

Abstract
Mitochondrial biogenesis is a complex process involving the coordinated expression of mitochondrial and nuclear genes, the import of the products of the latter into the organelle and turnover. The mechanisms associated with these events have been intensively studied in the last twenty years and our understanding of their details is much improved. Mitochondrial biogenesis requires the participation of calcium signaling that activates a series of calcium dependent protein kinases that in turn activate transcription factors and coactivators such as PGC-1α that regulates the expression of genes coding for mitochondrial components. In addition, mitochondrial biogenesis involves the balance of mitochondrial fission-fusion. Mitochondrial malfunction or defects in any of the many pathways involved in mitochondrial biogenesis can lead to degenerative diseases and possibly play an important part in aging.
Introduction
In eukaryotes, cellular respiration, energy production and other metabolic processes take place in specialized double membrane organelles, containing their own genome, the mitochondria. Mitochondria play a very important role in cell metabolism, not only by energy generation but also by being a major site for production of reactive oxygen species (ROS) and being a key player in apoptosis and calcium homeostasis. Malfunction of mitochondria has been associated with numerous degenerative diseases and aging [1, 2]. Table 1 shows some of the diseases associated with mitochondrial biogenesis that are discussed in this paper.
Table 1.
Diseases Associated with Mitochondrial Biogenesis
| Mutations | Diseases | References |
|---|---|---|
| mtDNA mutations | Multiple respiratory chain defects | Reviewed in [1, 2, 108] |
| Helicase TWINKLE mutations | Autosomal Dominant progressive external ophthalmoplegia (PEO). Sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO) | [9, 10] |
| Polymerase γ mutations | Catalytic subunit: PEO, SANDO, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Pol2 subunit : PEO | Reviewed in [8] |
| E1a subunit of pyruvate dehydrogenase (mutations in mitochondrial targeting sequencing) | Pyruvate dehydrogenase deficiency | [46] |
| Alanine/glyoxylate aminotransferase (mistargeting of peroxisomal protein to mitochondria) | Alanine/glyoxylate aminotransferase deficiency | [47] |
| Superoxide dismutase (defect in Import efficiency) | Severe alcoholic liver disease | [48] |
| Mutation in Tim8 (affects biogenesis of inner membrane translocase TIM23) | Deafness dystonia syndrome or Mohr- Tranebjaerg syndrome | [49] |
| Mutation on Pam18 (motor protein of TIM23 complex) | Dilated cardiomyopathy with Ataxia | [50] |
| HSP60 mutations | Spastic paraplegia-13 | [51] |
| Mitofusin 2 mutations | Charcot-Marie-Tooth 2A | [90] |
| Opa1 mutations | Autosomal dominant optic atrophy | [91, 92] |
The origin of this organelle in eukaryotes is explained by the endo-symbiont theory, which proposes that mitochondria originated from bacteria that were incorporated into a host cell and maintained during evolution [3, 4]. This hypothesis is backed by substantial experimental evidence, including the similarities between the bacterial and mitochondrial translational apparatus [5].
Mitochondrial DNA
Figure 1A illustrates the structure and expression of the human mitochondrial DNA (mtDNA), a double stranded circular molecule of 16,569 bp encoding 2 ribosomal RNAs, 22 transfer RNAs and 13 proteins that form part of the multi subunits complexes of the oxidative phosphorylation system (7 subunits of complex I, 1 subunit of complex III, 3 subunits of complex IV and 2 subunits of complex V).
Figure 1. Structure and expression of the human mitochondrial DNA.
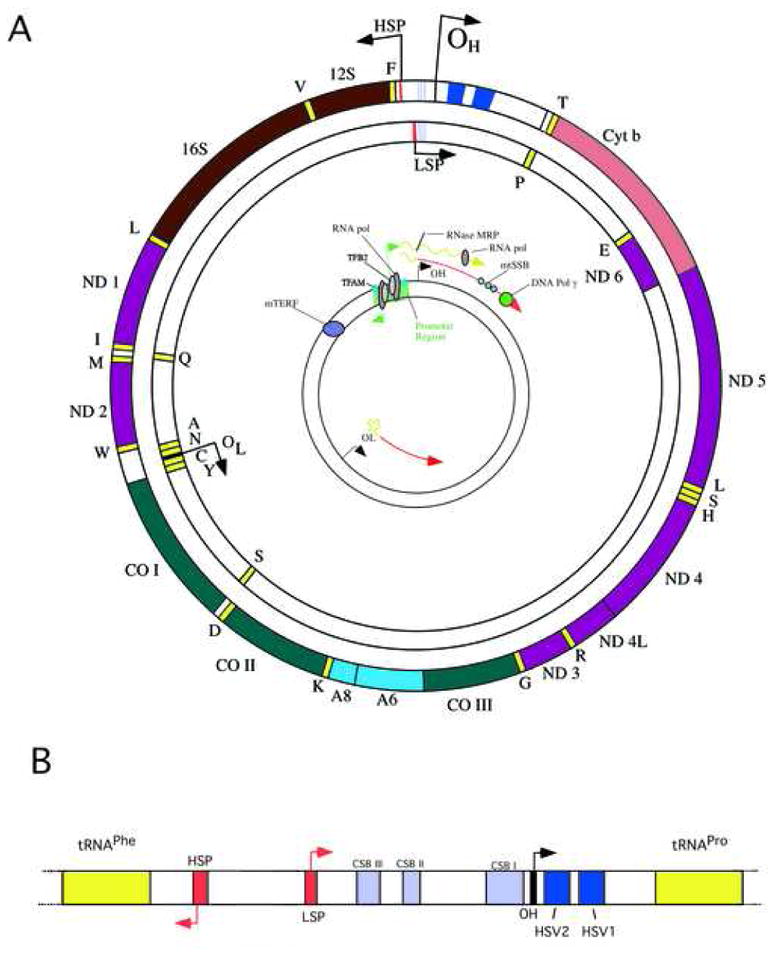
The 16,569 bp human mtDNA (panel A) showing 13 protein coding genes as well as 2 rRNA- and 22 tRNA-coding genes. Genes coding for subunits of complex I (ND1-ND6), complex III (Cyt b), complex IV (COX I-COX III) and complex V (A8 and A6) are shown by different colors. The insert on panel A illustrates one of the mechanisms proposed for mtDNA replication. It also shows a consensus model of polycyctronic transcription, including the approximately binding sites for the mitochondrial RNA polymerase, the mitochondrial transcription factor TFAM, the RNA processing enzyme RNAse MRP and the transcription termination factor mTERF. The origins of replication for the H- and L- (OH and OL) strands are also shown. The structure of the regulatory D-loop region is shown in panel B, including the approximate position of the conserved sequence boxes believed to play a role in replication and RNA primer processing. It also shows the location of the two hypervariable regions (HSV1 and HSV2) commonly used for evolutionary studies.
