotein signal peptidase [38]. Turnowsky and colleagues [39] demonstrated that over-expression of EnvM (FabI) protein from the plasmid-borne gene conferred resistance to diazaborine, a specific inhibitor for FabI. Tsay and coworkers [40] screened a thiolactomycin sensitive E. coli strain against an E. coli genomic library for recombinant plasmids that conferred resistance to thiolactomycin, an inhibitor for FabB protein (β-ketoacyl-acyl carrier protein synthase I). They isolated nine plasmids, all of which contain a functional fabB gene. Over-expression of FabB protein appeared to lead to increased resistance to thiolactomycin in E. coli [40]. Apfel and colleagues [41] demonstrated that over-expression of peptide deformylase in a cell reduced its susceptibility to inhibitors specific for the essential target, peptide deformylase.
Recently, there have been several reports of using target over-expression to confirm targets of drugs or inhibitors in a number of microorganisms. Over-expression of farnesyl diphosphate synthase (FDP synthase) in social amoeba Dictyostelium resulted in resistance to aminobisphosphate inhibitors [42]. Butcher and coworkers [43] constructed a pool of Saccharomyces cerevisiae strains each over-expressing a different protein. These strains were grown in the presence of rapamycin and the growth was monitored by microarray. Candidate target proteins of rapamycin were identified [43]. While the approaches by these groups [42, 43] are valuable and robust, they are limited only for use in identification of targets of antifungal compounds. In a recent report using a bacterial system (E. coli), multicopy suppressors were identified for antibacterial compounds as a method of determining the targets of the inhibitors and the mechanisms of resistance [29]. In this study [29], an E. coli random genomic library was screened for clones which could suppress antibacterial activities of inhibitors to a hyperpermeable strain of E. coli. As a result, the majority of suppressors were found to encode the multidrug efflux pump AcrB and a target of two lead compounds was identified as dihydrofolate reductase (DHFR), encoded by the folA gene [29]. This approach of screening genomic libraries for suppressor clones is a significant method of identifying targets of antibacterial inhibitors. However, due to the random nature of genomic libraries and of cloned inserts, it is difficult to determine if an essential gene is structurally intact or functionally expressed in a plasmid clone without extensive validation experiments.
Here we report on a first deliberate effort to clone individual essential genes from E. coli for the sole purpose of constructing an array of E. coli over-expression clones to identify cellular targets of potent antibacterial inhibitors generated from high throughput screening campaigns. As a proof-of-concept, we cloned eight essential genes in E. coli, observed essential protein over-expression in majority of the clones (seven out of eight) and confirmed elevated resistance of clones to their specific antibacterial inhibitors. We demonstrated proof of concept in identification of targets of two known inhibitors. Our results suggest that the approach is very likely a generic one and can provide a viable tool for identification of cellular targets of potent inhibitors and for studies of drug-target interactions. Additionally, with each essential gene clone sequence validated and over-expression demonstrated, we have the choices of two complimentary and corroborating assay formats for the identification of targets of potent inhibitors: the mixed clone method and the individual clone method. Both assay formats have their own advantages. The mixed clone method streamlines the number of growth vessels (microplate wells) used and may amplify even slightest fitness advantages of certain clones in a fitness test environment. The individually arrayed method is advantageous in that the well positions of every clone are known in advance, which enables a direct linkage of growth of a particular well to a target without genetic profiling of clones. In addition, both methods are amenable to robotic adaptation to a high throughput screening setting involving a large number of over-expressing clones and multiple potent antibacterial compounds. Once completed, this array of over-expression E. coli clones and the associated assays would facilitate discovery of novel antibiotics.
Acknowledgements
Funding for this project has been partially provided by a CSUPERB faculty-student collaborative research seed grant, the NIH RIMI Program (P20MD001824) and an NIH MBRS-SCORE grant (S06GM008101) to H. H. Xu. Technical assistance from Peter Chong, David Yang, Deisy Contreras and Marine Cholakyan is greatly appreciated. We are grateful to Anca Segal for sharing plasmid and to F. Zhou and N. McQueen for critical review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chopra I, Hodgson J, Metcalf B, Poste G. The search for antimicrobial agents effective against bacteria resistant to multiple antibiotics. Antimicrob Agents Chemother. 1997;41:497–503. doi: 10.1128/aac.41.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tenover FC. Development and spread of bacterial resistance to antimicrobial agents: an overview. Clin Infect Dis. 2001;33(Suppl 3):S108–15. doi: 10.1086/321834. [DOI] [PubMed] [Google Scholar]
- [3].Finch R. Bacterial resistance--the clinical challenge. Clin Microbiol Infect. 2002;8(Suppl 3):21–32. doi: 10.1046/j.1469-0691.8.s.3.3.x. discussion 33-5.
