

Abstract
Maintaining protein homeostasis is vital to cell viability, with numerous studies demonstrating a role for proteasome inhibition occurring during the aging of a variety of tissues, and presumably contributing to the disruption of cellular homeostasis during aging. In the present study we sought to elucidate the differences between neurons and astrocytes in regards to basal levels of protein synthesis, proteasome-mediated protein degradation, and sensitivity to cytoxicity following proteasome inhibitor treatment. In these studies we demonstrate that neurons have an increased vulnerability, as compared to astrocyte cultures, to proteasome inhibitor-induced cytotoxicity. No significant difference was observed between these two cell types in regards to the basal rates of protein synthesis, or basal rates of protein degradation, in the pool of short-lived proteins. Following proteasome inhibitor treatment neuronal crude lysates were observed to undergo greater increases in the levels of ubiquitinated and oxidized proteins, and selectively exhibited increased levels of newly synthesized proteins accumulating within the insoluble protein pool, as compared to astrocytes. Together, these data suggest a role for increased oxidized proteins and sequestration of newly synthesized proteins to the insoluble protein pool, as potential mediators of the selective neurotoxicity following proteasome inhibitor treatment. The implications for neurons exhibiting increased sensitivity to acute proteasome inhibitor exposure, and the corresponding changes in protein homeostasis observed following proteasome inhibition, are discussed in the context of both aging and age-related disorders of the nervous system.
Keywords: Neuron, Proteasome, Ubiquitin, Oxidative stress, Astrocyte
Introduction
The degradation of proteins is necessary to maintain homeostasis and allow cells successfully respond to cellular stressors, with an increasing number of studies demonstrating a role for altered proteolysis contributing to cellular dysfunction in response to aging (1–4). In particular, a number of studies have suggested a role for impairment of the proteasome proteolytic pathway as a mediator of cell dysfunction and pathogenesis in response to aging and a number of age-related diseases (5–11). Despite such progress it remains largely unknown which protein alterations are responsible for mediating the deleterious effects of proteasome inhibition in the brain during aging and age-related disorders of the brain.
Proteins differ significantly in regards to their rates of turnover, although most commonly they are divided into groups of short- (minutes) and long-lived proteins (>12 hours), with the proteasome proteolytic pathway implicated in primarily short-lived protein degradation (12–14). Inhibition of proteasome activity has been demonstrated to activate both pro- and anti-apoptotic pathways depending on the cell type and experimental paradigm examined (15–20). Additionally, studies have shown that cells differ in regards to their susceptibility to cell death following proteasome inhibition, and that some cell types may even be protected from apoptosis by inhibition of the proteasome proteolytic pathway (21–24). The basis for these differential effects of proteasome inhibition is almost certain to occur as the result of differential effects on the proteome, but to date has never been elucidated for any cell type.
In this manuscript we identify for the first time that primary rat neurons are more susceptible to the toxicity of proteasome inhibitor treatment, as compared to primary rat astrocyte cultures. This increase in susceptibility does not appear to be due to alterations in the gross rates of basal short lived protein synthesis or short-lived protein degradation. What is observed for the first time is that neurons undergo increased levels of oxidized proteins as compared to astrocyte cultures, following proteasome inhibition, with neurons also selectively exhibiting increased accumulation of newly synthesized proteins to the insoluble protein pool following proteasome inhibition. Taken together, these data implicate a role for proteasome-mediated increases in oxidized proteins, and the accumulation of newly synthesized proteins to the insoluble protein pool, as potential mediators of the selective vulnerability of neurons to the toxicity of proteasome inhibitors.
Materials and Methods
Materials
The antibodies to β-actin (SC-47778) and ubiquitin (SC - 8017) were purchased from Santa Cruz Biotechnology Company (Santa Cruz, CA, USA). MG132 was purchased from EMD Chemicals (Gibbstown, NJ, USA). The BCA reagent was purchased from Thermo Scientific, Inc. (Waltham, Illinois, USA). Oxyblot kit was purchased from Millipore Company (Billerica, MA, USA). 35S methionine is purchased from Perkins-Elmer (Cat# NEG009A500UC; Shelton, CT, USA). All the chemicals including Hoechts, H 33342 (bisBenzamide trihydrochloride) staining, trichloroacetic acid, Triton X-100, protease inhibitor mix, EDTA, DNase I and cyclohexamide were purchased from Sigma-Aldrich, Corp. (St. Louis, MO, USA). All electrophoresis and immunoblot reagents were purchased from Bio-Rad Laboratories (Hercules, CA, USA). All cell culture supplies were obtained from GIBCO Life Sciences (Gaithersburg, MD, USA). The proteasome substrate Suc-Leu-Leu-Val-Tyr-AMC (for measurement of chymotrypsin-like activity) was purchased from Bachem Inc. (Torrance, CA, USA). The BCA and micro-BCA reagents were purchased from Thermo Scientific, Inc. (IL, USA). The black 96 well plates for reading chymotrypsin assays were purchased from Corning Incorporated (NY, USA).