The mammalian mtDNA contains few non-coding sequences, the largest being the D-loop or displacement-loop, which contains promoters and origins of replication (Figure 1B). Protein coding genes have no intronic regions [6]. Alkaline gradient centrifugation experiments allowed the separation of the mtDNA double strands into a heavy (H-strand) and a light (L-strand) due to their differential content of guanosine and cytidine. The H-strand encodes for the 2 rRNAs, 12 of the polypeptides and 14 of the tRNAs whereas the L-strand encodes for only one of the polypeptides (ND6) and 8 tRNAs. The genetic code of mitochondria differs from the nuclear universal code. The TGA codon codes for tryptophan instead of stop, the AGA and AGG code for stop instead of arginine and the ATA codes for methionine instead of isoleucine [6].
The mtDNA is associated with several proteins packed in structures denominated nucleoids, which are also associated with the inner mitochondrial membrane. In the last few years progress has been made on identifying the composition of these structures. Several studies showed that yeast nucleoids include proteins that bind DNA and are associated with replication and transcription such as the mitochondrial transcription factor A (TFAM), helicase TWINKLE, polymerase γ (Polγ) and the mitochondrial single-strand binding protein (mtSSB); proteins associated with metabolism such as the E2 subunit of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, aconitase; and some cytoskeletal proteins [7] Mutations in components of the replication machinery are associated with mitochondrial depletion syndromes (reviewed by [8]).
Mutations in the PEO1 gene encoding the helicase TWINKLE have been reported in some patients with autosomal dominant progressive external ophthalmoplegia (PEO) and in one patient with sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO) [9, 10]. In addition, mutations in the catalytic subunit of polymerase γ (have been associated with a wide range of disease such as PEO, SANDO, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), parkinsonism and Alpers syndrome (review by [8]) whereas mutations in the gene POL2 encoding one of the subunits of Polγ, is associated with autosomal dominant PEO [11].
Recently, Wang and Bogenhagen [12] found a novel protein, DHX30 (DEAH-box helicase) to be present in nucleoids purified from HeLa cells [12], In addition, the authors also found ATAD3, a member of the AAA+-ATPases, associated with nucleoids. Further studies in rat liver mitochondria by He et al. [13] found that ATAD3 binds mtDNA, preferentially in the D-loop region, and it appears to be involved in nucleoid organization and segregation [13, 14].
Despite these advances, understanding of the composition and structure of mitochondrial nucleoids is far from complete. There are several proteins associated with mtDNA metabolism that are not easily detectable in nucleoids. These include the mitochondrial transcription factors B1 and B2 and regulators of transcription from the mTERF family [12]. The mitochondrial transcription factors B1 and B2 have been found to be required for mtDNA transcription (at least one of them) and may also have a role in methylation of DNA or RNA [15]. TFAM seems to be present at relatively high levels, and by itself is able to organize the mtDNA in “nucleoid-like” structures [16].
Mitochondrial DNA replication and transcription
The mtDNA replication is linked to the transcription of the light strand promoter (LSP) that serves as an RNA primer to initiate DNA replication at the origin of replication of the H-strand (OH) located in the D-loop (inset on figure 1A) [17]. To form the RNA primer, transcription of LSP is initiated by the mitochondrial RNA polymerase (POLRMT) and then to produce a 3’ end of these RNA primer, an endoribonuclease mitochondrial RNA processing (MRP) is required [18, 19]. The mtDNA replication is carried out by POLγ, with 3’–5’ exonuclease and 5’-deoxyribose phosphate lyase activities, assisted by the helicase TWINKLE that unwinds the DNA duplex, and by single strand binding proteins (mtSSB) that keep the DNA in a single stranded form.
The exact mechanism for DNA replication is still unknown but two models have been proposed. The first model proposes that the replication of mtDNA is unidirectional and asynchronous (inset in figure 1A) [20, 21]. The synthesis of the leading-strand starts at the OH and proceeds displacing the parental heavy strand until it reaches and exposes the origin of replication of the other strand (OL), that is activated and the replication of the lagging- strand starts in the opposite direction. The resulting two new DNA molecules are ligated to form the circular double strand mtDNA [20]. The second model involves the coupled replication of both leading and lagging strands [22].
The mtDNA transcription starts from the promoters present in each strand (HSP and LSP) producing polycistronic RNAs. In the HSP there are two sites of initiation, the H1 site, that produces a short transcript containing the 12S RNA, 16S RNA and the tRNAleu, and the H2 site, that produces a polycistronic transcript of the length of the full genome, whereas in the LSP there is only one initiation site [23–25]. The transcription is carried out by a mitochondrial RNA polymerase (POLRMT) assisted by a mitochondrial transcription factor A (TFAM), that recruits POLRMT to the promoter site, and by mitochondrial transcription factor B1 or B2 (TFB1M and TFB2M; Figure 1A). The termination of the H1 short transcript is dependent on the mitochondrial transcription terminator factor (MTERF) ([26] reviewed in [27, 28]). Recently, Park et al. [29] identified the function of a new transcription terminator factor, MTERF3, as a negative regulator of transcription [29].
Mitochondrial Protein Import
Mitochondrial proteomics in humans has estimated approximately 1500 proteins with a very small portion of these proteins encoded by the mtDNA [30, 31]. Therefore, the majority of the mitochondrial proteins are encoded by the nuclear DNA and imported into the organelle. Progress on the knowledge of the components and the mechanisms of protein transport into the mitochondria have been spearheaded by studies performed in fungi (Neurospora crassa and Saccharomyces cerevisiae). In many instances, the mammalian counterparts have not been identified yet.
Distribution of mitochondrial proteins into the appropriate compartment in the organelle is a very complex process and the targeting signals for sorting are not obvious. Depending on the mitochondrial location of the protein (matrix, inner mitochondrial membrane, intermembrane space or outer mitochondrial membrane) there are different import pathways. We briefly describe each scenario but for specific details refer to recent reviews [32, 33].
Matrix proteins: these proteins are synthesized in cytoplasmic ribosomes as precursors containing a targeting signal located in the amino terminal end. This signal is about 10–80 amino acids long with no consensus in the primary structure and composed of hydrophobic and positively charged amino acids that can form amphipathic helices. Once the protein is inside the mitochondria this amino terminal sequence is cleaved by mitochondrial processing peptidases to generate the mature form of the imported protein. There are cases where the targeting sequence is located internally or in the carboxy terminal end of the protein. Protein precursors synthesized in the cytoplasm are associated with cytoplasmic chaperones (e.g. Hsp70 and Hsp90), which keep them in an unfolded state and competent for translocation posttranslationally into the mitochondria, while other proteins seem to be imported cotranslationally [34].
