

Abstract
Background
The TRPA1 receptor is directly activated by a wide range of chemicals including many endogenous molecules relevant for esophageal pathophysiology. We addressed the hypothesis that the TRPA1 agonists differentially activate esophageal nociceptive subtypes depending on their embryological source (neural crest or epibranchial placodes).
Methods
Single cell RT-PCR and whole cell patch clamp recordings were performed on the vagal neurons retrogradely labeled from the guinea pig esophagus. Extracellular recordings were made in the isolated innervated esophagus preparation ex vivo.
Key results
Single cell RT-PCR revealed that the majority of the nodose (placodes-derived) and jugular (neural crest-derived) TRPV1-positive esophageal nociceptors express TRPA1. Single fiber recording showed that the TRPA1 agonists allyl-isothiocyanate (AITC) and cinnamaldehyde were effective in inducing robust action potential discharge in the nerve terminals of nodose nociceptors, but had far less effect in jugular nociceptors (approximately fivefold less). Higher efficacy of the TRPA1 agonists to activate nodose nociceptors was confirmed in the isolated esophagus-labeled vagal neurons in the whole cell patch clamp studies. Similarly to neural crest-derived vagal jugular nociceptors, the spinal DRG nociceptors that are also neural crest-derived were only modestly activated by allyl-isothiocyanate.
Conclusions & Inferences
We conclude that the TRPA1 agonists are substantially more effective activators of the placodes-derived than the neural crest-derived esophageal nociceptors. Our data predict that in esophageal diseases the presence of endogenous TRPA1 activators will be preferentially signaled by the vagal nodose nociceptors.
INTRODUCTION
The heartburn and esophageal pain resistant to acid suppression are prevalent clinical problems (1). Such conditions, often classified as non-erosive esophagitis, functional heartburn or non-cardiac chest pain, are characterized by limited esophageal inflammation and poor responsiveness to available therapies (2, 3). The mechanisms underlying the heartburn and pain in these circumstances are incompletely understood, but involve the activation of the damage-sensing afferent nerves (nociceptors) in the esophagus. Thus, better understanding of the activation of the esophageal nociceptors is essential for progress in this area (4).
The TRPA1 channel is one of the leading drug targets for the treatment of pain (5,6). This is largely because of its preferential expression in peripheral nociceptors and its activation by an unusually wide range of endogenous molecules associated with inflammation and tissue damage. The endogenous TRPA1 activators relevant for esophageal physiology are exemplified by certain products of oxidative and nitrosative stress (7–9) occurring in the esophagus exposed to refluxed acid (10, 11). Other TRPA1 activators including many reactive molecules commonly produced by immune cells (12) are also likely produced in the esophagus in pathological circumstances. Previous studies demonstrated that TRPA1 contributes to mechanotransduction and sensitization in subsets of esophageal afferent nerves (13, 14), but the TRPA1-mediated activation of esophageal nociceptors has not been analyzed.
The afferent nociceptive innervation of the esophagus is complex (15). A large population of the esophageal nociceptors is derived from the embryonic neural crest: the spinal nociceptors with the neurons in dorsal root ganglia (DRG) and the vagal jugular nociceptors with the neurons in vagal jugular ganglia. The remaining nociceptors are derived from embryonic epibranchial placodes and have their neurons located in vagal nodose ganglia. The neural crest- and placodes-derived esophageal nociceptive subtypes share defining nociceptive properties (discriminative mechanical responsiveness and expression of the capsaicin receptor TRPV1)(16), but differ in a number of activation mechanisms, notably the purinergic and serotoninergic transduction (17–19). The esophageal nociceptive subtypes are predicted to regulate different aspects of esophageal nociception (15).
In a preliminary study we noted that TRPA1 agonists were relatively ineffective activators of the vagal neural crest-derived esophageal nociceptors. However, published studies showed that TRPA1 agonists effectively stimulate many vagal presumably placodes-derived bronchopulmonary nociceptors (20, 21). Based on these observations we developed a hypothesis that TRPA1 agonists are substantially more effective to activate the placodes-derived than the neural crest-derived nociceptors in the esophagus. Differential responsiveness of esophageal nociceptive subtypes to TRPA1 agonists would influence the reflexes and sensations elicited by endogenous TRPA1 activators in the innervated esophageal tissue.