Establishment and maintenance of primary neuron and astrocyte cultures: treatment with MG132
Neuronal cultures were established as described previously by our laboratory (25). Briefly, primary rat cortical neuronal cells were cultured from E18 Sprague-Dawley rats and maintained in 5% CO2 at 37°C in MEM or Neurobasal medium containing 5% fetal bovine serum (heat inactivated), N2 supplement, B27 supplement, and 1% antibiotic. Cells were used in experiments between days 6–9 (Neurons) and 12–15 (Astrocytes) post plating. Rat astrocyte cultures were established from E18 Sprague-Dawley rats as described previously by our laboratory (22). Astrocytes were maintained in 5% CO2 at 37°C in MEM medium containing 5% fetal bovine serum (heat inactivated), N2 supplement and 1% antibiotic solution. All animals were utilized in accordance with IACUC approved protocols at the Pennington Biomedical Research Center. For the analysis of protein ubiquitination and oxidation levels following proteasome inhibition, the primary cultures of neurons and astrocytes were treated with MG132 (0.1, 1 and 10 μM) for 15 hours and the pelleted cells were frozen at −80°C until further use.
Analysis of cell viability
Cell survival was determined by quantification of apoptotic and necrotic nuclei using Hoechts 344 staining as described previously (26). Briefly, Neuronal cells, treated with MG132 (0, 0.1, 1 and 10 μM) for 15 hours, were stained with the fluorescent DNA-binding dye Hoechts 344 at a concentration of 1μg/μl, and the percentage of viable cells were determined by counting the number of dead cells (condensed and fragmented nuclei) using a fluorescence microscope equipped with a 32X objective. Additional confirmation of cell viability was determined using MTT reduction as a measure of cell viability as reported previously (27).
Analysis of proteasome activity
Assays for chymotrypsin-like activity of proteasomes were performed using fluorogenic Succinyl-Leu-Leu-Val-Tyr-AMC as a substrate. All assays were carried out in 250 μl of activity assay buffer in a 96 well black assay plate with clear bottom as described previously by our laboratory [29]. Each set came from 6 dishes of from primary cell cultures and each set were assessed in triplicate. Following 1 hr incubation with MG132 the amount of proteasome activity was assayed in 23 μg lysate with the 26S proteasome activity assayed in the presence of 2 mM ATP whereas 20S proteasome activity was carried out in the presence of 0.02% SDS (which activates 20S proteasome activity and partially inhibits the 26S proteasome activity as described previously. The reaction mixture, following the addition of 50 μM of substrate, was incubated for 45 min at 37 °C and the fluorescence of the released AMC product was measured in PerkinElmer plate reader at an emission wavelength of 355 nm and an excitation wavelength of 460 nm. Relative proteasome activities were represented as percent control.
Analysis of ubiquitinated and oxidized protein levels
Protein carbonyl levels were analyzed using Oxyblot kit (Millipore) as described by the manufacturer. Briefly, 10 μg of protein from cell lysates or 1% triton soluble and insoluble fraction were derivatized with DNP and then the derivatized products were detected by the Western blot analysis as described by the manufacturer. Amounts of ubiquitinated protein in 10 μg of crude lysates or 1% triton soluble and insoluble pools were detected by Western blotting analysis using antibodies against ubiquitin.
Analysis of protein synthesis and degradation
For the analysis of proteasome mediated degradation of proteins in primary neuronal and astrocytes, cells were radio labeled with 35S methionine for 5 min or one hour as indicated and the proteasome dependent degradation was studied by chase for different time points in the presence or absence of a proteasome inhibitor,10μM MG132,. Cells, that were plated in 60mm dishes, were labeled with 35S methionine (30 μCurie) in locke’s buffer containing 154 mM NaCl, 5.6 mM KCl, 2.3 mM CaCl2, 1mM MgCl2, 3.6 mM NaHCO3, 5mM HEPES pH-7.2 plus 1mg/ml glucose. After labeling, the cells were chased for indicated time points in culture medium containing 10μg/mL cyclohexamide. The cells that were pulsed and chased in the presence of 10μM MG132 were pre incubated in the culture medium with 10μM MG132 for 1 hour, just before the labeling. After the pulse and chase, the cells were scraped in cell lysis buffer and then the proteins were precipitated with 10% TCA by incubating on ice for one hour. The precipitated protein was centrifuged at 10,000×g for 10 minutes and the counts of the insoluble material (pellet) were measured in Beckman scintillation counter.
