

Abstract
A study of two dihydroxybenzoic acid isomers shows that computational methods can be used to predict hydrate formation, the compound : water ratio and hydrate crystal structures. The calculations also help identify a novel hydrate found in the solid form screening that validates this study.
Changes in hydration states of crystalline compounds throughout the manufacturing process often cannot be avoided. The identification of hydrates (crystals that incorporate water into the structure) and their hydration/dehydration conditions is essential in the development of pharmaceutical compounds. The presence of water in the crystal lattice may lead to very different physicochemical properties, and so influence the bioavailability of a drug compound.1 Hydrate formation is a widespread phenomenon; it is believed that it might occur for up to three quarters of pharmaceutical compounds2 and is observed for a third of organic molecules.3–5 The probability of water inclusion is particularly high when charged groups (salts) or polar groups, such as carbonyl ( ), ether (C–O–C), hydroxyl (O–H) and primary amine (N–H) groups are present,2 which is usually the case in pharmaceuticals.
), ether (C–O–C), hydroxyl (O–H) and primary amine (N–H) groups are present,2 which is usually the case in pharmaceuticals.
Even for the types of molecules prone to hydrate formation, it is not certain that any hydrates will be formed, let alone the stoichiometry (the compound : water ratio). For example, 2,4-dihydroxybenzoic acid (2,4-DHB) forms two hydrates, a stable hemihydrate (QIVTUK)6 and a metastable monohydrate whose structure could not be determined experimentally.7 In contrast, the 2,5 isomer of dihydroxybenzoic acid (2,5-DHB) forms no hydrates, as shown by the accompanying solid form screen (ESI†) which found four novel solvates.
Multiple analytical techniques have to be used to characterise the solid state of any compound. Over the last decade methodologies for computationally generating crystal structures have progressed considerably,8 becoming a useful complement to experimental solid form screening.9,10 crystal structure prediction calculations are now being successfully performed on multi-component systems, such as co-crystals,8,11 solvates12–16 and salts.17,18 Evaluating whether the co-crystal or solvate will crystallise and its stoichiometry is an additional challenge. There have been successes in rationalising the experimentally observed stoichiometries for co-crystals19 and acetic acid solvates.13,14 Only four attempts to predict stoichiometric hydrates‡ have been undertaken,15,16,20 including the hydrate set as a target in the last blind test of crystal structure prediction.21 In order to complete and understand the differing hydration behaviour seen in our screens for solid state forms of the pharmaceutically relevant molecules 2,4- and 2,5-DHB (ESI†), we perform the first computational organic hydrate study which considers different stoichiometries.
Hypothetical hemi-, mono- and dihydrate crystal structures were generated in an extensive search22 and the lattice energies evaluated accurately,23 allowing for minor conformational change24 and molecular polarisation25 within the crystal, as detailed in the ESI.† The stability of the different hydrate stoichiometries for both isomers relative to the anhydrate and ice was assessed from the stabilisation energy per water molecule (Estab, Fig. 1). Assuming that hydrate formation is thermodynamically driven and that this can be estimated by relative lattice energies, we can predict which of the hydrates are more stable than the anhydrate by comparing Estab with the lattice energy of ice. Using the same computational model, Elatt (ice) varies from −66.61 to −70.11 kJ mol−1 for the ordered polymorphs II, VIII, XI and XV (ESI†).26–29 This leads to a comparison with a range of lattice energies rather than a single value.
Fig. 1.

Definition of the molecules and the starting conformations used in the searches for possible hydrate structures, and the definition of the stabilisation energy Estab from the lattice energy§ of each predicted hydrate structure and the given lattice energy of the experimentally most stable DHB anhydrous polymorph (AH) as determined by thermal and slurrying measurements (ESI†). Hydrate formation is thermodynamically expected if Estab < Elatt (ice).
Fig. 2 shows that 2,5-DHB is expected not to form a hydrate, but that 2,4-DHB can form both mono- and hemihydrates, but not a dihydrate, in agreement with the extensive experimental screen.
Fig. 2.

Crystal energy landscape of dihydroxybenzoic acid hydrates plotted by stabilisation energy per water molecule in the lattice, Estab (Fig. 1). Each symbol denotes a crystal structure (Tables S9 and S10, ESI†) which is a lattice energy minimum. The range of ice polymorph energies is in yellow. Only the most stable structures are shown, so the upper region of the hemihydrate landscape omits many 2,4-DHB structures.