METHODS
All experiments described in this study were approved by the Johns Hopkins Animal Use and Care Committee and Jessenius Medical School Ethic Committee.
Retrograde labeling of the afferent neurons projecting into the esophagus was performed as described previously (18, 19). In a brief ketamine (50mg/kg) and xylazine (2.5mg/kg) anesthesia, the cervical esophagus was surgically exposed and the retrograde tracer DiI (0.1% in 10% DMSO in sterile saline) was injected into the esophageal wall (1–2) sites up to the total volume 5–10µl. The vagal nodose, vagal jugular and spinal DRG (T1–T4) ganglia were harvested 10–15 days later, enzymatically dissociated and plated on coverslips. The neurons were used within 6h for picking single neurons (RT-PCR analysis) or within 24h for electrophysiology (patch clamp).
Single cell RT-PCR was performed as described previously (17, 19, 22). First strand cDNA was synthesized from single esophagus-labelled afferent neurons by using the Super-Script(tm) III CellsDirect cDNA Synthesis System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. Cell Picking: Coverslips with dissociated neurons were constantly superfused with simple buffer and DiI-labeled identified by fluorescence microscopy were individually harvested into a glass-pipette (tip diameter 50–150 µm). The pipette tip with the cell was broken in a PCR tube containing resuspension buffer (1µl, CellsDirect system) and RNAse Inhibitor (RNAseOUT, 2 U l−1, Invitrogen), immediately frozen and stored at −20°C. Only the neurons free of debris and attached cells were collected. From one coverslip, one to five cells were collected. A sample of the bath solution was collected in some coverslips for no-template (bath) controls. RT-PCR: Samples were defrosted, lysed (10 min at 75°C), treated with DNAse I, poly(dT) and random hexamer primers (Roche Applied Bioscience) were added. The samples were reverse transcribed by adding SuperscriptIII RT for cDNA synthesis. In some experiments a portion of the volume (50–75%) was reverse transcribed with RT, whereas RT was omitted in the remaining sample used in the following as RNA control. 3 to 4 µl of each sample (cDNA, RNA control or bath control, respectively) were used for PCR amplification by the HotStar Taq Poymerase Kit (Qiagen) according to the manufacturer's recommendations (final volume 20µl). After an initial activation step at 95°C (15 min), cDNAs were amplified by 50 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 1 min followed by a final extension at 72°C for 10 min. Products were then visualized in ethidum-bromide stained 1.5 % agarose gels. The custom-synthesized intron-spanning primers (Invitrogen) were used (5’-forward-3’, 5’-reverse-3’, predicted product size, NCBI Reference Sequence number) : TRPA1 (TTAGCAACTGCCTCTGCATC, TACCAGCGCCTTGATCTCTT, 177 bp, NM_001198770.1), TRPV1 (CAGAGAGCCATCACCATCCT, GGGACCAGGGCAAAGTTC, 284 bp, AY729017). Genomic products for these primers predicted by in silico PCR (UCSC Genome Bioinformatics) are >1500bp and would be clearly distinguishable if amplified.