Triton X-100 fractionation of cell lysates and analysis
Triton X-100 fractionation was done as described by Qian et al., (28) with minor modifications. We have used triton X-100 method followed by TCA precipitation to detect the accumulation and distribution of recently synthesized proteins in triton X-100 soluble and insoluble fractions, in the absence or presence of proteasome inhibitor, MG132. Briefly, neuronal and astrocyte cells were labeled for 1hour in locke’s buffer with 35S methionine in the presence or absence of MG132 as described above and chased in the cell culture medium with 10μg/mL cyclohexamide for the indicated time points. After that the cells were collected by scraping with the extraction buffer (150μl) containing 50mM Tris- HCl, pH-7.4, 150mM NaCl, and 1mM EDTA, 1% triton X-100, protease inhibitor cocktail and DNase I (buffer A). The protein lysate was incubated for 30 minutes with shaking at 4°C. Then the triton X-100 soluble and insoluble fractions were separated by centrifugation for 20 minutes at 14,000 ×g and precipitated with 1350 μL of 11% TCA to get the final concentration of TCA to ~10%. TCA insoluble counts in soluble and insoluble fractions of triton X-100 were measured using scintillation counter.
The levels of ubiquitinated and oxidized proteins in triton X-100 soluble and insoluble fractions were also analyzed by western blotting. Briefly, cells were pelleted after the incubation in cyclohexamide for indicated time points, in the presence or absence of MG132, exactly as described above except for labeling of cells with 35S methionine. The pelleted cells were suspended in extraction buffer (buffer A) and incubated for 30 minutes with shaking at 4°C and then the triton X-100 soluble and insoluble fractions were separated by centrifugation. The protein levels in triton X-100 soluble and insoluble fractions (after 10X dilution with buffer A without Triton X-100) were estimated and equal amounts of protein were used for the analysis of ubiquitination and oxidation levels using the western blotting.
Protein estimation
Protein concentration of the cell lysates was estimated using BCA (Thermo Scientific) reagent as described by the manufacturer.
Western blotting
The protein samples were analyzed by SDS-PAGE and immunoblotted with specified antibodies as described previously by our laboratory (29).
Results
Neuronal cells exhibit increased vulnerability to the toxicity of proteasome inhibitors
In order to begin to elucidate the relative susceptibilities of primary rat neuronal and astrocyte cultures to the toxicity of proteasome inhibitors, we conducted studies analyzing the severity of proteasome inhibition and induction of cell death, following treatment with the proteasome inhibitor MG132. In these studies we observed that proteasome inhibitor treatment resulted in a robust dose dependent impairment in 26S and 20S chymotrypsin-like activity in both neurons and astrocytes (Fig. 1). Interestingly, the level of impairment was nearly identical in both cell types (Fig 1). We next analyzed the levels of neuron death following treatment with MG132 (Fig. 2), and observed that every dose of MG132 induced significant levels of neuron death, and the highest dose utilized promoting nearly 90% decrease in neuronal viability. In contrast, astrocytes appeared largely resistant to the toxicity of proteasome inhibitor treatment, with only the highest doses of proteasome inhibitors promoting approximately 10% cell death (Fig. 2). Nearly identical results were obtained with the proteasome inhibitor MG115 (data not shown). Taken together, these data indicate that neuronal cells are significantly more vulnerable to the acute cytotoxicity induced by proteasome inhibitors.
Figure 1. Proteasome inhibitors induce impairment of 20S and 26S proteasomes in both neurons and astrocytes.

Primary rat astrocytes and neurons analyzed for sensitivity to proteasome inhibition following exposure to MG132. Activities of 20S and 26S were determined 1 hr following exposure to proteasome inhibitor MG132 for 1 hour, with 20S and 26S proteasome activity measured as described in methods. Data is representative of results from 2 separate experiments.
Figure 2. Proteasome inhibition induces more cell death in primary rat neurons as compared to astrocytes.

Cells were treated with increasing concentrations of the proteasome inhibitor MG132 and analyzed for cell viability 24 hours post treatment. Neurons were observed to have significantly higher levels of cell death in response to proteasome inhibitor administration (A) as compared to astrocyte cultures (B). Results using morphological criteria as well as nuclear condensation/fragmentation gave nearly identical results. Data are presented as the mean and S.E.M. of results from 3 different sets of independent experiments (5 dishes per experiment).
Neuronal cells exhibit a significantly higher level of oxidized, but not ubiquitinated, proteins following proteasome inhibitor treatment
Increases in ubiquitinated and oxidized proteins have been reported to occur in response to proteasome inhibition (30–32), but the comparison between neurons and astrocytes in regards to the severity of ubiquitinated and oxidized protein accumulation following proteasome inhibition, has not been reported previously. In our analysis we observed that proteasome inhibitor treatment resulted in a similar level of ubiquitinated protein accumulation in neurons and astrocytes (Fig. 3A). Neurons exhibited a larger degree of oxidized protein accumulation following proteasome inhibition (Fig. 3B), as compared to astrocytes.