The import of the matrix proteins is mediated by two translocase complexes, one located in the outer mitochondrial membrane (translocase of the outer membrane or TOM) and one in the inner mitochondrial membrane (translocase of the inner membrane or TIM23) that function in a concerted manner. The TOM complex is formed by seven proteins (Tom70, Tom40, Tom22, Tom20, Tom7, Tom6 and Tom5) with specialized functions: 1) surface receptor function (Tom70, Tom22 and Tom20) that recognize the precursor protein and interacts with cytosolic chaperones, and 2) import pore function (Tom40, Tom5, Tom6 and Tom7) that can form up to 3 pores in the holocomplex. Once the protein has crossed the TOM complex it is recognized by the TIM23 complex that is formed by Tim50, Tim23, Tim21, Tim17 and by the motor components that drive the translocation (Tim44, Tim16/Pam16, Tim14/Pam18, Mge1 and mtHsp70). This translocation process is dependent on ATP and a membrane potential. The final step in the matrix import consists in an active mtHSP70 function. Although it is not clear if the pulling of the preprotein into the matrix by mtHSP70 is done by an active conformational change or by a Brownian movement ratchet [35], recent evidence has favored the latter [36].
Inner membrane proteins: there are 3 different ways for inner membrane proteins to be translocated and inserted into the inner membrane. 1) Proteins that contain several transmembrane domains (with negative charges), are synthesized with a presequence and translocated via TOM-TIM23 to the matrix where the presequence is cleaved and they are “exported” into the membrane by Oxa1 [37]. 2) Proteins that contain one transmembrane domain (usually with more charged and hydrophobic residues in the carboxy terminus, than the proteins inserted via Oxa1) are translocated through the TOM complex, pass to the TIM23 complex, get arrested, transferred laterally and integrated into the inner membrane where the targeting sequence is cleaved. 3) Proteins that contain hydrophobic residues and an even number of transmembrane domains are first translocated by the TOM complex and then are intercepted by the small Tim proteins (small polypeptides Tim 9, Tim10 and Tim12, containing Cx3C motifs that form hexamers) in the inner membrane space [38] and then transferred to another translocase, the TIM22 complex in the inner membrane that inserts the protein into the membranes in a membrane potential dependent manner. The TIM22 complex is formed by 3 membrane proteins Tim54, Tim22 and Tim18 [39].
Import of outer membrane proteins: many of these proteins contain a β-barrel structure and they use the TOM complex to pass through the outer membrane, then are guided by the small Tim proteins to another complex located also in the outer membrane, the translocase of outer membrane β-barrel proteins (TOB) or sorting and assembly machinery (SAM) complex (TOB/SAM), that inserts the protein into the membrane. The TOB/SAM complex is formed by Tom55 (also called Sam50 or Tom50), Tob38 (Sam35 or Tom38) and by Mas37 (Sam37 or Tom37) [40, 41].
Import of intermembrane space proteins: some proteins contain two signal sequences allowing the insertion into the inner membrane via TOM-TIM23 complex and then the mature protein is formed by proteolysis and released into the inner membrane, whereas other proteins are translocated by the TOM complex and then folded in the intermembrane space by the help of Mia40 and Erv1 [42–45]. Figure 2 summarizes the current knowledge of the mitochondrial import machinery.
Figure 2. Protein import into mitochondria.
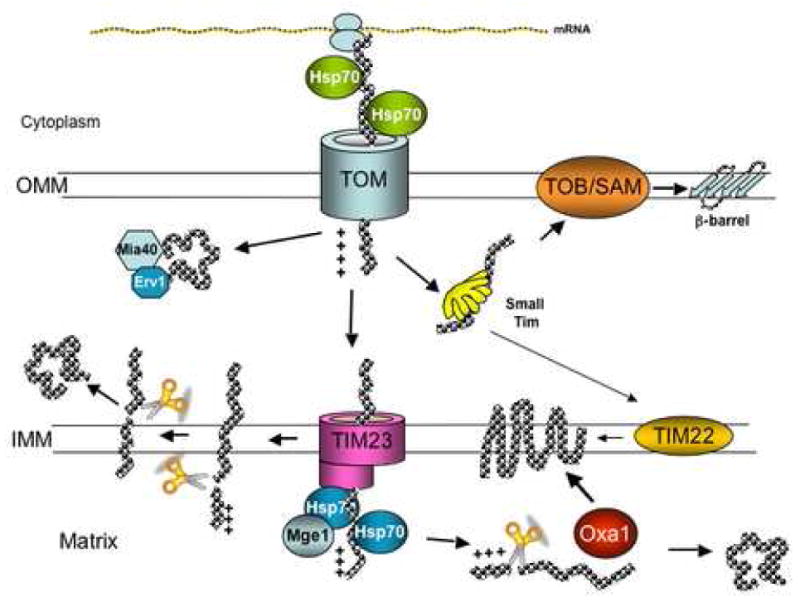
Nuclear encoded proteins are imported into mitochondria by different pathways depending on their final destination. Matrix proteins, synthesized as precursors containing a targeting sequence in the amino terminus are translocated through TOM and TIM23-Hsp70/Mge1 complexes and once in the matrix the targeting sequence is cleaved by mitochondrial proteases to render the mature form. Inner membrane proteins are translocated by TOM and TIM23 but get arrested in the latter, transferred laterally into the membrane and have their targeting sequence cleaved. Other inner membrane proteins are imported into the matrix by TOM and TIM23 and inserted into the inner membrane with the help of Oxa1. In addition, very hydrophobic inner membrane proteins with even number of transmembrane domains are translocated through TOM, then passed to TIM22 via small Tim proteins and inserted into the membrane. Intermembrane space (IMS) proteins are transported via TOM-TIM23 and laterally transferred to inner membrane, where the presequence is cleaved and the protein is released into the intermembrane space. Other IMS proteins are translocated through the TOM complex and then folded in the intermembrane space with the help of Mia40 and Erv1 chaperones. Outer membrane proteins are translocated through the TOM complex, guided by the small Tim proteins to the TOB/SAM complex and inserted into the membrane. See text for more details.
There are several diseases associated with errors in the import of mitochondrial proteins such as pyruvate dehydrogenase deficiency (mutations in the mitochondrial targeting sequence of subunit E1α [46]), Alanine/glyoxylate aminotransferase deficiency (peroxisomal protein that is mistargeted into the mitochondria [47]), severe alcoholic liver disease (caused by defects in the import efficiency of the genetic dimorphic manganese superoxide dismutase [48]), Deafness Dystonia Syndrome or Mohr-Tranebjaerg Syndrome (caused by mutations in one of the small Tim proteins, Tim8, that result in an impaired biogenesis of the TIM23 complex [49]), Dilated cardiomyopathy with Ataxia (caused by defects in Pam18, one of the motor proteins of the TIM23 complex [50]) and Spastic Paraplegia-13 (caused by defects in the mitochondrial Hsp60 causing disruption in protein folding in the matrix [51]). For details see review by MacKenzie and Payne [52].