Extracellular recording was performed as described previously (16, 18, 19). Ex vivo single fiber recordings were made from vagal or DRG neurons with mechanosensitive nerve terminals in the esophagus in isolated, perfused, vagally- or spinally-innervated guinea pig esophagus preparations. The tissue and the ganglia were secured in separate chambers and superfused (4–6 ml/min) with indomethacin (3 µM) containing Krebs solution (118mM NaCl, 5,4mM KCl, 1mM NaH2PO4, 1.2mM MgSO4, 1.9 mM CaCl2, 25 mM NaHCO3, 11mM dextrose, gassed with 95%O2/5% CO2, pH=7.4, 35°C). The silver/silver chloride return electrode and earthed pellet were placed in the recording compartment. The aluminosilicate glass microelectrode (2 MΩ) filled with 3M sodium chloride was micromanipulated into a studied ganglion. The recorded signal was amplified (Microelectrode AC amplifier 1800, A-M Systems) and filtered (low cut-off, 0.3 kHz; high cut-off, 1 kHz) and analyzed on Apple computer using the software TheNerveOfIt (sampling frequency 33 kHz; PHOCIS, Baltimore, MD, US). Isobaric esophageal distention was induced by increasing intraluminal pressure by using custom-made gravity-driven pressure generator. An afferent fiber with a mechanosensitive receptive field in the esophagus was identified by the response to esophageal distention and focal mechanical stimulation (von Fray hair). The chemicals diluted in Krebs solution were delivered to the esophagus in the external perfusion for indicated periods of time. The nerve activity (action potential discharge) was monitored continuously and analyzed in 1 s bins (Hz) and 10 s bins (mean±SEM). The peak frequency (Hz) was defined as the maximal 1s bin action potential discharge. The sustained activation was better reflected by evaluating the maximum discharge in 10s bins. The response to a pharmacological/chemical stimulus was considered positive if it evoked action potential discharge with peak frequency at least 3 Hz (in the fibers with no baseline activity or a peak frequency at least three times the frequency of baseline activity in fibers with baseline activity). The baseline activity was recorded 5–15 min just prior to application of a pharmacological stimulus. Only the nerve fibers with confirmed drug access by a positive response to at least one agonist were included in the analysis. Paired or unpaired T-test was used as appropriate. As detailed previously, the nociceptive vagal afferent nerve fibers were identified by the linear non-saturating response to esophageal distention up to the noxious levels (100mmHg)(Yu et al., 2005; Yu et al., 2008) combined with the responsiveness to capsaicin tested in the majority of the nerve fibers at the end of experiment. The vehicle used to deliver the TRPA1 agonists was DMSO in the maximum concentration of 0.03%. DMSO had no effect on the activity of the esophageal nociceptive fibers. In nodose nociceptors (10 min baseline, 10 min 0.2%DMSO) the peak action potential discharge was 1.8±0.4Hz (baseline) vs. 2.6±0.6 Hz (DMSO) (n=9, NS), and maximum discharge in 10s bins was 3.4±1.3/10s (baseline) vs. 4.2±1.0/10s (DMSO) (n=9, NS). In jugular nociceptors (15 min baseline, 15 min 0.03%DMSO) the peak action potential discharge was 1.6±0.4Hz (baseline) vs. 1.9±0.6Hz (DMSO) (n=8, NS) and maximum discharge in 10s bins was 3.8±1.3/10s (baseline) vs. 3.1±1.0/10s (DMSO) (n=8, NS).
Whole cell patch clamp recordings were performed as described previously (17–19). In order to preserve intracellular signaling pathways, a gramicidin-perforated whole-cell patch-clamp technique was employed. The recordings were performed using Multiclamp 700A amplifier and Axograph 4.9 software. The pipette (1.5–3MΩ) was filled with a pipette solution composed of KCl (140 mM), CaCl2 (1mM), MgCl2 (2mM), EGTA (11mM), HEPES (10mM) and dextrose (10mM) titrated to pH 7.3 with KOH (304 mosmol l−1) containing gramicidin 2–3.5µg/ml (was dissolved in DMSO 1mg/ml and mixed with the pipette solution just prior to each recording). After forming a gigaohm seal, cell membrane potential was held at −60mV. The inclusion criteria were the series resistance < 30MΩ and the membrane resistance (determined from a series of negative current steps) >100 MΩ. In voltage clamp mode recordings were made after whole cell capacitance compensation. During the experiments, the cells were continuously superfused (6ml/min) with Locke’s solution (35–37°C); composed of (mM): 136 NaCl, 5.6 KCl, 1.2 MgCl2, 2.2 CaCl2, 1.2 NaH2PO4, 14.3 NaHCO3 and 10 dextrose gassed with 95% O2–5% CO2 (pH 7.3–7.4). The data are presented as mean±SEM of inward current density (inward current normalized for the cell capacitance, pA/pF). In some experiments during the response to supramaximal concentration of capsaicin (1µM) a sharp drop of series resistance to <5MΩ was measured indicating conversion of perforated to ruptured recording. Inasmuch as the series resistance influences the measurement of the current amplitude, these results were excluded from the quantitative analysis of the capsaicin response.