Figure 3. The levels of oxidized proteins, but not ubiquitinated proteins, are increased to larger extent in neurons as compared to astrocytes following proteasome inhibition.

Rat primary cortical neurons and astrocyte cultures were analyzed for the levels of ubiquitinated (A) and oxidized proteins (B) following treatment with proteasome inhibitors. Cells were treated with increasing concentrations of the proteasome inhibitor 10 μM MG132 and analyzed for ubiquitinated and oxidized protein levels following proteasome inhibitor treatment for 15 hours. Neurons were observed to have more severe increases in oxidized proteins following proteasome inhibitor treatment. Data are representative of results from three separate experiments.
Short-lived protein synthesis, protein turnover, and Heat shock protein induction following proteasome inhibitor treatment
A potential basis for the increased cytotoxicity observed in neuronal cells could be based on differences in the relative rates of protein synthesis being higher in neuronal cells, and the basal rate of protein degradation being slower in neuronal cells, as compared to astrocyte cultures. In our studies we observed that neurons and astrocytes exhibited similar levels of short-lived protein synthesis and degradation (Fig. 4). Next we analyzed the basal levels of key heat shock proteins (Hsps) in neurons and astrocytes, as well as the Hsp levels following treatment with MG132. In both cell types there was observed to be an increase in Hsp40 and Hsp70 following proteasome inhibitor treatment (Fig. 5). Interestingly, the amount of Hsp40 was observed to be significantly lower in neurons as compared to astrocyte cultures (Fig 5). The levels of Hsp90 were not significantly altered in neurons and astrocytes treated with the proteasome inhibitor MG132 (Fig 5).
Figure 4. The levels of short-lived protein synthesis and degradation are similar in neurons and astrocytes.

Rat primary cortical neurons and astrocyte cultures were analyzed for protein synthesis following a 5 minute pulse of 35S-methionine. The levels of short lived protein degradation were analyzed in neuron and astrocyte cultures following 5 minute pulse of 35S-methionine and corresponding increasing lengths of chase period. Results indicate that neurons and astrocytes have similar levels of short lived protein synthesis (A) and short lived protein degradation (B). Data are presented as the mean and S.E.M. of results from 3 independent set of experiments with 4 dishes for each time point in an experiment.
Figure 5. Heat shock proteins in neurons and astrocytes following proteasome inhibition.
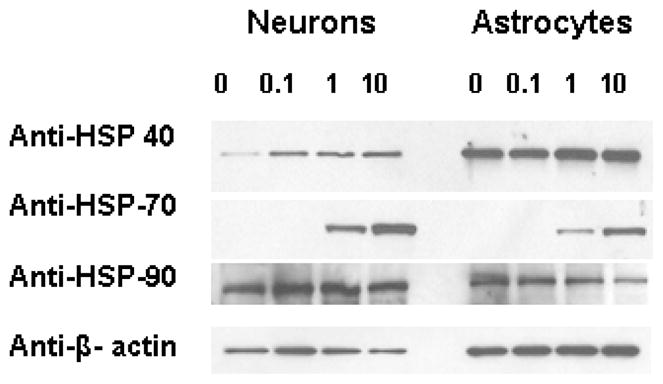
Lysates from rat primary cortical neurons and astrocyte cultures were analyzed by Western blotting for the levels of heat shock protein induction following proteasome inhibition for 15 hours. Antibodies against the Hsp40, Hsp70 or Hsp90 were used in the analysis. Beta actin was used to show the equal loading of protein lysates. Data represent the 3 independent set of experiments done under similar conditions.
Neuronal cells exhibit selective increase in recently synthesized proteins into insoluble protein pool following proteasome inhibition
Because the short-lived protein pool is believed to be predominantly degraded by the proteasome, and neurons were observed to be more vulnerable to the toxicity of proteasome inhibition, we next sought to identify if there were significant differences between neurons and astrocytes in regards to how the short lived protein pool responds following treatment with proteasome inhibitors. In pulse and chase studies we observed that neurons, but not astrocytes, underwent an increased accumulation of newly synthesized proteins within the insoluble protein pool (Fig 6), following proteasome inhibitor treatment. No significant alteration in the level or turnover of short lived proteins within the soluble protein pool was observed between neurons and astrocytes (data not shown). In contrast to our findings with crude lysates, we observed that neurons exhibited significantly higher levels of high molecular weight ubiquitinated protein in insoluble protein pool (Fig. 7) as compared to astrocytes. Additionally, no differences were observed in oxidized protein levels between astrocytes and neurons in triton x-100 soluble and insoluble fractions (Fig. 8), which directly contrasts with our findings in crude lysates. It should be noted here that neuronal cells showed higher gross levels of oxidation as compared with astrocytes only in conditions following prolonged incubation (15Hours) with proteasome inhibitor (Fig. 3B).