Mitochondrial-Nuclear Communication
Responses to different stimuli such as changes in temperature, nutritional state, energy production, hormonal changes as well as normal homeostasis in mammals, require the action of both mitochondrial and nuclear genomes. These responses involve signals from the nucleus to mitochondria as well as from mitochondria to the nucleus. The nuclear to mitochondrial signal involves the activation of certain transcription factors that regulate the expression of mitochondrial genes leading to mitochondrial biogenesis, whereas the mitochondrial to nucleus signals (also referred to as “retrograde signaling”) sense the mitochondrial functional state (e.g. loss of function by decrease in membrane potential, defects in oxidative phosphorylation, lower ATP levels, uncoupling, mtDNA mutations and hypoxia) [53]. In addition, mitochondrial stress response to heat shock or reduced levels of certain proteins can also trigger a nuclear response [54]. The mechanisms involved in the retrograde signaling are not well understood, although in yeast a number of transcriptional activators mediating this signaling pathway have been identified [55]. As described below, calcium participates in both antero- and retro-grade mitochondrial nuclear signaling in response to stimuli or stress (recently reviewed in [56]).
Mitochondrial Biogenesis
Although the complete pathway controlling mitochondrial biogenesis has not been elucidated, great progress in identifying key players has been obtained in the last few years. The expression of mitochondrial proteins encoded in the nuclear genome participating in oxidative phosphorylation, heme biosynthesis, mitochondrial protein import, and mtDNA transcription and replication, is regulated by transcription factors and transcriptional coactivators. The most prevalent transcription factors activating promoters of mitochondrial genes are the nuclear respiratory factor 1 and 2 (NRF-1 and NRF-2) [57, 58] and the estrogen-related receptor (ERRα) that work in concert with transcriptional coactivators of the peroxisome proliferator-activated receptor γ-coactivator-1 (PGC-1) family, PGC-1α, PGC-1β and PRC (PGC-1 related coactivator) [59]. This family of coactivators regulates several metabolic pathways such as cellular respiration, thermogenesis and hepatic glucose metabolism (reviewed in [60–62]). Although these coactivators stimulate mitochondrial biogenesis, PGC-1α is mainly involved in the regulation of gluconeogenesis and PGC-1β in the regulation of β-oxidation of fatty acids [63, 64]. Srivastava and Moraes [65] showed that overexpression of PGC-1α and PGC–1β was associated with an improvement of respiration in cells with deleterious mtDNA mutations [65].
Physiological mechanisms involved in PGC-1α regulation have been extensively studied in the last few years. Transgenic mice overexpressing PGC-1α showed mitochondrial proliferation in skeletal muscle and switch in fiber type composition from the more prominent type II (glycolytic) to type I (oxidative) [66]. In skeletal muscle, endurance exercise induces an increase in mitochondrial mass that is mediated by the increase in intracellular calcium levels during fiber contraction [67]. This process is regulated by the orchestrated expression of both nuclear and mitochondrial genes and the main pathway involved includes the activation and action of PGC-1α.
Increased levels of intracellular calcium activates cytoplasmic protein kinases such as calcium/calmodulin-dependent protein kinase (CaMK) or protein kinase C (PKC) that in turn stimulate the expression of several nuclear and mitochondrial genes [68]. Studies in vitro in isolated rat epitrochlearis muscle where an intracellular increase of calcium was achieved by incubation with low concentrations of caffeine (concentrations of 3.5 mM caffeine were low enough to not induce muscle contraction but to increase PGC-1α levels) and using specific Kinase inhibitors showed that the activation of CaMK occurs upstream of the activation of p38 mitogen-activated protein kinase (p38 MAPK) [69]. Moreover, p38 MAPK is responsible for phosphorylating, activating and inducing PGC-1α expression [69, 70]. In addition, Jager et al. [71] showed that AMP-activated protein kinase (AMPK) directly binds and phosphorylates PGC-1α [71]. The induction of PGC-1α expression is also mediated through ATF-2 (activating transcription factor 2) that binds to the PGC-1α promoter in the CREB (cyclic AMP-response binding protein) element binding site [72, 73]. Figure 3 shows a general diagram of the calcium signaling involved in mitochondrial biogenesis.
Figure 3. Calcium signaling participating in Mitochondrial Biogenesis.
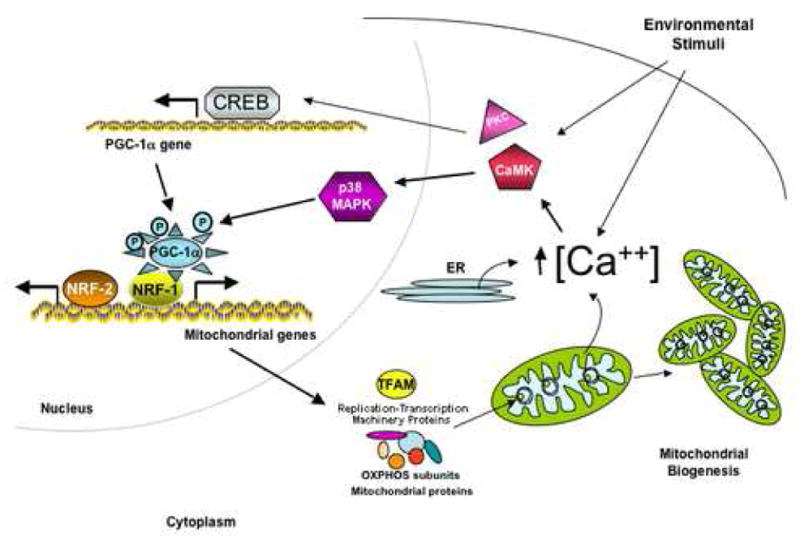
Extracellular stimuli such as nutrient deprivation, changes in temperature or exercise increase intracellular concentrations of calcium that stimulates different kinases (depending on the stimuli) that can activate transcription by stimulating specific transcription factors (e.g. CREB). Certain kinases also activate the transcriptional coactivator PGC-1α by directly phosphorylating it or by activating its transcription. PGC-1α stimulates transcription factors such as nuclear respiratory factor NRF1 and NRF2 to activate transcription of several mitochondrial genes including proteins necessary for transcription and replication of mtDNA, proteins of the electron transport chain, mitochondrial protein import, TCA cycle and β-oxidation of fatty acids that will lead to mitochondrial biogenesis.
The effect of calcium on the expression of mitochondrial proteins is not confined to muscle. Au et al. [74] had shown the increase of COX1, ATPase6, SDH and TFAM (transcript as well as protein levels) in human granulosa cells treated with the calcium ionophore A23187 and this effect was inhibited by EGTA [74]. Likewise, Mercy et al [75] using mtDNA depleted (rho0) cell lines, L929 (mouse fibroblast) and 143B (human osteosarcoma) showed that mitochondrial biogenesis is mediated by calcium signaling through CREB [75]. In addition to phosphorylation, another post-translational modification that regulates PGC-1α activity has been identified. Namely, deacetylation mediated by SIRT1 during fasting promotes the expression of mitochondrial genes involved in lipid oxidation [76].