- [4].Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997;40:135–6. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- [5].Montecalvo MA, Horowitz H, Gedris C, Carbonaro C, Tenover FC, Issah A, Cook P, Wormser GP. Outbreak of vancomycin-, ampicillin-, and aminoglycoside-resistant Enterococcus faecium bacteremia in an adult oncology unit. Antimicrob Agents Chemother. 1994;38:1363–7. doi: 10.1128/aac.38.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Butler JC, Hofmann J, Cetron MS, Elliott JA, Facklam RR, Breiman RF. The continued emergence of drug-resistant Streptococcus pneumoniae in the United States: an update from the Centers for Disease Control and Prevention’s Pneumococcal Sentinel Surveillance System. J Infect Dis. 1996;174:986–93. doi: 10.1093/infdis/174.5.986. [DOI] [PubMed] [Google Scholar]
- [7].Lyytikainen O, Koljalg S, Harma M, Vuopio-Varkila J. Outbreak caused by two multi-resistant Acinetobacter baumannii clones in a burns unit: emergence of resistance to imipenem. J Hosp Infect. 1995;31:41–54. doi: 10.1016/0195-6701(95)90082-9. [DOI] [PubMed] [Google Scholar]
- [8].Projan SJ, Shlaes DM. Antibacterial drug discovery: is it all downhill from here? Clin Microbiol Infect. 2004;10(Suppl 4):18–22. doi: 10.1111/j.1465-0691.2004.1006.x. [DOI] [PubMed] [Google Scholar]
- [9].Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- [10].Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–74. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- [11].Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Danchin A, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–56. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- [12].Haney SA, Alksne LE, Dunman PM, Murphy E, Projan SJ. Genomics in anti-infective drug discovery--getting to endgame. Curr Pharm Des. 2002;8:1099–118. doi: 10.2174/1381612023394845. [DOI] [PubMed] [Google Scholar]
- [13].Gil R, Silva FJ, Pereto J, Moya A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev. 2004;68:518–37. doi: 10.1128/MMBR.68.3.518-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Arigoni F, Talabot F, Peitsch M, Edgerton MD, Meldrum E, Allet E, Fish R, Jamotte T, Curchod ML, Loferer H. A genome-based approach for the identification of essential bacterial genes. Nat Biotechnol. 1998;16:851–6. doi: 10.1038/nbt0998-851. [DOI] [PubMed] [Google Scholar]
- [15].Freiberg C, Wieland B, Spaltmann F, Ehlert K, Brotz H, Labischinski H. Identification of novel essential Escherichia coli genes conserved among pathogenic bacteria. J Mol Microbiol Biotechnol. 2001;3:483–9. [PubMed] [Google Scholar]
- [16].Jana M, Luong TT, Komatsuzawa H, Shigeta M, Lee CY. A method for demonstrating gene essentiality in Staphylococcus aureus. Plasmid. 2000;44:100–4. doi: 10.1006/plas.2000.1473. [DOI] [PubMed] [Google Scholar]
- [17].Zhang L, Fan F, Palmer LM, Lonetto MA, Petit C, Voelker LL, St John A, Bankosky B, Rosenberg M, McDevitt D. Regulated gene expression in Staphylococcus aureus for identifying conditional lethal phenotypes and antibiotic mode of action. Gene. 2000;255:297–305. doi: 10.1016/s0378-1119(00)00325-5. [DOI] [PubMed] [Google Scholar]
- [18].Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Venter JC. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 1999;286:2165–9. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- [19].Reich KA, Chovan L, Hessler P. Genome scanning in Haemophilus influenzae for identification of essential genes. J Bacteriol. 1999;181:4961–8. doi: 10.1128/jb.181.16.4961-4968.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Akerley BJ, Rubin EJ, Novick VL, Amaya K, Judson N, Mekalanos JJ. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc Natl Acad Sci U S A. 2002;99:966–71. doi: 10.1073/pnas.012602299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D’Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabasi AL, Oltvai ZN, Osterman AL. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185:5673–84. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thanassi JA, Hartman-Neumann SL, Dougherty TJ, Dougherty BA, Pucci MJ. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 2002;30:3152–62. doi: 10.1093/nar/gkf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kobayashi K, Ehrlich SD, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100:4678–83. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ji Y, Zhang B, Van, Horn SF, Warren P, Woodnutt G, Burnham MK, Rosenberg M. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science. 2001;293:2266–9. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- [25].Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, C KG, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu Zy ZY, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol. 2002;43:1387–400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- [26].DeVito JA, Mills JA, Liu VG, Agarwal A, Sizemore CF, Yao Z, Stoughton DM, Cappiello MG, Barbosa MD, Foster LA, Pompliano DL. An array of target-specific screening strains for antibacterial discovery. Nat Biotechnol. 2002;20:478–83. doi: 10.1038/nbt0502-478. [DOI] [PubMed] [Google Scholar]