Figure 6. Proteasome inhibition resulted in increased accumulation of recently synthesized proteins in Triton x-100 insoluble fraction of neurons, as compared with astrocytes.

Rat primary neurons and astrocytes were pulsed for one hour with 35S-methionine and chased for indicated time points in the presence or absence of proteasome inhibitor, MG132. Whole cell lysates were separated in to triton X-100 soluble and insoluble fractions and TCA insoluble radioactivity of these fractions was measured as described in methods. Proteasome inhibition resulted in the accumulation of higher levels of recently synthesized short lived proteins in triton X-100 insoluble fractions of primary neurons (A) when compared with the astrocytes (B). Data are presented as the mean and S.E.M. of results from 3 different sets of independent experiments.
Figure 7. Proteasome inhibition resulted in increased accumulation of recently synthesized proteins, that are ubiquitinated, in triton x-100 insoluble pool of neurons as compared with astrocytes.

Rat primary Neurons and astrocytes were collected after treatment with cyclohexamide for indicated time points in the presence or absence of proteasome inhibitor, MG132, as described in methods. Whole cell lysates were fractionated by Triton X-100 and the amounts of ubiquitinated proteins in Triton X-100 soluble and insoluble fractions were analyzed using western blot analysis. Results indicate the increased accumulation of recently synthesized proteins, that are higher molecular weight ubiquitinated proteins, in triton x-100 insoluble pool of neurons (A), as compared with astrocytes (B), following proteasome inhibition.
Figure 8. Primary Neuron and astrocytes showed no differences in the levels of oxidized proteins in triton x-100 soluble and insoluble fractions following proteasome inhibition.

Rat primary neurons and astrocytes were collected after treatment with cyclohexamide for indicated time points in the presence or absence of proteasome inhibitor, MG132, as described in methods. Whole cell lysates were fractionated by triton X-100 and the amounts of oxidized proteins in triton X-100 soluble and insoluble fractions were analyzed as described in methods. Results did not show differences in the levels of oxidized proteins in triton x-100 insoluble and soluble fractions between neurons (A) and astrocytes (B) following proteasome inhibition.
Discussion
Our studies identify for the first time that primary rat neuron cultures undergo a greater degree of cytotoxicity following proteasome inhibitor treatment as compared to primary rat astrocyte cultures. These data suggest that neurons may be preferentially affected by the proteasome inhibition reported to occur in the brain during aging and age-related diseases of the nervous system (30). In this model the negative effects of proteasome inhibition in the brain during aging and age-related diseases, are likely mediated by the direct effect of proteasome inhibition on neurons, as compared to indirect neurotoxicity mediated by the effects of proteasome inhibition on non-neuronal cells. Because a primary role for the proteasome is to degrade short-lived proteins, it is likely that the toxicity of proteasome inhibition results from the ability of select proteins or pools of proteins to accumulate and promote cytotoxic events in cells. However, the basis by which such alterations in the proteome promote cytotoxicity, in particular cell type specific and/or selective cytotoxicity, remains to be elucidated experimentally. It is also important to point out that the present study utilizes primary cell cultures to understand the basis for neuronal and glial alterations in the brain, and may not fully recapitulate key aspects of brain physiology. Because of this the data need to be carefully evaluated in conjunction with additional in vivo reports to develop a comprehensive understanding for perturbations observed in the brain. Additionally, it is interesting to note that our study includes primary cells of mitotic (astrocytes) and post-mitotic cells (neurons), and demonstrates that post-mitotic cells are more sensitive to the effects of proteasome inhibitors as compared to mitotic cells, which is consistent with previous reports in the literature on this topic. Therefore, it is important to consider in conditions where proteasome inhibitors are used as chemotherapeutic agents that they may promote significant and deleterious toxicity to bystander cells, and in particular post-mitotic cells like neurons and cardiomyocytes. Such toxicity may therefore promote constraints on the utility of proteasome inhibitors as chemotherapeutic agents, which need to be carefully considered ad evaluated.