The structure of the known 2,4-DHB hemihydrate (Fig. 3) was found as second in energy (Fig. 2), although the difference between Estab (Fig. 1) and Elatt for the most stable ice polymorph, VIII, is small. The global minimum of the hemihydrate search is closely related to the most stable anhydrate structure (Fig. S7, ESI†).
Fig. 3.

Overlay of the predicted (green) and experimentally determined 2,4-DHB hemihydrate (red), with a rmsd15¶ of 0.218 Å.
Several 2,4-DHB monohydrates were predicted to be thermodynamically feasible, and could be used to propose a structure for the unsolved, metastable monohydrate (Fig. 4). The experimental powder X-ray diffraction pattern of the monohydrate matches that simulated from the most stable structure as well as the experimental hemihydrate powder pattern matches that simulated from the corresponding calculated hemihydrate (Fig. 5). Further spectroscopic and X-ray diffractometric|| evidence (ESI†) confirms that the dimerised carboxylic acid group and acid⋯water interactions (Fig. 4), lattice parameters and space group of the most stable computed structure correspond to the monohydrate. However, since the searches with different p-OH proton conformations (Fig. 1) also generated low energy structures with very similar diffraction patterns (Fig. 5), differing only in the positions of the p-OH and adjacent water protons but not in the packing (Fig. S11, ESI†), we cannot eliminate the possibility of variations in the proton positions or proton disorder. Characterising the proton positions in the experimental structure is not currently possible, because the metastability prevents the growth of crystals suitable for single crystal diffraction studies.
Fig. 4.
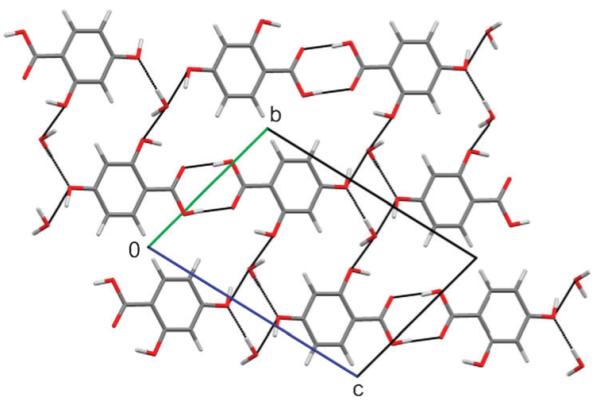
The most stable computed structure of 2,4-DHB monohydrate. Black lines indicate the hydrogen bond interactions.
Fig. 5.
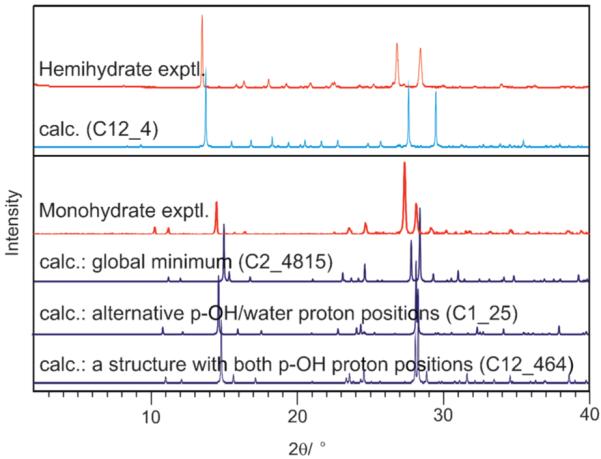
Experimental powder X-ray diffraction patterns (hemi- and monohydrate) obtained at room temperature compared with simulated patterns (λ = 1.5418 Å) for the calculated structures (Table S10, ESI†). For the monohydrate, the diffractogram of the proposed structure (Fig. 4) is contrasted with other structures that differ only in the p-OH and adjacent water proton positions (ESI†, 6.3).
The two isomers differ in the hydrogen bonding motifs of the computed hydrate structures. The carboxylic acid dimer is found in nearly all 2,4-DHB low-energy hydrate structures, whereas two thirds of the 2,5-DHB low-energy hydrates have water molecules hydrogen bonded to the acid group. This is significant because both isomers form the expected carboxylic acid dimer in their anhydrous forms. Despite the structural differences, the hydrates of the two isomers are comparable in lattice energy (Tables S9 and S10, ESI†). The dominant reason for the different hydrate formation propensity (Fig. 2) is that the hydrate forming isomer, 2,4-DHB, is less stable and less dense in the anhydrous state than the 2,5-DHB isomer.