The following drugs were used (source, stock solution concentration, solvent): allyl-isothiocyanate (Sigma, 1M, dimethylsulfoxide), cinnamaldehyde (Sigma, 1M, dimethylsulfoxide), capsaicin (Sigma, 10mM, ethanol) and AP-18 (Biomol, 0.1M, dimethylsulfoxide ). Stock solutions were stored at −20°C. The drugs were diluted to their final concentrations in Krebs solution for extracellular experiments and Locke’s solution for patch clamp experiments shortly before use.
RESULTS
Single cell RT-PCR studies
First we investigated whether the TRPA1 receptor is expressed in esophageal nociceptors. Nodose and jugular neurons projecting the nociceptors (capsaicin-sensitive nerve fibers) into the esophagus were identified by retrograde tracing and expression of the capsaicin receptor TRPV1 by single cell RT-PCR (17, 19). TRPA1 was detected in ≈95% (16/17) of the nodose and ≈75% (20/27), of the jugular esophagus-specific TRPV1-positive neurons (Fig. 1). These data show that the majority of the nodose and jugular nociceptors in the guinea pig esophagus express TRPA1. In control experiments a portion of a single neuron sample was processed without reverse transcriptase (RT- controls). No TRPA1 signal was detected in any of the RT- controls from the TRPA1-positive neurons (n=11). No signal was detected from the samples of bath fluid superfusing the neurons during cell collection (n=3) and from water controls (n=6).
Figure 1. The majority of the nodose and jugular esophageal nociceptors express TRPA1.
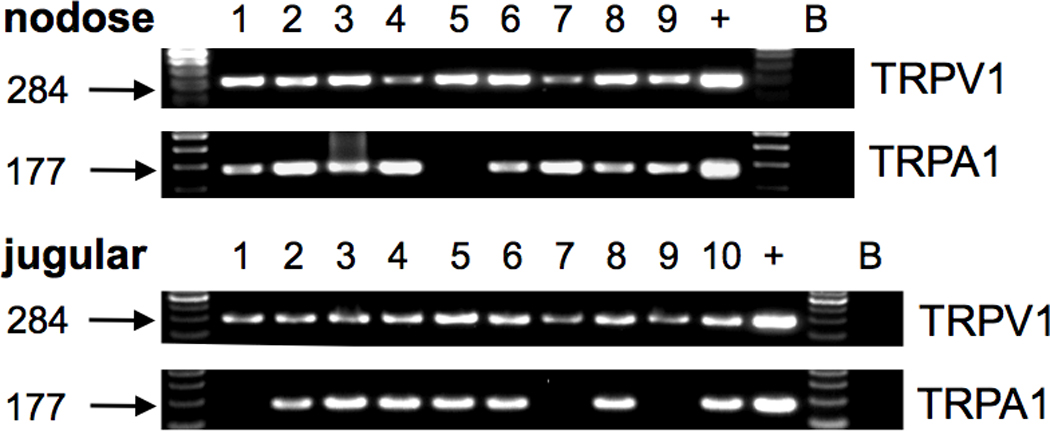
Examples of the single cell RT-PCR on the TRPV1-positive (nociceptive) nodose and jugular neurons retrogradely labeled from the esophagus. Individual neurons are numbered. TRPA1 was expressed in ≈95% (16/17) of the nodose and ≈75% (20/27) of the jugular TRPV1-positive neurons (NS, χ2-test). +, whole nodose ganglion cDNA (positive control), B, sample of superfusing bath solution containing no cell (negative control).