In this study we also demonstrate for the first time (using crude lysates) that proteasome inhibition promotes an increase in oxidized protein levels (increased carbonyls) in neuronal cells relative to astrocyte cultures. This data provides further support linking proteasome inhibition to the increased levels of oxidative stress observed during aging and age-related diseases of the brain. One primary question has been whether oxidized proteins result in proteasome inhibition, or whether proteasome inhibition promotes the accumulation of oxidized proteins in cells. Our data suggests that it is likely the inhibition of the proteasome that results in the accumulation of oxidized proteins in neurons, with such increases being relatively specific to neuron cultures as compared to astrocytes. This preferential increase in gross oxidized protein levels in the neuronal cells likely occurs as the result of high basal levels of oxidative stress being present in neuronal cultures relative to astrocytes, or may be due to neuronal cells being less able to mount a sufficient heat shock protein response in order to effectively cope with the levels of oxidized protein following inhibition of the proteasome. Interestingly, we have previously shown that Hsp40 levels are sufficient to modulate sensitivity to proteasome inhibitor toxicity and oxidative stress in neural cells (10), with neurons exhibiting a lower amount of Hsp40 as compared to astrocytes in the current study.
An additional important and novel observation in this study arises from our identification that following proteasome inhibition neurons exhibit a robust increase in newly synthesized proteins within the insoluble protein pool, as compared to astrocytes. This observation may be of fundamental importance to understanding the role of the proteasome in regulating neuronal homeostasis and neuropathogenesis. For example, these data raise the possibility that in neurons the proteasome plays a much more direct and significant role in rapidly degrading newly synthesized proteins, as compared to astrocyte cultures. Additionally, these data indicate that in the face of proteasome inhibition newly synthesized proteins themselves are more prone to becoming insoluble in neurons as compared to astrocyte cultures. An increase in ubiquitinated proteins within the high molecular weight fraction of insoluble protein pool of neurons, following proteasome inhibition, suggest that the newly synthesized proteins have been ubiquitinated so as to mark them to be removed by the ubiquitin proteasome system. Such data while correlative, provide additional support for the ubiquitin proteasome system in neurons being involved in the rapid bulk removal of newly synthesized proteins, and that inhibition of the proteasome results in their rapid accumulation in to insoluble protein pool.
Newly synthesized proteins would not be expected to have undergone all the necessary folding and post-translational processing that are required for the generation of functional proteins. Without such processing these proteins would be expected to be extremely hydrophobic and prone to undergoing promiscuous interactions with other polypeptides and proteins, which could be important in the promotion of the observed increases in protein insolubility. Additionally, these data may be important to explaining the increase in protein aggregation and inclusion formation that is known to occur in neurons following proteasome inhibition. Previous studies from our laboratory and others have demonstrated that proteasome inhibition results in rapid alterations in ribosome function and nucleolar homeostasis in a variety of cell types including neurons (25, 33–34). Interestingly in neurons these effects were observed to be rapid, and reversible with removal of proteasome inhibitors. This raises the possibility that the observations in the present study may therefore be due to proteasome inhibition promoting increased levels of translational errors, generation of aberrant polypeptides, and/or errors in the processing proteins as they come off of the ribosome complex. The ability of the proteasome to selectively promote these presumably aberrant proteins in neurons as compared to astrocytes suggest that they may be due to the proteasome being directly involved in the regulation of the ribosome via promoting the turnover of an as yet to be identified component of the ribosome complex.
Previous studies have identified the presence of a pool of proteins which are known as defective ribosome products (DRiPs) (28, 35), which are believed to be rapidly degraded by the proteasome-proteolytic pathway. These defective ribosome products are believed to occur as the result of misfolding or translational error, and are presumed to be rapidly degraded as they are synthesized by the ribosome. Defective ribosome products, and their degradation by the proteasome, have been studied primarily in terms of their potential role in antigen presentation (36–38). However, the presumed increased hydrophobicity of these polypeptides and proteins which allows for their isolation and analysis, may implicate a role for defective ribosome products as potential modulators in the cytotoxic effects of proteasome inhibition in multiple cell types (even non immune cells). It is interesting to note that the methodologies used to isolate DRiPs were employed for Figures 4 and 5 in the present study (28), suggesting that DRiPs may be present in both neurons and astrocytes.
How could an increase in oxidized and insoluble protein result in the increase in cytotoxicity within neurons? Firstly, previous studies have shown that even though two cell types exhibit similar levels of proteasome inhibition following proteasome inhibitor treatment, they can vary wildly in the amount of cytotoxicity elicited (22). There are many direct and indirect routes by which this could occur, and further exploration of each of the many potential pathways is likely to be important in understanding the potential role of proteasome inhibition as a mediator of neuron dysfunction in aging and age-related diseases of the brain. For example, current dogma suggests that oxidative stress contributes to cellular dysfunction (39–44) and in large part via the accumulation of oxidized proteins promoting an imbalance in multiple cellular functions due to the presumed loss of function that occurs as the result of protein oxidation. Our data suggest that interactions between global increases in oxidized protein levels, an insolubility of newly synthesized proteins in neurons, serves as a novel basis by which proteasome inhibition promotes neuronal toxicity. In this model there is not only the loss of protein function (from loss of newly synthesized proteins into an insoluble pool), but also the genesis of insoluble proteins in neurons which are capable of promoting neuron death via direct as well as indirect pathways.