The control of mitochondrial biogenesis during the cell cycle in mammalian cells remains unknown and controversial. Recently, Lee et al. [77] investigated mitochondria in synchronized cultures of HeLa cells at different points during the cell cycle and showed evidence that mitochondrial morphology varies during the cell cycle. During interphase, the typical mitochondrial tubular network was observed with heterogeneity in mitochondrial size and morphology, but progression into the mitotic phase disrupted this network and the mitochondrial population became more homogeneous [77]. Morphological changes of mitochondria during the cell cycle have been reported previously [78]. The tubular network seen during interphase was associated with microtubules whereas in mitosis, the fragmented mitochondria no longer interacted with microtubules but rather with actin. The mitochondrial mass and membrane potential increased during the progression of G1 to mitosis and after cell division these parameters were reduced again. The levels of mtDNA also increased in G1/S to G2 transition concomitant with increase of NRF-1 levels although the levels of TFAM and PRC were not altered [77].
Mitochondrial biogenesis, can also be stimulated by other pathways like reactive oxygen species [79], nitric oxide [80, 81] and hypoxia [82], which can be caused by a mitochondrial disorder or ischemic insults.
During heat shock or in the case of a respiratory deficiency caused by the decrease of one subunit of one of the respiratory complexes, an accumulation of unassembled subunits may occur, leading mitochondria into stress signals and a cellular unfolded protein response (UPR) whereby the cell responds by increasing the amount of proteins required for quality control (chaperones or proteases) [54].
Mitochondrial biogenesis is commonly stimulated in mitochondrial disorders, a group of diseases with heterogeneous array of clinical manifestations. These disorders are associated with either mutations in the mtDNA or in the nuclear DNA and often affect tissues with high energetic demands such as brain, heart and skeletal muscle [1, 2]. In addition, alterations of mtDNA (both large deletions and point mutations) have been observed in aging [83, 84], although the functional significance of these is still unclear. One of the hallmarks of oxidative phosphorylation dysfunction is the abnormal mitochondrial proliferation as a compensatory mechanism for the deficiency. This mitochondrial proliferation is commonly described as “ragged red fibers” in muscle [1] , but it is also observed in some other tissues [85] .
Mitochondrial Fusion and Fission
Mitochondria are dynamic organelles that undergo fusion and fission as part of their normal function. These two events are coordinated and necessary for proper morphology and function and seem to play critical roles during development, cell division and apoptosis. Fusion and fission are interrelated with the distribution of mtDNA within the cell as well as with their inheritance into daughter cells. In yeast, fusion defects cause loss of mtDNA and respiratory function [86]. Most of our knowledge in this field is due to studies performed in S. cerevisiae, Drosophila melanogaster and Caenorhabditis elegans.
The first fusion protein discovered was the fuzzy onion protein (Fzo) in Drosophila [87]. Fzo1 is an outer mitochondrial transmembrane GTPase that together with Mgm1 and Ugo1 are involved in mitochondrial fusion. In yeast, defects in Mgm1 lead to loss of mtDNA and respiratory deficiency [88]. In mammalians, proteins homologous to Fzo1 and Mgm1 have been identified, and correspond to mitofusins (Mfn1 and Mfn2) and OPA1 respectively [89]. In humans, Charcot-Marie-Tooth 2A and Autosomal Dominant Optic Atrophy, two neurodegenerative diseases involving defects in peripheral and retinal ganglia neurons, are caused by mutations in these two genes responsible for mitochondrial fusion, Mfn2 and OPA1 respectively [90–92].
Although the physiological role of mitochondrial fusion is not known, a conditional knockout mouse lacking Mfn2 in cerebellum showed neuronal degeneration where Purkinje cells lacked proper dendritic extensions, had respiratory function deficiency and abnormal mitochondrial distribution [93].
In yeast, mitochondrial fission involves the action of a dynamin- related protein Dmn1, with GTPase activity, as well as Fis1 and Mdv1. Fis1, an outer mitochondrial membrane protein, in association with Mdv1 recruits Dmn1 to the mitochondrial membrane at the site of division. Dmn1 is able to self assemble, by GTP-dependent conformational changes, into spiral structures that wrap around the mitochondria constricting the membrane resulting in fission (Figure 4) [89]. The human homologues of Dmn1 and Fis1 are Drp1 and hFis1 respectively, but so far no homolog of Mdv1 has been identified. Although the exact mechanism is still unknown, mitochondrial fission seems to be regulated by endophilin B (belonging to a family of self assembling proteins shown to remodel membranes in endocytosis [94] and by sumoylation of Drp1, which protects the protein from degradation [95] . In addition, mitochondrial fusion and fission have been shown to regulate calcium signaling in cells. Overexpression of Drp1, caused a fragmentation of the mitochondrial network that inhibited normal intramitochondrial calcium waves [96]. The general composition of the fusion/fission apparatus is described in figure 4.
Figure 4. Mitochondrial Fission and Fusion.
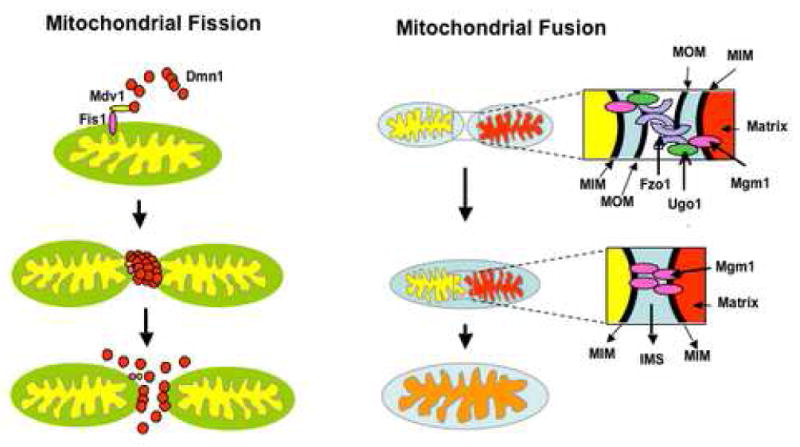
In yeast, mitochondria fission involves the action of Dmn1, that can self assemble into polymeric spirals and is recruited into the mitochondrial membrane by Fis1 and Mdv1. Dmn1 polymers wrap around the organelle and constrict the membrane until fission occurs. In humans, Drp1 and hFis1 are the homologs of Dmn1 and Fis1 whereas no homolog of Mdv1 has been found yet. Mitochondrial fusion involves the interaction of Fzo1/Ugo1 molecules located in the outer membrane of two mitochondria until outer membrane fuses, and then inner membrane fusion occurs through the interaction of Mgm1 molecules. In mammals, there are two homologs of Fzo1, the mitofusins 1 and 2 (Mfn1 and Mfn2) whereas the homolog of Mgm1 is OPA1. MIM, mitochondrial inner membrane: MOM, mitochondrial outer membrane. See text for more details. The cartoon is based on models described in reference [80].