Extracellular recordings studies
Next we investigated the activation of the nerve terminals of esophageal nociceptors by TRPA1 agonists. In order to avoid cross-desensitization of the response to TRPA1 agonists by the TRPV1 activation (23), the TRPA1 agonists were always applied prior to capsaicin. As detailed in Methods the vehicle for TRPA1 agonists (DMSO) had no effect on the activity of esophageal nociceptors. The response to a tested agonist was considered positive if the agonist evoked action potential discharge with peak frequency at least 3 Hz (in the fibers with no baseline activity or a peak frequency at least three times the frequency of baseline activity in fibers with baseline activity). The TRPA1 agonist allyl-isothiocyanate (AITC, 100µM) evoked a robust action potential discharge with peak frequency averaging 33.5±4.5 Hz in 9 of 11 nodose nociceptors (Fig. 2, p<0.01 compared to peak frequency of baseline discharge 1.9±1.3 Hz, paired T-test). In the AITC-unresponsive nodose nociceptors the drug access to the nerve terminal was confirmed by the response to capsaicin (1µM). The response to AITC (100µM) consisted of either a sustained action potential discharge (example shown in Fig. 2A) or frequent robust bursts of action potentials. The response to AITC (100µM) in nodose nociceptors commenced after a variable delay averaging 5±1 min (Fig. 2A). Similar delays attributable to drug equilibration and diffusion in the tissue were consistently observed with activating chemicals in our innervated esophagus preparation (16, 19). Lower concentrations of AITC did not have a consistent stimulating effect. AITC (30µM) evoked no activation in 4 nodose nociceptors, but induced a robust activation (peak frequency 21Hz) in the remaining nodose nociceptor. The TRPA1 antagonist AP-18 (24, 25) completely prevented AITC-induced activation. Thus, AITC (100µM, 15 min) evoked no activation over the baseline in the presence of AP-18 (30µM, including 30 min pre-incubation) (peak discharge 1.8+0.5 Hz and 1.7±0.6 Hz, respectively, n=5, NS). The inhibitory effect of AP-18 was at least partially reversible. After 60–90 min wash AITC (100µM) induced a robust activation in all 5 nodose nociceptors tested (peak discharge 17±3Hz). AP-18 (30µM) had no effect on the mechanical response of nodose nociceptors (esophageal distention to 60 mmHg for 20s evoked 56±15 and 51±14 action potentials in the absence and presence of 30µM AP-18, respectively, n=6, NS). These results provide the evidence that the response of nodose nociceptors to AITC in the concentration of 100µM is mediated by the TRPA1 receptor.
Figure 2. The TRPA1 agonists are more effective in activating the nodose than the jugular esophageal nociceptors.
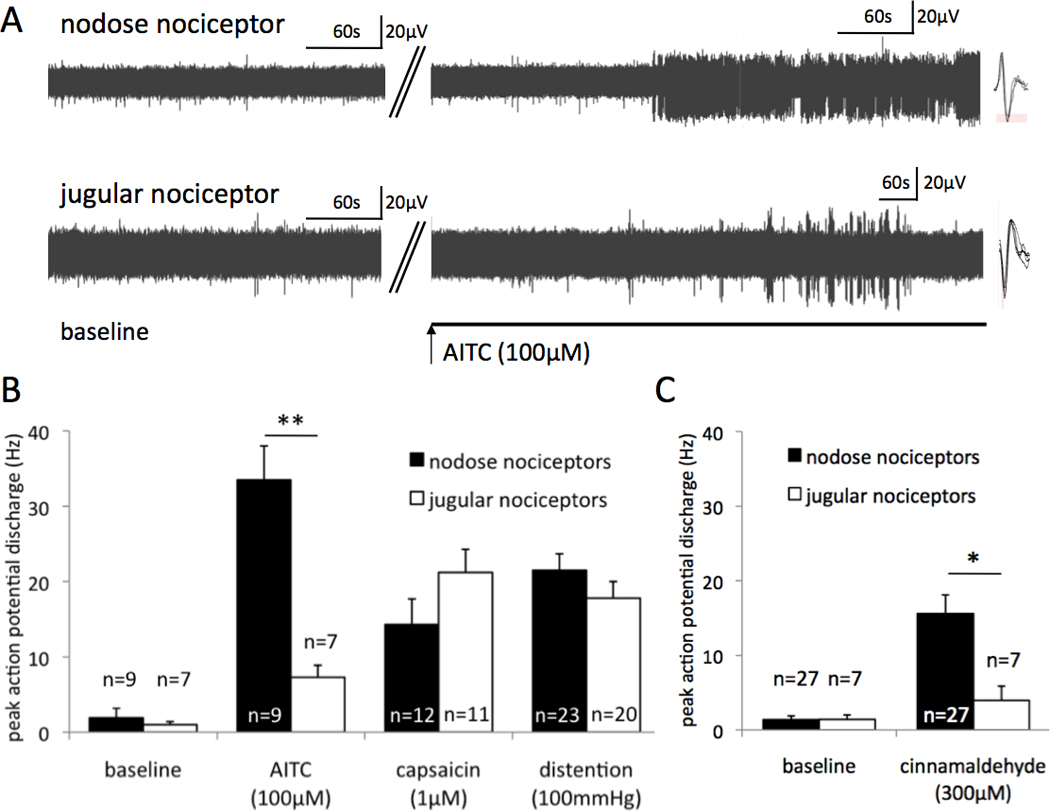