References
- 1.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez KA, Gaczynska M, Osmulski PA. Molecular mechanisms of proteasome plasticity in aging. Mech Ageing Dev. 2010;131:144–55. doi: 10.1016/j.mad.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chondrogianni N, Gonos ES. Proteasome dysfunction in mammalian aging: steps and factors involved. Exp Gerontol. 2005;40:931–938. doi: 10.1016/j.exger.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Ramanujan VK, Herman BA. Aging process modulates nonlinear dynamics in liver cell metabolism. J Biol Chem. 2007;282:19217–19226. doi: 10.1074/jbc.M700572200. [DOI] [PubMed] [Google Scholar]
- 5.Ding Q, Dimayuga E, Keller JN. Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid Redox Signal. 2006;8:163–172. doi: 10.1089/ars.2006.8.163. [DOI] [PubMed] [Google Scholar]
- 6.Farout L, Friguet B. Proteasome function in aging and oxidative stress: implications in protein maintenance failure. Antioxid Redox Signal. 2006;8:205–216. doi: 10.1089/ars.2006.8.205. [DOI] [PubMed] [Google Scholar]
- 7.van Tijn P, Hol EM, van Leeuwen FW, Fischer DF. The neuronal ubiquitin-proteasome system: murine models and their neurological phenotype. Prog Neurobiol. 2008;85:176–193. doi: 10.1016/j.pneurobio.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Dahlmann B. Role of proteasomes in disease. BMC Biochem. 2007;8(Suppl 1):S3. doi: 10.1186/1471-2091-8-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray DA, Tsirigotis M, Woulfe J. Ubiquitin, proteasomes, and the aging brain. Sci Aging Knowledge Environ. 2003;34:re6. doi: 10.1126/sageke.2003.34.re6. [DOI] [PubMed] [Google Scholar]
- 10.Ding Q, Keller JN. Proteasome inhibition in oxidative stress neurotoxicity: implications for heat shock proteins. J Neurochem. 2001;77:1010–1017. doi: 10.1046/j.1471-4159.2001.00302.x. [DOI] [PubMed] [Google Scholar]
- 11.Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, Mayer RJ, Layfield R. Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:9902–9906. doi: 10.1073/pnas.170173897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuertes G, Villarroya A, Knecht E. Role of proteasomes in the degradation of short-lived proteins in human fibroblasts under various growth conditions. Int J Biochem Cell Biol. 2003;35:651–664. doi: 10.1016/s1357-2725(02)00382-5. [DOI] [PubMed] [Google Scholar]
- 13.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 14.Medicherla B, Goldberg AL. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. Cell Biol. 2008;182:663–673. doi: 10.1083/jcb.200803022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srimatkandada P, Loomis R, Carbone R, Srimatkandada S, Lacy J. Combined proteasome and Bcl-2 inhibition stimulates apoptosis and inhibits growth in EBV-transformed lymphocytes: a potential therapeutic approach to EBV-associated lymphoproliferative diseases. Eur J Haematol. 2008;80:407–418. doi: 10.1111/j.1600-0609.2008.01044.x. [DOI] [PubMed] [Google Scholar]
- 16.Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Ottosson A, Ji C, Feng X, Nordenskjold M, Henter JI, Fadeel B, Zheng C. Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses NKp46-mediated cytotoxicity. Haematologica. 2009;94:470–478. doi: 10.3324/haematol.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280:14189–141202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 19.Yang W, Monroe J, Zhang Y, George D, Bremer E, Li H. Proteasome inhibition induces both pro- and anti-cell death pathways in prostate cancer cells. Cancer Lett. 2006;243:217–227. doi: 10.1016/j.canlet.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 20.Yew E, Cheung NS, Choy MS, Qi RZ, Lee AY, Peng ZF, Melendez AJ, Manikandan J, Koay ES, Chiu LL, Ng WL, Whiteman M, Kandiah J, Halliwell B. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: a microarray analysis. J Neurochem. 2005;94:943–956. doi: 10.1111/j.1471-4159.2005.03220.x. [DOI] [PubMed] [Google Scholar]
- 21.van Leyen K, Siddiq A, Ratan RR, Lo EH. Proteasome inhibition protects HT22 neuronal cells from oxidative glutamate toxicity. J Neurochem. 2005;92:824–830. doi: 10.1111/j.1471-4159.2004.02915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shringarpure R, Catley L, Bhole D, Burger R, Podar K, Tai YT, Kessler B, Galardy P, Ploegh H, Tassone P, Hideshima T, Mitsiades C, Munshi NC, Chauhan D, Anderson KC. Gene expression analysis of B-lymphoma cells resistant and sensitive to bortezomib. Br J Haematol. 2006;134:145–156. doi: 10.1111/j.1365-2141.2006.06132.x. [DOI] [PubMed] [Google Scholar]