- [27].Freiberg C, Brunner NA, Schiffer G, Lampe T, Pohlmann J, Brands M, Raabe M, Habich D, Ziegelbauer K. Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity. J Biol Chem. 2004;279:26066–73. doi: 10.1074/jbc.M402989200. [DOI] [PubMed] [Google Scholar]
- [28].Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358–61. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- [29].Li X, Zolli-Juran M, Cechetto JD, Daigle DM, Wright GD, Brown ED. Multicopy suppressors for novel antibacterial compounds reveal targets and drug efflux susceptibility. Chem Biol. 2004;11:1423–30. doi: 10.1016/j.chembiol.2004.08.014. [DOI] [PubMed] [Google Scholar]
- [30].Chopra I. Over-expression of target genes as a mechanism of antibiotic resistance in bacteria. J Antimicrob Chemother. 1998;41:584–8. doi: 10.1093/jac/41.6.584. [DOI] [PubMed] [Google Scholar]
- [31].Krause M, Ruckert B, Lurz R, Messer W. Complexes at the replication origin of Bacillus subtilis with homologous and heterologous DnaA protein. J Mol Biol. 1997;274:365–80. doi: 10.1006/jmbi.1997.1404. [DOI] [PubMed] [Google Scholar]
- [32].Sambrook J, Russell DW. Molecular cloning: a laboratory manual. third ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- [33].Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Hogenauer G, Turnowsky F. Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli. J Biol Chem. 1994;269:5493–6. [PubMed] [Google Scholar]
- [34].Cohen G, Jacob F. Inhibition of the synthesis of the enzymes participating in the formation of tryptophan in Escherichia coli. C R Hebd Seances Acad Sci. 1959;248:3490–2. [PubMed] [Google Scholar]
- [35].Reitz RH, Slade HD, Neuhaus FC. The biochemical mechanisms of resistance by streptococci to the antibiotics D-cycloserine and O-carbamyl-D-serine. Biochemistry. 1967;6:2561–70. doi: 10.1021/bi00860a038. [DOI] [PubMed] [Google Scholar]
- [36].Morris S, Bai GH, Suffys P, Portillo-Gomez L, Fairchok M, Rouse D. Molecular mechanisms of multiple drug resistance in clinical isolates of Mycobacterium tuberculosis. J Infect Dis. 1995;171:954–60. doi: 10.1093/infdis/171.4.954. [DOI] [PubMed] [Google Scholar]
- [37].Alcaide F, Pfyffer GE, Telenti A. Role of embB in natural and acquired resistance to ethambutol in mycobacteria. Antimicrob Agents Chemother. 1997;41:2270–3. doi: 10.1128/aac.41.10.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tokunaga M, Loranger JM, Wu HC. Isolation and characterization of an Escherichia coli clone overproducing prolipoprotein signal peptidase. J Biol Chem. 1983;258:12102–5. [PubMed] [Google Scholar]
- [39].Turnowsky F, Fuchs K, Jeschek C, Hogenauer G. envM genes of Salmonella typhimurium and Escherichia coli. J Bacteriol. 1989;171:6555–65. doi: 10.1128/jb.171.12.6555-6565.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tsay JT, Rock CO, Jackowski S. Overproduction of beta-ketoacyl-acyl carrier protein synthase I imparts thiolactomycin resistance to Escherichia coli K-12. J Bacteriol. 1992;174:508–13. doi: 10.1128/jb.174.2.508-513.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Apfel CM, Locher H, Evers S, Takacs B, Hubschwerlen C, Pirson W, Page MG, Keck W. Peptide deformylase as an antibacterial drug target: target validation and resistance development. Antimicrob Agents Chemother. 2001;45:1058–64. doi: 10.1128/AAC.45.4.1058-1064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sugden CJ, Roper JR, Williams JG. Engineered gene over-expression as a method of drug target identification. Biochem Biophys Res Commun. 2005;334:555–60. doi: 10.1016/j.bbrc.2005.06.117. [DOI] [PubMed] [Google Scholar]
- [43].Butcher RA, Bhullar BS, Perlstein EO, Marsischky G, LaBaer J, Schreiber SL. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat Chem Biol. 2006;2:103–9. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]