Ex vivo single unit extracellular recordings were made from the nodose and jugular nociceptors innervating the esophagus. (A) Representative traces of the baseline activity and the action potential discharge induced by allyl isothiocyanate (AITC) in a nodose nociceptor and a jugular nociceptor. Insets: Individual action potential shapes. (B) The magnitude of the response to AITC (in AITC-responsive nociceptors), capsaicin and high level of esophageal distention (100mmHg) in the nodose and jugular nociceptors. The baseline was quantified for AITC experiments (paired). **p<0.01. (C) The magnitude of the activity evoked by cinnamaldehyde averaged in all nociceptors tested. *p<0.05
For comparison, we evaluated the response of nodose nociceptors to two effective noxious stimuli, the supramaximal concentration of capsaicin (1µM) and supra-physiological (noxious) esophageal distention (100mmHg). Figure 2B shows that in the nodose nociceptors the TRPA1-mediated activation by AITC (100µM) is approximately twice as effective as these stimuli. The high efficacy of AITC was even more obvious when more sustained activation quantified as the maximum 10s bin discharge was analyzed (i.e. a maximal number of action potentials over the baseline in a 10s bin, see Methods for details). The maximum response to AITC (100µM, n=9), capsaicin (1µM, n=12) and distention (100mmHg, n=23) was 154±37, 62±14 and 69±9 action potentials/10s, respectively. AITC (100µM) caused a modest mechanical desensitization in the responsive nodose nociceptors reducing the distention-induced activation on average by ≈35% (distention to 60 mmHg for 20s evoked 61±15 and 43±12 action potentials prior and after perfusion with AITC, n=9, p<0.01). This mechanical desensitization was partially reversible (not shown).
In contrast to its effect on nodose nociceptors, AITC had relatively modest effect on the jugular nociceptors (Fig. 2). AITC (100µM) induced overt action potential discharge (peak frequency 7.3±1.6 Hz) in 7/10 jugular nociceptors (p<0.01 compared to peak frequency of baseline discharge 1.0±0.4Hz, paired T-test). In the AITC-unresponsive jugular fibers the drug access to the nerve terminal was confirmed by the response to capsaicin (1µM). The response to AITC in jugular fibers commenced after a variable delay averaging 8±1 min (NS compared to the delay in nodose nociceptors) and typically consisted of an irregular low frequency discharge sometimes combined with irregular bursts of action potentials smaller than in nodose nociceptors (Fig. 2A). This lower efficacy is also reflected by a modest maximum 10s bin discharge in response to AITC (100µM, 15±4 action potentials/10s over baseline, n=7) substantially smaller than in the nodose nociceptors (p<0.01). For comparison, the maximum 10s bin discharge in jugular nociceptors induced by capsaicin (1µM) and esophageal distention (100mmHg) was 60±9 (n=11) and 67±15 (n=20) action potentials/10s, respectively. Increasing the concentration of AITC (300–1000µM) did not induce a significantly more robust response in the jugular nociceptors (n=5, not shown). Thus AITC (100µM) was about fivefold less effective to activate jugular than nodose nociceptors.
