

Abstract
Fluorescent ligands for the heat shock protein 90 (Hsp90) were synthesized containing either fluorescein isothiocyanate (FITC), 4-nitrobenzo[1,2,5]oxadiazole (NBD) or the red shifted dye sulforhodamine 101 (Texas Red) conjugated to PU-H71. Two of the compounds, PU-H71-FITC2 (9) and PU-H71-NBD1 (8), were shown to be suitable for fluorescence-activated flow cytometry and fluorescence microscopy. Thus these molecules serve as useful probes for studying Hsp90 in heterogeneous live cell populations.
Keywords: Fluorescent Hsp90 inhibitor, Purine, PU-H71, Flow cytometry, Fluorescence microscopy
Heat shock protein 90 (Hsp90) is a chaperone which functions to properly fold numerous proteins to their active conformation. Many of its clients play a prominent role in disease onset and progression in several pathologies, such as in cancer and neurodegeneration,1–3 and currently there is significant interest in the translation of Hsp90 inhibitors in the treatment of cancers and other diseases.4, 5
While the Hsp90 protein is essentially identical in sequence in all human cells, the use of Hsp90 inhibitors in treatment is justified by the selective biological activity of these inhibitors against stressed, pathologically altered, cells.2 As a means to justify the observed therapeutic index, a high-affinity binding of certain Hsp90 inhibitors for cancer cells and for neurodegenerative disease afflicted neurons was reported.6, 7 While the biochemical nature of this selective binding remains poorly understood, it is hypothesized that co-chaperones and post-translational modifications alter the affinity of small molecules for the Hsp90 chaperone complexes in a cell-by-cell manner.2, 8 Such heterogeneity in Hsp90 presentation suggests that means to investigate the affinity of select Hsp90 inhibitors for the target in live cells are needed.
Fluorescence-activated flow cytometry, remains a method of choice for enumerating, purifying and analyzing cells.9, 10 In fact, a multitude of measurements can be performed now by flow cytometry, and recent technical advances allow these measurements to be made simultaneously on individual cells within heterogeneous populations.11 Such multiparameter analysis is quite powerful as it provides more data from less sample, a key consideration when patient samples are limited. Multiparameter analysis also allows more accurate identification of populations, by excluding unwanted cells that bind some reagents.9, 10 The method is thus optimal for analyzing the binding of Hsp90 ligands, when fluorescently labeled, to distinct cell populations.
Fluorescently labeled ligands have historically had a wide variety of uses in biology and pharmacology,12 and offer the advantage of retaining the pharmacological properties of the unlabeled ligand. In addition to in vitro investigations of ligand-receptor binding, small molecule fluorescent probes allow for real time and non-invasive monitoring of the interaction between the target and the ligand in living cell populations, such as by means of flow cytometry.
Fluorescently labeled inhibitors of Hsp90 have already been reported, with analogs of geldanamycin (GM-FITC,13 GM-Bodipy,13 GM-cy3b14) as well as pyrazole 1 (fluorescein analog, VER-00045864)15 used as ligands in fluorescence polarization assays (Figure 1). A cell-impermeable GM-FITC derivative was used to identify cell surface Hsp90 by fluorescence microscopy.16 Hsp90 however, is mainly a cytoplasmic protein with cell surface expression detected only in certain cells.1, 2 Fluorescent probes are thus needed to analyze both intracellular and cell surface Hsp90.
Figure 1.

Structures of known Hsp90 inhibitors.
Fluorescent dyes absorb light at certain wavelengths and in turn emit their fluorescence energy at a higher wavelength. Each dye has a distinct emission spectrum, which can be exploited for multicolor analysis by flow cytometry. Among the most used are fluorescein isothiocyanate (FITC), 4-nitrobenzo[1,2,5]oxadiazole (NBD) or the red shifted dye sulforhodamine 101 (Texas Red). FITC and NBD are detected in the FL1 channel on most instruments and are also a good choice for fluorescence microscopy (excitation 495 and 466 nM and emission 519 and 539 nM, respectively), whereas Texas Red is detected in FL3 on single laser instruments (excitation 589 nM and emission 615 nM).