We have reported the first successful crystal structure prediction of the compound : water ratio of stoichiometric hydrates. Moreover, a structure for the 2,4-DHB monohydrate could be proposed which is consistent with extensive experimental data. The experimentally observed stability order of the two 2,4-DHB hydrate stoichiometries is not reproduced. Improving the calculations to give more accurate free energies, including possible proton disorder,26 would help increase confidence in thermodynamic versus possible kinetic arguments for which hydrates are observed. However, the calculation of the crystal energy landscapes of the potential hydrate structures of these isomers provide considerable support and rationalisation of the extensive experimental hydrate screening.
Supplementary Material
Acknowledgments
DEB acknowledges financial support from the Austrian Science Fund (FWF, project No. J2897-N17). The authors would like to thank Ulrich J. Griesser for access to the instrumentation in the University of Innsbruck. Other resources are from EPSRC funding of Control and Prediction of the Organic Solid State www.cposs.org.uk.
Notes
Electronic supplementary information (ESI) available: Polymorph screen, identification and characterisation of 2,4- and 2,5-DHB solid forms, computer model for lattice energies, the crystal energy landscapes. See DOI: 10.1039/c1cc10762c
Stoichiometric hydrates (solvates) are regarded as molecular compounds and contain a fixed solvent : compound ratio, in contrast to non-stoichiometric solvates, a type of inclusion compounds.27
Lattice energy is the energy difference between the molecules in the lattice and when infinitely separated in their most stable conformation.
The reproduction of the crystal structures was evaluated by the optimal root-mean square overlay of all non-hydrogen atoms in a 15 molecule coordination cluster (rmsd15).28
PXRD data were collected at room temperature on a Panalytical X’Pert PRO diffractometer using Cu-Kα1,2 radiation (2θ = 2–40°, step size = 0.013 Å, 40 s per step). Refined unit cell parameters of 2,4-DHB monohydrate a, b, c (Å) = 3.8037, 8.9263, 11.5873; α, β, γ (°) = 75.177, 89.016, 81.601; Pawley χ2 = 1.954 (Rietica29). The PXRD data quality did not allow a structure determination or refinement of atomic positions from the proposed structures (ESI†).
references
- 1.Khankari RK, Grant DJW. Thermochim. Acta. 1995;248:61–79. [Google Scholar]
- 2.Infantes L, Chisholm J, Motherwell S. CrystEngComm. 2003;5:480–486. [Google Scholar]
- 3.Brittain HG. Polymorphism in Pharmaceutical Solids. Informa Healthcare; New York: 2009. [Google Scholar]
- 4.Threlfall TL. Analyst. 1995;120:2435–2460. [Google Scholar]
- 5.Stahly GP. Cryst. Growth Des. 2007;7:1007–1026. [Google Scholar]
- 6.Horneffer V, Dreisewerd K, Ludemann HC, Hillenkamp F, Lage M, Strupat K. Int. J. Mass Spectrom. 1999;187:859–870. [Google Scholar]
- 7.Braun DE, Karamertzanis PG, Arlin JB, Florence AJ, Kahlenberg V, Tocher DA, Griesser UJ, Price SL. Cryst. Growth Des. 2011;11:210–220. doi: 10.1021/cg101162a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Day GM, Cooper TG, Cruz-Cabeza AJ, Hejczyk KE, Ammon HL, Boerrigter SXM, Tan J, Della Valle RG, Venuti E, Jose J, Gadre SR, Desiraju GR, Thakur TS, van Eijck BP, Facelli JC, Bazterra VE, Ferraro MB, Hofmann DWM, Neumann M, Leusen FJJ, Kendrick J, Price SL, Misquitta AJ, Karamertzanis PG, Welch GWA, Scheraga HA, Arnautova YA, Schmidt MU, van de Streek J, Wolf A, Schweizer B. Acta Crystallogr., Sect. B. 2009;65:107–125. doi: 10.1107/S0108768109004066. [DOI] [PubMed] [Google Scholar]