Mitochondrial Turnover
Defective mitochondria have to be eliminated to maintain cellular homeostasis. Menzies and Gold [97] estimated that the turnover of mitochondria in liver, heart and brain is 9.3, 17.5 and 24.4 days respectively [97]. Autophagy appears to be the main pathway for mitochondrial turnover (mitophagy) although it has been shown that ubiquitin mediated proteolysis plays a major role in the elimination of the paternal mitochondria during fertilization [98, 99].
Mitophagy consist in the sequestration of the defective organelle into a double membrane phagosome that is later fused with lysosomes forming autolysosomes, where the contents are degraded and can be recycled (reviewed in [100, 101]). In yeast, several genes involved in autophagy (ATG genes) have been described and their mechanism of action requires two novel conjugation systems that are somehow interrelated (reviewed in [102]). In one of the systems, Atg12, a small protein, is conjugated to a lysine on Atg5 by Atg7 and Atg10, in a way similar to the E1 and E2 enzymes of the ubiquitin system, and once Atg5 is tagged with Atg12 it forms a complex with Atg16 [103]. In the other conjugation system, Atg8, also a small protein, is conjugated to phosphatidylethanolamine (PE, membrane phospholipid) by Atg7 and Atg3 [104]. In addition to the conjugated systems, two protein kinase complexes are required for autophagy. The protein kinase complexes are the Atg1 kinase complex and the phosphatidylinositol 3 kinase (PI3K) complex both formed by several proteins. The four systems described above (protein conjugation and kinase complexes) work in concert to produce the autophagosome.
The mitophagy process starts by the recruitment and transient association of the conjugated protein (Atg5-Atg12 conjugated) into a precursor vesicle (PE-Atg8 conjugated) that elongates along the perimeter of the mitochondrion until both ends fuse forming the double membrane of the autophagosome. The PI3K complex is required for the initial process of autophagosome formation and its inhibition stops autophagy [105]. The function of the Atg1 kinase is not very clear but appears to control the autophagy response induced by nutrient deprivation [106].
The process of how an individual mitochondrion is recognized for degradation is not known but appears that there are some proteins such as Uth1 (mitochondrial outer membrane protein) involved in the selectivity of the process [107]. For more details, this topic (calcium and mitochondria autophagy) is covered in another chapter of this special issue of Cell Calcium.
Conclusions
Mitochondrial biogenesis is a very complex cellular process that requires the coordination of several mechanisms involving nuclear-mitochondrial communication, mitochondrial protein expression and import, mtDNA gene expression, assembly of multi subunit enzyme complexes, regulation of mitochondrial fission and fusion as well as mitochondrial turnover in response to various stimuli. Disruption of any of these processes can lead to defective mitochondrial function and therefore to a disease state. Many of the pathways participating in mitochondrial biogenesis require the participation of calcium as a signaling molecule, as discussed in more detail in the subsequent chapters of this book.
Acknowledgments
Our work was supported by grants from the National Institutes of Health (NINDS, NCI and NEI), and the Muscular Dystrophy Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–68. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 2.Wallace D. Mitochondrial defects in neurodegenerative disease. Mental Retardation and Developmental Disabilities Research Reviews. 2001;7:158–166. doi: 10.1002/mrdd.1023. [DOI] [PubMed] [Google Scholar]
- 3.Margulis L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp Soc Exp Biol. 1975:21–38. [PubMed] [Google Scholar]
- 4.Margulis L, Bermudes D. Symbiosis as a mechanism of evolution: status of cell symbiosis theory. Symbiosis. 1985;1:101–24. [PubMed] [Google Scholar]
- 5.Shutt TE, Gray MW. Bacteriophage origins of mitochondrial replication and transcription proteins. Trends Genet. 2006;22:90–5. doi: 10.1016/j.tig.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 7.Kaufman BA, Newman SM, Hallberg RL, Slaughter CA, Perlman PS, Butow RA. In organello formaldehyde crosslinking of proteins to mtDNA: identification of bifunctional proteins. Proc Natl Acad Sci U S A. 2000;97:7772–7. doi: 10.1073/pnas.140063197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Copeland WC. Inherited Mitochondrial Diseases of DNA Replication. Annual Review of Medicine. 2008:59. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–31. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 10.Hudson G, Deschauer M, Busse K, Zierz S, Chinnery PF. Sensory ataxic neuropathy due to a novel C10Orf2 mutation with probable germline mosaicism. Neurology. 2005;64:371–373. doi: 10.1212/01.WNL.0000149767.51152.83. [DOI] [PubMed] [Google Scholar]
- 11.Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland WC, Chinnery PF. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–34. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem. 2006;281:25791–802. doi: 10.1074/jbc.M604501200. [DOI] [PubMed] [Google Scholar]
- 13.He J, Mao C-C, Reyes A, Sembongi H, Di Re M, Granycome C, Clippingdale AB, Fearnley IM, Harbour M, Robinson AJ, Reichelt S, Spelbrink JN, Walker JE, Holt IJ. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J Cell Biol. 2007;176:141–146. doi: 10.1083/jcb.200609158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holt IJ, He J, Mao CC, Kirk-Up JD, Martinsson P, Sembongi H, Reyes A, Spelbrink JN. Mammalian mitochondrial nucleoids: Organizing an independently minded genome. Mitochondrion. 2007 doi: 10.1016/j.mito.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Cotney J, Wang Z, Shadel GS. Relative abundance of the human mitochondrial transcription system and distinct roles for h-mtTFB1 and h-mtTFB2 in mitochondrial biogenesis and gene expression. Nucleic Acids Res. 2007;35:4042–54. doi: 10.1093/nar/gkm424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang DD, Clayton DA. Priming of human mitochondrial DNA replication occurs at the light-strand promoter. Proc Natl Acad Sci U S A. 1985;82:351–5. doi: 10.1073/pnas.82.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang DD, Fisher RP, Clayton DA. Roles for a promoter and RNA processing in the synthesis of mitochondrial displacement-loop strands. Biochim Biophys Acta. 1987;909:85–91. doi: 10.1016/0167-4781(87)90029-7. [DOI] [PubMed] [Google Scholar]
- 19.Chang DD, Clayton DA. A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science. 1987;235:1178–84. doi: 10.1126/science.2434997. [DOI] [PubMed] [Google Scholar]
- 20.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 21.Brown TA, Cecconi C, Tkachuk AN, Bustamante C, Clayton DA. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 2005;19:2466–2476. doi: 10.1101/gad.1352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt IJ, Lorimer HE, Jacobs HT. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–24. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 23.Montoya J, Christianson T, Levens D, Rabinowitz M, Attardi G. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc Natl Acad Sci U S A. 1982;79:7195–9. doi: 10.1073/pnas.79.23.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montoya J, Gaines GL, Attardi G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell. 1983;34:151–9. doi: 10.1016/0092-8674(83)90145-9. [DOI] [PubMed] [Google Scholar]