This work investigates the synthesis of various fluorescently labeled derivatives of PU-H71 (2), a purine-scaffold inhibitor of Hsp9017 (Figure 1) and describes their biological application as probes for studying Hsp90 by fluorescence-activated flow cytometry and fluorescence microscopy. Several Hsp90 inhibitors based on the purine-scaffold, including BIIB021, MPC-3100, PU-H71 and Debio 0932 (formerly CUDC-305) are currently in clinical development for cancers.18, 19
For the development of small dye-labeled ligands, the selection of an optimal fluorophore and its site of attachment are of high relevance. Particularly in small molecules the introduced dye can crucially affect the biochemical and pharmacologic characteristics of the ligand. According to the X-ray crystal structure of PU-H71 (2) bound to Hsp90,20 the N9-alkylamino chain of the ligand is oriented towards solvent. As a result of this, as well as previous SAR, each of the compounds synthesized here contains the fluorescent label attached to the N9 position. Three types of derivatives of PU-H71 with different linkers were labeled with either FITC, NBD or Texas Red (TR).
In the first type, a six carbon spacer was appended to the constituent amine (Scheme 1). We had previously used this linker to attach PU-H71 to solid support and showed that 3 retains good affinity for Hsp90.21 3 was reacted with FITC in DMF/Et3N to give 4 (PU-H71-FITC1) in 40% yield following purification by HPLC (Scheme 1). PU-H71-Texas Red (5; PU-H71-TR) was synthesized by the reaction of 3 with sulforhodamine 101 sulfonyl chloride in DMF to give 5 in 61% yield following purification by HPLC (Scheme 1). In the case of the NBD analog, bromide 6 was reacted with 7 in DMF to give 8 (PU-H71-NBD1) in 47% yield (Scheme 1). 621 and NBD derivative 722 were prepared as previously described.
Scheme 1.

Reagents and conditions: (a) FITC, Et3N, DMF, rt, 12 h, 40%; (b) Texas Red sulfonyl chloride, DMF, 0-10°C, 12 h, 61%; (c) DMF, rt, 20 h, 47%.
In the second type, we took advantage of the secondary amine present in PU-H7123 and reacted it directly with FITC or NBD-Cl to give 9 (PU-H71-FITC2) (72%) or 10 (PU-H71-NBD2) (40%), respectively (Scheme 2).
Scheme 2.

Reagents and conditions: (a) FITC, Et3N, DMF, rt, 5 h, 72%; (b) NBD-Cl, Et3N, DMF, rt, 12 h, 40%.
In the third type, desisopropyl-PU-H71 13 was reacted with FITC or NBD-Cl to give 14 (PU-H71-FITC3) (74%) or 15 (PU-H71-NBD3) (42%), respectively (Scheme 3). 13 was synthesized by N9-alkylation of 11 with N-(3-bromopropyl)-phthalimide and subsequent removal of phthalimide with hydrazine (Scheme 3). It was hypothesized that attachment of the dye directly to the amine in both types 2 and 3 derivatives would result in more cell permeable analogs, because in contrast to type 1 derivatives, these lack an ionizable amine. Furthermore, type 2 derivatives contain an isopropyl group in place of a hydrogen as in type 3 derivatives, which makes them more lipophilic and may enhance their cell permeability.
Scheme 3.

Reagents and conditions: (a) N-(3-bromopropyl)-phthalimide, Cs2CO3, DMF, rt, 34%; (b) hydrazine hydrate, MeOH, CH2Cl2, rt, 64%; (c) FITC, Et3N, DMF, rt, 12 h, 74%; (d) NBD-Cl, Et3N, DMF, rt, 12 h, 42%.
Cell permeable probes are favored because fixation/permeabilization methods used for the detection of intracellular antigens by flow cytometry often result in the destruction of cellular morphology and surface immunoreactivity, properties useful in flow cytometry for the characterization of cells in heterogeneous populations. Thus, it is of particular interest to find cell-permeable ligands that interact with the target in live cells without the requirement of fixation and permeabilization steps.