- 9.Price SL. Acc. Chem. Res. 2009;42:117–126. doi: 10.1021/ar800147t. [DOI] [PubMed] [Google Scholar]
- 10.Price SL. Adv. Drug Delivery Rev. 2004;56:301–319. doi: 10.1016/j.addr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Karamertzanis PG, Kazantsev AV, Issa N, Welch GWA, Adjiman CS, Pantelides CC, Price SL. J. Chem. Theor. Comput. 2009;5:1432–1448. doi: 10.1021/ct8004326. [DOI] [PubMed] [Google Scholar]
- 12.Cruz-Cabeza AJ, Day GM, Motherwell WDS, Jones W. J. Am. Chem. Soc. 2006;128:14466–14467. doi: 10.1021/ja065845a. [DOI] [PubMed] [Google Scholar]
- 13.Cruz-Cabeza AJ, Day GM, Jones W. Chem.–Eur. J. 2008;14:8830–8836. doi: 10.1002/chem.200800668. [DOI] [PubMed] [Google Scholar]
- 14.Cruz-Cabeza AJ, Karki S, Fabian L, Friscic T, Day GM, Jones W. Chem. Commun. 2010;46:2224–2226. doi: 10.1039/b922955h. [DOI] [PubMed] [Google Scholar]
- 15.van Eijck BP, Kroon J. Acta Crystallogr., Sect. B. 2000;56:745. doi: 10.1107/s0108768100000276. [DOI] [PubMed] [Google Scholar]
- 16.Hulme AT, Price SL. J. Chem. Theor. Comput. 2007;3:1597–1608. doi: 10.1021/ct700045r. [DOI] [PubMed] [Google Scholar]
- 17.Karamertzanis PG, Price SL. J. Phys. Chem. B. 2005;109:17134–17150. doi: 10.1021/jp052465n. [DOI] [PubMed] [Google Scholar]
- 18.Karamertzanis PG, Anandamanoharan PR, Fernandes P, Cains PW, Vickers M, Tocher DA, Florence AJ, Price SL. J. Phys. Chem. B. 2007;111:5326–5336. doi: 10.1021/jp068530q. [DOI] [PubMed] [Google Scholar]
- 19.Habgood M, Price SL. Cryst. Growth Des. 2010;10:3263–3272. [Google Scholar]
- 20.Anderson KM, Day GM, Paterson MJ, Byrne P, Clarke N, Steed JW. Angew. Chem., Int. Ed. 2008;47:1058–1062. doi: 10.1002/anie.200703785. [DOI] [PubMed] [Google Scholar]
- 21.Bardwell DA, Adjiman CS, Ammon HL, Arnautova EA, Bartashevich E, Boerrigter SXM, Braun DE, Cruz-Cabeza AJ, Day GM, Della Valle RG, Desiraju GR, van Eijck BP, Facelli JC, Ferrao MD, Grillo D, Habgood M, Hofmann DWM, Hofmann F, Jose J, Karamertzanis PG, Kazantsev AV, Kendrick J, Kuleshova LN, Leusen FJJ, Maleev A, Misquitta AJ, Mohamed S, Needs RJ, Neumann MA, Nikylov D, Orendt AM, Pal R, Pantelides CC, Pickard CJ, Price LS, Price SL, Scheraga HA, van de Streek J, Thakur TS, Tiwari S, Venuti E, Zhitkov I. Acta Crystallogr., Sect. B. 2011 in preparation. [Google Scholar]
- 22.Karamertzanis PG, Pantelides CC. Mol. Phys. 2007;105:273–291. [Google Scholar]
- 23.Price SL, Leslie M, Welch GWA, Habgood M, Price LS, Karamertzanis PG, Day GM. Phys. Chem. Chem. Phys. 2010;12:8478–8490. doi: 10.1039/c004164e. [DOI] [PubMed] [Google Scholar]
- 24.Kazantsev AV, Karamertzanis PG, Adjiman CS, Pantelides CC. ch. 1. In: Adjiman CS, Galindo A, editors. Molecular System Engineering. WILEY-VCH Verlag GmbH & Co.; Weinheim: 2010. pp. 1–42. [Google Scholar]
- 25.Cooper TG, Hejczyk KE, Jones W, Day GM. J. Chem. Theor. Comput. 2008;4:1795–1805. doi: 10.1021/ct800195g. [DOI] [PubMed] [Google Scholar]
- 26.Habgood M, Grau-Crespo R, Price SL. Phys. Chem. Chem. Phys. 2011 doi: 10.1039/c1cp20249a. in press. [DOI] [PubMed] [Google Scholar]
- 27.Griesser UJ. In: Polymorphism: In the Pharmaceutical Industry. Hilfiker Rolf., editor. Wiley-VCH; Germany: 2006. pp. 211–233. [Google Scholar]
- 28.Chisholm JA, Motherwell S. J. Appl. Crystallogr. 2005;38:228–231. [Google Scholar]
- 29.Hunter B. Rietica—a visual Rietveld program. International Union of Crystallography; 1998. Commission on Powder Diffraction Newsletter No. 20. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.