- 25.Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–35. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 26.Martin M, Cho J, Cesare AJ, Griffith JD, Attardi G. Termination Factor-Mediated DNA Loop between Termination and Initiation Sites Drives Mitochondrial rRNA Synthesis. Cell. 2005;123:1227–1240. doi: 10.1016/j.cell.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 27.Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–99. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 28.Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–25. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 29.Park CB, Asin-Cayuela J, Camara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument-Bromage H, Tempst P, Falkenberg M, Gustafsson CM, Larsson NG. MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell. 2007;130:273–85. doi: 10.1016/j.cell.2007.05.046. [DOI] [PubMed] [Google Scholar]
- 30.Lopez MF, Kristal BS, Chernokalskaya E, Lazarev A, Shestopalov AI, Bogdanova A, Robinson M. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis. 2000;21:3427–40. doi: 10.1002/1522-2683(20001001)21:16<3427::AID-ELPS3427>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 31.Calvo S, Jain M, Xie X, Sheth SA, Chang B, Goldberger OA, Spinazzola A, Zeviani M, Carr SA, Mootha VK. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. 2006;38:576–82. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 32.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–49. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 33.van der Laan M, Rissler M, Rehling P. Mitochondrial preprotein translocases as dynamic molecular machines. FEMS Yeast Res. 2006;6:849–61. doi: 10.1111/j.1567-1364.2006.00134.x. [DOI] [PubMed] [Google Scholar]
- 34.Neupert W. Protein import into mitochondria. Annu Rev Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 35.Schneider HC, Berthold J, Bauer MF, Dietmeier K, Guiard B, Brunner M, Neupert W. Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature. 1994;371:768–74. doi: 10.1038/371768a0. [DOI] [PubMed] [Google Scholar]
- 36.Sato T, Esaki M, Fernandez JM, Endo T. Comparison of the protein-unfolding pathways between mitochondrial protein import and atomic-force microscopy measurements. Proc Natl Acad Sci U S A. 2005;102:17999–8004. doi: 10.1073/pnas.0504495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartl FU, Ostermann J, Guiard B, Neupert W. Successive translocation into and out of the mitochondrial matrix: targeting of proteins to the intermembrane space by a bipartite signal peptide. Cell. 1987;51:1027–37. doi: 10.1016/0092-8674(87)90589-7. [DOI] [PubMed] [Google Scholar]
- 38.Webb CT, Gorman MA, Lazarou M, Ryan MT, Gulbis JM. Crystal structure of the mitochondrial chaperone TIM9.10 reveals a six-bladed alpha-propeller. Mol Cell. 2006;21:123–33. doi: 10.1016/j.molcel.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 39.Sirrenberg C, Bauer MF, Guiard B, Neupert W, Brunner M. Import of carrier proteins into the mitochondrial inner membrane mediated by Tim22. Nature. 1996;384:582–5. doi: 10.1038/384582a0. [DOI] [PubMed] [Google Scholar]
- 40.Paschen SA, Waizenegger T, Stan T, Preuss M, Cyrklaff M, Hell K, Rapaport D, Neupert W. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature. 2003;426:862–6. doi: 10.1038/nature02208. [DOI] [PubMed] [Google Scholar]
- 41.Wiedemann N, Kozjak V, Chacinska A, Schonfisch B, Rospert S, Ryan MT, Pfanner N, Meisinger C. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 2003;424:565–71. doi: 10.1038/nature01753. [DOI] [PubMed] [Google Scholar]
- 42.Naoe M, Ohwa Y, Ishikawa D, Ohshima C, Nishikawa S, Yamamoto H, Endo T. Identification of Tim40 that mediates protein sorting to the mitochondrial intermembrane space. J Biol Chem. 2004;279:47815–21. doi: 10.1074/jbc.M410272200. [DOI] [PubMed] [Google Scholar]
- 43.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–69. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 44.Terziyska N, Lutz T, Kozany C, Mokranjac D, Mesecke N, Neupert W, Herrmann JM, Hell K. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett. 2005;579:179–84. doi: 10.1016/j.febslet.2004.11.072. [DOI] [PubMed] [Google Scholar]
- 45.Rissler M, Wiedemann N, Pfannschmidt S, Gabriel K, Guiard B, Pfanner N, Chacinska A. The essential mitochondrial protein Erv1 cooperates with Mia40 in biogenesis of intermembrane space proteins. J Mol Biol. 2005;353:485–92. doi: 10.1016/j.jmb.2005.08.051. [DOI] [PubMed] [Google Scholar]
- 46.Takakubo F, Cartwright P, Hoogenraad N, Thorburn DR, Collins F, Lithgow T, Dahl HH. An amino acid substitution in the pyruvate dehydrogenase E1 alpha gene, affecting mitochondrial import of the precursor protein. Am J Hum Genet. 1995;57:772–80. [PMC free article] [PubMed] [Google Scholar]
- 47.Danpure CJ, Cooper PJ, Wise PJ, Jennings PR. An enzyme trafficking defect in two patients with primary hyperoxaluria type 1: peroxisomal alanine/glyoxylate aminotransferase rerouted to mitochondria. J Cell Biol. 1989;108:1345–52. doi: 10.1083/jcb.108.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutton A, Khoury H, Prip-Buus C, Cepanec C, Pessayre D, Degoul F. The Ala16Val genetic dimorphism modulates the import of human manganese superoxide dismutase into rat liver mitochondria. Pharmacogenetics. 2003;13:145–57. doi: 10.1097/01.fpc.0000054067.64000.8f. [DOI] [PubMed] [Google Scholar]
- 49.Koehler CM, Leuenberger D, Merchant S, Renold A, Junne T, Schatz G. Human deafness dystonia syndrome is a mitochondrial disease. Proc Natl Acad Sci U S A. 1999;96:2141–6. doi: 10.1073/pnas.96.5.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43:385–93. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansen JJ, Durr A, Cournu-Rebeix I, Georgopoulos C, Ang D, Nielsen MN, Davoine CS, Brice A, Fontaine B, Gregersen N, Bross P. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet. 2002;70:1328–32. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacKenzie JA, Payne RM. Mitochondrial protein import and human health and disease. Biochim Biophys Acta. 2007;1772:509–23. doi: 10.1016/j.bbadis.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002;21:7839–49. doi: 10.1038/sj.onc.1205983. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. Embo J. 2002;21:4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Z, Butow RA. A Transcriptional Switch in the Expression of Yeast Tricarboxylic Acid Cycle Genes in Response to a Reduction or Loss of Respiratory Function. Mol Cell Biol. 1999;19:6720–6728. doi: 10.1128/mcb.19.10.6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–22. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- 57.Virbasius CA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 1993;7:2431–2445. doi: 10.1101/gad.7.12a.2431. [DOI] [PubMed] [Google Scholar]
- 58.Virbasius JV, Virbasius CA, Scarpulla RC. Identity of GABP with NRF-2, a multisubunit activator of cytochrome oxidase expression, reveals a cellular role for an ETS domain activator of viral promoters. Genes Dev. 1993;7:380–392. doi: 10.1101/gad.7.3.380. [DOI] [PubMed] [Google Scholar]