To investigate which of the above synthesized fluorescently labeled PU-H71 derivatives retained the cell permeability profile of the parent compound PU-H71, we next examined the cellular permeability of these Hsp90 probes in human acute myelogenous leukemia (AML) cell lines, MV4-11 and MOLM-13. Of the seven fluorescent derivatives of PU-H71, we find that only PU-H71-FITC2 (9) and PU-H71-NBD1 (8) have these required characteristics (Figure 2). Specifically, we show efficient staining of live cells by these two derivatives (Figure 2A) as well as biological activity in these cells indicative of target (Hsp90) inhibition (Figure 2B,C). In particular, we show that both PU-H71-FITC2 (9) and PU-H71-NBD1 (8) decrease the viability of MOLM-13 cells (Figure 2B), effect associated with degradation of Hsp90-client proteins such as mutant FLT3 and Raf-1 (Figure 2C) indicating intracellular Hsp90 inhibition in these cancer cells.1–3
Figure 2.
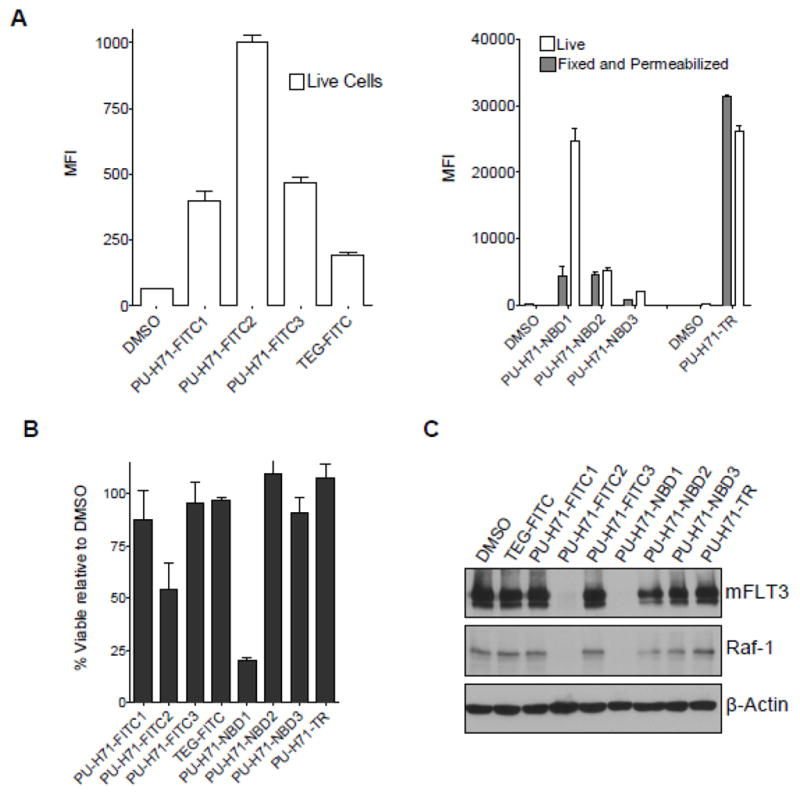
(A) MOLM-13 cells were treated with the indicated PU-H71-fluorescent derivatives (1 μM) at 37°C for 4h. Their binding to live cells (DAPI negative) were measured by flow cytometry and shown as mean fluorescence intensity (MFI). (B) MOLM-13 cells were treated with the indicated PU-H71-fluorescent derivatives (1 μM) at 37°C for 24h. Their viability was determined by DAPI exclusion. (C) MOLM-13 cells were treated with the indicated PU-H71-fluorescent derivatives (1 μM) for 24h. The steady-state level of the Hsp90 client proteins mFLT3 and Raf-1 was analyzed by Western blot. β-Actin was used to normalize for equal protein loading.
Furthermore, confocal fluorescence microscopy of leukemia cells stained with PU-H71-FITC2 (9) showed prominent intracellular localization (Figure 3A). In these experiments, DAPI was used as a viability dye to discriminate between viable and non-viable cells. This dye is impermeable in live cells at the tested concentration, but permeates non-viable cells and binds specific regions of DNA. DAPI is excited in most instruments with a UV laser. Similar data were generated with PU-H71-NBD1 (8) (not shown).