- 23.Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schroter M, Becherer JD, Irmler M, Tschopp J, Martinou JC. Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 1996;15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- 24.Goldbaum O, Vollmer G, Richter-Landsberg C. Proteasome inhibition by MG-132 induces apoptotic cell death and mitochondrial dysfunction in cultured rat brain oligodendrocytes but not in astrocytes. Glia. 2006;53:891–901. doi: 10.1002/glia.20348. [DOI] [PubMed] [Google Scholar]
- 25.Ding Q, Dimayuga E, Markesbery WR, Keller JN. Proteasome inhibition induces reversible impairments in protein synthesis. FASEB J. 2006;20:1055–1063. doi: 10.1096/fj.05-5495com. [DOI] [PubMed] [Google Scholar]
- 26.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruman I, Bruce AJ, Bredesen DE, Waeg G, Mattson MP. Evidence that 4-Hydroxynonenal Mediates Oxidative Stress- Induced Neuronal Apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian SB, Bennink JR, Yewdell JW. Quantitating defective ribosome products. Methods Mol Biol. 2005;301:271–281. doi: 10.1385/1-59259-895-1:271. [DOI] [PubMed] [Google Scholar]
- 29.Dasuri K, Zhang L, Ebenezer P, Liu Y, Fernandez-Kim SO, Keller JN. Aging and dietary restriction alter proteasome biogenesis and composition in the brain and liver. Mech Ageing Dev. 2009;130:777–783. doi: 10.1016/j.mad.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dasuri K, Ebenezer P, Zhang L, Fernandez-Kim SO, Bruce-Keller AJ, Markesbery WR, Keller JN. Increased protein hydrophobicity in response to aging and Alzheimer disease. Free Radic Biol Med. 2010;48:1330–1337. doi: 10.1016/j.freeradbiomed.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- 32.Powell SR, Wang P, Divald A, Teichberg S, Haridas V, McCloskey TW, Davies KJ, Katzeff H. Aggregates of oxidized proteins (lipofuscin) induce apoptosis through proteasome inhibition and dysregulation of proapoptotic proteins. Free Radic Biol Med. 2005;38:1093–1101. doi: 10.1016/j.freeradbiomed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Ding Q, Cecarini V, Keller JN. Interplay between protein synthesis and degradation in the CNS: physiological and pathological implications. Trends Neurosci. 2007;30:31–36. doi: 10.1016/j.tins.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Stavreva DA, Kawasaki M, Dundr M, Koberna K, Muller WG, Tsujimura-Takahashi T, Komatsu W, Hayano T, Isobe T, Raska I, Misteli T, Takahashi N, McNally JG. Potential roles for ubiquitin and the proteasome during ribosome biogenesis. Mol Cell Biol. 2006;26:5131–5145. doi: 10.1128/MCB.02227-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yewdell JW. Serendipity strikes twice: the discovery and rediscovery of defective ribosomal products (DRiPS) Cell Mol Biol (Noisy-le-grand) 2005;51:635–641. [PubMed] [Google Scholar]
- 36.Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 37.Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157:1823–1826. [PubMed] [Google Scholar]
- 38.Yewdell JW, Schubert U, Bennink JR. At the crossroads of cell biology and immunology: DRiPs and other sources of peptide ligands for MHC class I molecules. J Cell Sci. 2001;114:845–851. doi: 10.1242/jcs.114.5.845. [DOI] [PubMed] [Google Scholar]
- 39.Moreira PI, Santos MS, Oliveira CR, Shenk JC, Nunomura A, Smith MA, Zhu X, Perry G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol Disord Drug Targets. 2008;7:3–10. doi: 10.2174/187152708783885156. [DOI] [PubMed] [Google Scholar]
- 40.Giasson BI, Ischiropoulos H, Lee VM, Trojanowski JQ. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer’s and Parkinson’s diseases. Free Radic Biol Med. 2002;32:1264–1275. doi: 10.1016/s0891-5849(02)00804-3. [DOI] [PubMed] [Google Scholar]
- 41.Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ, Perry G. Oxidative stress and neurodegeneration. Ann N Y Acad Sci. 2005;1043:545–552. doi: 10.1196/annals.1333.062. [DOI] [PubMed] [Google Scholar]
- 42.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith MA, Sayre LM, Monnier VM, Perry G. Oxidative posttranslational modifications in Alzheimer disease. A possible pathogenic role in the formation of senile plaques and neurofibrillary tangles. Mol Chem Neuropathol. 1996;28:41–48. doi: 10.1007/BF02815203. [DOI] [PubMed] [Google Scholar]
- 44.Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal. 2005;7:685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]