- 59.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81–9. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 60.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–83. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 61.Chabi B, Adhihetty PJ, Ljubicic V, Hood DA. How is mitochondrial biogenesis affected in mitochondrial disease? Med Sci Sports Exerc. 2005;37:2102–10. doi: 10.1249/01.mss.0000177426.68149.83. [DOI] [PubMed] [Google Scholar]
- 62.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 63.Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–8. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 64.Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck-Nielsen H, Groop L, Vaag A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J Clin Invest. 2004;114:1518–26. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Srivastava S, Barrett JN, Moraes CT. PGC-1alpha/beta upregulation is associated with improved oxidative phosphorylation in cells harboring nonsense mtDNA mutations. Hum Mol Genet. 2007;16:993–1005. doi: 10.1093/hmg/ddm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 67.Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol. 1976;38:273–91. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- 68.Freyssenet D, Di Carlo M, Hood DA. Calcium-dependent regulation of cytochrome c gene expression in skeletal muscle cells. Identification of a protein kinase c-dependent pathway. J Biol Chem. 1999;274:9305–11. doi: 10.1074/jbc.274.14.9305. [DOI] [PubMed] [Google Scholar]
- 69.Wright DC, Geiger PC, Han DH, Jones TE, Holloszy JO. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:18793–9. doi: 10.1074/jbc.M611252200. [DOI] [PubMed] [Google Scholar]
- 70.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–82. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 71.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280:19587–93. doi: 10.1074/jbc.M408862200. [DOI] [PubMed] [Google Scholar]
- 73.Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24:3057–67. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Au HK, Yeh TS, Kao SH, Shih CM, Hsieh RH, Tzeng CR. Calcium-dependent up-regulation of mitochondrial electron transfer chain gene expressions in human luteinized granulosa cells. Fertil Steril. 2005;84(Suppl 2):1104–8. doi: 10.1016/j.fertnstert.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 75.Mercy L, Pauw A, Payen L, Tejerina S, Houbion A, Demazy C, Raes M, Renard P, Arnould T. Mitochondrial biogenesis in mtDNA-depleted cells involves a Ca2+-dependent pathway and a reduced mitochondrial protein import. Febs J. 2005;272:5031–55. doi: 10.1111/j.1742-4658.2005.04913.x. [DOI] [PubMed] [Google Scholar]
- 76.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J. 2007;26:1913–23. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee S, Kim S, Sun X, Lee JH, Cho H. Cell cycle-dependent mitochondrial biogenesis and dynamics in mammalian cells. Biochem Biophys Res Commun. 2007;357:111–7. doi: 10.1016/j.bbrc.2007.03.091. [DOI] [PubMed] [Google Scholar]
- 78.Arakaki N, Nishihama T, Owaki H, Kuramoto Y, Suenaga M, Miyoshi E, Emoto Y, Shibata H, Shono M, Higuti T. Dynamics of mitochondria during the cell cycle. Biol Pharm Bull. 2006;29:1962–5. doi: 10.1248/bpb.29.1962. [DOI] [PubMed] [Google Scholar]
- 79.Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem. 2007;282:2355–62. doi: 10.1074/jbc.M608009200. [DOI] [PubMed] [Google Scholar]
- 80.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–9. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 81.Leary SC, Shoubridge EA. Mitochondrial biogenesis: which part of “NO” do we understand? Bioessays. 2003;25:538–41. doi: 10.1002/bies.10298. [DOI] [PubMed] [Google Scholar]
- 82.McLeod CJ, Pagel I, Sack MN. The Mitochondrial Biogenesis Regulatory Program in Cardiac Adaptation to Ischemia--A Putative Target for Therapeutic Intervention. Trends in Cardiovascular Medicine. 2005;15:118–123. doi: 10.1016/j.tcm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 83.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–9. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 84.Khaidakov M, Heflich RH, Manjanatha MG, Myers MB, Aidoo A. Accumulation of point mutations in mitochondrial DNA of aging mice. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2003;526:1–7. doi: 10.1016/s0027-5107(03)00010-1. [DOI] [PubMed] [Google Scholar]
- 85.Diaz F, Garcia S, Hernandez D, Regev A, Rebelo A, Oca-Cossio J, Moraes CT. Pathophysiology and Fate of Hepatocytes in a Mouse Model of Mitochondrial Hepatopathies. Gut. 2007 doi: 10.1136/gut.2006.119180. Published Online First: 19 October 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–92. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 87.Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–9. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- 88.Jones BA, Fangman WL. Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev. 1992;6:380–389. doi: 10.1101/gad.6.3.380. [DOI] [PubMed] [Google Scholar]
- 89.Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–80. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- 90.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battologlu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 91.Alexander C, Votruba M, Pesch UEA, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 92.Delettre C, Lenaers G, Griffoin J-M, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 93.Chen H, McCaffery JM, Chan DC. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 94.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–39. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–5. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 96.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 97.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–9. [PubMed] [Google Scholar]
- 98.Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature. 1999;402:371–2. doi: 10.1038/46466. [DOI] [PubMed] [Google Scholar]
- 99.Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitinated sperm mitochondria, selective proteolysis, and the regulation of mitochondrial inheritance in mammalian embryos. Biol Reprod. 2000;63:582–90. doi: 10.1095/biolreprod63.2.582. [DOI] [PubMed] [Google Scholar]
- 100.Mijaljica D, Prescott M, Devenish RJ. Different fates of mitochondria: alternative ways for degradation? Autophagy. 2007;3:4–9. doi: 10.4161/auto.3011. [DOI] [PubMed] [Google Scholar]
- 101.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–6. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 103.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 104.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 105.Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3-kinase--Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J. 2003;376:577–86. doi: 10.1042/BJ20030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Young ARJ, Chan EYW, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 107.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–74. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 108.Cottrell DA, Blakely EL, Johnson MA, Borthwick GM, Ince PI, Turnbull DM. Mitochondrial DNA mutations in disease and ageing. Novartis Found Symp. 2001;235:234–43. doi: 10.1002/0470868694.ch19. discussion 243–6. [DOI] [PubMed] [Google Scholar]