Figure 3.

(A) MV4-11 cells were treated with 1 μM PU-H71-FITC2 (9) at 37°C for 24 h. Cells were then fixed, and stained with Na+/K+-ATPase α 1 antibody and goat anti-rabbit Alexa Fluor 568. Images showing live cells (DAPI (blue) negative) with PU-H71-FITC2 (green) binding alongside cell surface marker Na+/K+-ATPase α 1 staining (red) were captured by confocal microscopy. (B) A primary acute myeloid leukemia sample was pre-treated with the indicated dose of PU-H71 or vehicle (Untreated) for 24 h. Post-treatment cells were treated with 1 μM PU-H71-FITC2 or TEG-FITC. Binding of PU-H71-FITC2 and TEG-FITC to the cells was evaluated by flow cytometry and is represented as mean fluorescence intensity (MFI). TEG-FITC is shown as a non-specific binding control. CD45 vs. SSC gating was used to distinguish binding to blast (malignant cells) or lymphocytes (normal cells) from the primary specimens.
Flow cytometry is commonly used to separate and distinguish different cell populations in normal and malignant hematopoiesis by the use of specific markers. As an example, blast cells are often quantified and characterized by dim CD45 staining (CD45dim), in contrast to the circulating non-blast cell populations, which are bright for CD45 staining (CD45hi).24 These cells, sorted and separated by the presence of their identifying markers, we show here can also be stained for the target, Hsp90, with PU-H71-FITC2 (Figure 3B). In accord with previous reports indicating the selective binding of PU-H71 to tumor cell Hsp90,23 PU-H71-FITC2 preferentially stained the malignant cell (blasts) and not the normal cell (lymphocytes) population in a primary acute myeloid leukemia sample (Figure 3B).
In summary, we present here the synthesis of several fluorescently labeled conjugates of the purine-scaffold Hsp90 inhibitor PU-H71. Furthermore, we show that PU-H71-FITC2 (9) and PU-H71-NBD1 (8) permeate live cells and bind to the target. Specifically, we show that PU-H71-FITC2 and PU-H71-NBD1 stain live cells (Figure 2A), reduce the viability of leukemia cells (Figure 2B), inhibit the intracellular Hsp90 as indicated by degradation of Hsp90 client proteins (Figure 2C), are localized intracellularly as indicated by confocal microscopy (Figure 3A) and bind specifically to tumor versus normal cell Hsp90 as indicated by flow cytometry (Figure 3B), to provide ample evidence that these probes permeate the cell and bind specifically to the tumor Hsp90 target, similarly to PU-H71. In conclusion, these fluorescent derivatives of PU-H71 can be applied as probes for fluorescence-activated flow cytometry or as tools for monitoring real-time interaction of Hsp90 with the target by fluorescence microscopy.
Supplementary Material
Acknowledgments
This work was supported in part by W.H. Goodwin and A. Goodwin and the Commonwealth Cancer Foundation for Research, The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center (MSKCC), the Translational and Integrative Medicine Research Fund of MSKCC, SPORE Pilot Award and Research & Therapeutics Program in Prostate Cancer, Hirshberg Foundation for Pancreatic Cancer Research, 1R01CA155226-01, U01AG032969-01A1, R21AG028811, 3P30CA008748-44S2. G.C. is funded by Leukemia and Lymphoma Society LLS#6114-10 & LLS#6330-11, the Byrne Fund, the Geoffrey Beene Cancer Research Center of MSKCC, grant UL1RR024996 of the Clinical and Translational Science Center at Weill Cornell Medical College and Susan G. Komen for the Cure. T.T. is funded by Susan G. Komen for the Cure and the Department of Defense, Breast Cancer Research Program. M.L.G is funded by the National Institutes of Health through the NIH Director’s New Innovator Award Program, 1 DP2 OD007399-01 and is a V-Foundation Scholar. We also thank Dr. George Sukenick and Dr. Hui Liu of the NMR Analytical Core Facility at MSKCC for expert mass spectral analysis.
Footnotes
Supplementary data (experimental methods including synthesis and characterization of compounds and biological protocols) associated with this article can be found in the online version, at.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:761. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 2.Workman P, Burrows F, Neckers L, Rosen N. Ann N Y Acad Sci. 2007;1113:202. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 3.Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Mol Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taldone T, Gozman A, Maharaj R, Chiosis G. Curr Opin Pharmacol. 2008;8:370. doi: 10.1016/j.coph.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janin YL. Drug Discovery Today. 2010;15:342. doi: 10.1016/j.drudis.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. Nature. 2003;425:407. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 7.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Hutton M, Burrows F, Petrucelli L. J Clin Invest. 2007;117:648. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiosis G, Neckers L. ACS Chem Biol. 2006;1:279. doi: 10.1021/cb600224w. [DOI] [PubMed] [Google Scholar]
- 9.Peters JM, Ansari MQ. Arch Pathol Lab Med. 2011;135:44. doi: 10.5858/2010-0387-RAR.1. [DOI] [PubMed] [Google Scholar]
- 10.Covey TM, Cesano A. Best Pract Res Clin Haematol. 2010;23:319. doi: 10.1016/j.beha.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Jennings CD, Foon KA. Blood. 1997;90:2863. [PubMed] [Google Scholar]
- 12.Leopoldo M, Lacivita E, Berardi F, Perrone R. Drug Discov Today. 2009;14:706. doi: 10.1016/j.drudis.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Llauger-Bufi L, Felts SJ, Huezo H, Rosen N, Chiosis G. Bioorg Med Chem Lett. 2003;13:3975. doi: 10.1016/j.bmcl.2003.08.065. [DOI] [PubMed] [Google Scholar]
- 14.Moulick K, Clement CC, Aguirre J, Kim J, Kang Y, Felts S, Chiosis G. Bioorg Med Chem Lett. 2006;16:4515. doi: 10.1016/j.bmcl.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 15.Howes R, Barril X, Dymock BW, Grant K, Northfield CJ, Robertson AG, Surgenor A, Wayne J, Wright L, James K, Matthews T, Cheung KM, McDonald E, Workman P, Drysdale MJ. Anal Biochem. 2006;350:202. doi: 10.1016/j.ab.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 16.Tsutsumi S, Scroggins B, Koga F, Lee MJ, Trepel J, Felts S, Carreras C, Neckers L. Oncogene. 2008;27:2478. doi: 10.1038/sj.onc.1210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taldone T, Chiosis G. Curr Top Med Chem. 2009;9:1436. doi: 10.2174/156802609789895737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel HJ, Modi S, Chiosis G, Taldone T. Expert Opin Drug Discov. 2011;6:559. doi: 10.1517/17460441.2011.563296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papac DI, Patton JS, Reeves L, Bulka K, DeMie L, Roman O, Bradford C, Kim S-H, Tangallapally R, Trovato R, Markovitz B, Bajji A, Wettstein D, Baichwal V, Mather G. 102nd AACR annual meeting, abstract number 3233; Orlando, Fl. April 2–6, 2011. [Google Scholar]
- 20.Immormino RM, Kang Y, Chiosis G, Gewirth DT. J Med Chem. 2006;49:4953. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- 21.Taldone T, Zatorska D, Patel PD, Zong H, Rodina A, Ahn JH, Moulick K, Guzman ML, Chiosis G. Bioorg Med Chem. 2011;19:2603. doi: 10.1016/j.bmc.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taliani S, Simorini F, Sergianni V, La Motta C, Da Settimo F, Cosimelli B, Abignente E, Greco G, Novellino E, Rossi L, Gremigni V, Spinetti F, Chelli B, Martini C. J Med Chem. 2007;50:404. doi: 10.1021/jm061137o. [DOI] [PubMed] [Google Scholar]
- 23.He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, Wu N, Immormino RM, Gewirth DT, Chiosis G. J Med Chem. 2006;49:381. doi: 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 24.Vial JP, Lacombe F. Methods Cell Biol. 2001;64:343. doi: 10.1016/s0091-679x(01)64021-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.