Similar to AITC, another TRPA1 agonist cinnamaldehyde was notably more effective in stimulating nodose than jugular nociceptors. Cinnamaldehyde (300µM) evoked robust activation in 22 of 27 nodose nociceptors. The peak action potential discharge in the cinnamaldehyde-postive nodose nociceptors during cinnaldehyde perfusion and baseline was 18.8±2.7Hz and 1.1±0.3Hz, respectively (p<0.01, n=22). The maximum 10s bin discharge over baseline in response to cinnamaldehyde (300µM) was 82±17 action potentials/10s. In contrast to nodose nociceptors, cinnamaldehyde (300µM) evoked positive response (defined by peak frequency at least 3 Hz in the fibers with no baseline activity or a peak frequency at least three times the frequency of baseline activity in fibers with baseline activity, see Methods) only in 2 of 7 jugular nociceptors (the peak discharge in these two fibers was 3Hz and 15Hz over peak baseline 1Hz and 0Hz, respectively). The proportion of the jugular nociceptors responsive to 300µM cinnamaldehyde (2/7) was significantly smaller than the proportion of nodose nociceptors (22/27) (p<0.05, χ2-test). For the purpose of quantitative comparison the response was averaged in all nodose (n=27) and jugular (n=7) nociceptors tested (Fig. 2C). In 4 jugular nociceptors increasing the concentration of cinnamaldehyde to 1000µM did not uncover a more robust response (not shown). In contrast to the difference in the response to TRPA1 agonists, nodose and jugular nociceptors responded similarly to control stimuli capsaicin (1µM) and esophageal distention to 100mmHg (Fig. 2B) consistent with our previous reports (16, 18). Thus, the marked difference in the responsiveness of nodose vs. jugular nociceptors to TRPA1 agonists was not noted with other nociceptive stimuli.
Extracellular calcium may inhibit the responses to TRPA1 agonists by causing desensitization of TRPA1 channel upon TRPA1-mediated calcium entry (23, 26). We therefore investigated if lowering the extracellular calcium influences response of the jugular nociceptive fibers. Low extracellular calcium has been reported to increases the excitability in jugular afferent nerve fibers (27). Indeed, omitting calcium in the superfusing Krebs bicarbonate solution resulted in approximately 3-fold increase in the response of jugular nociceptors to esophageal distention (n=4). However, the response to AITC (100µM) was not increased. The peak action potential discharge was 11±2 Hz (n=4) in the Krebs buffer without calcium compared to 7±2Hz in the presence of calcium, n=8, NS, unpaired T-test). These data argue against the possibility that extracellular calcium contributes to the lower apparent response of jugular nociceptors to the TRPA1 agonists.
Patch clamp studies
In a complex esophageal tissue the magnitude of the response of the nerve fiber terminals to TRPA1 agonists may be influenced by factors such as diffusion rate, nerve terminal area and location, or secondary modulators produced by local cells affected by TRPA1 agonists. In order to address these issues and gain further mechanistic insight we evaluated the response to TRPA1 agonists in the isolated cell bodies of esophageal nociceptors. The neurons projecting the capsaicin-sensitive nerve fibers into the esophagus were identified by retrograde labeling and the response to capsaicin (1µM) added at the end of experiment. Consistent with our findings in the nerve terminals we observed that the TRPA1 agonist AITC induced substantially larger responses in the nodose than in the jugular esophageal nociceptive neurons (Fig. 3). The peak current density of the inward currents induced by AITC (100µM) in the nodose neurons was significantly larger than in the jugular neurons. In contrast, the response to maximally-effective concentration of capsaicin (1µM) was similar between the nodose and jugular neurons consistent with our previous studies (18, 19). These data indicate that the difference between the responsiveness of the nodose C-fibers and jugular nerve fibers is intrinsic to the afferent nerves.
Figure 3. The TRPA1 agonist AITC is more effective in activating the nodose than the jugular esophageal nociceptive neurons.
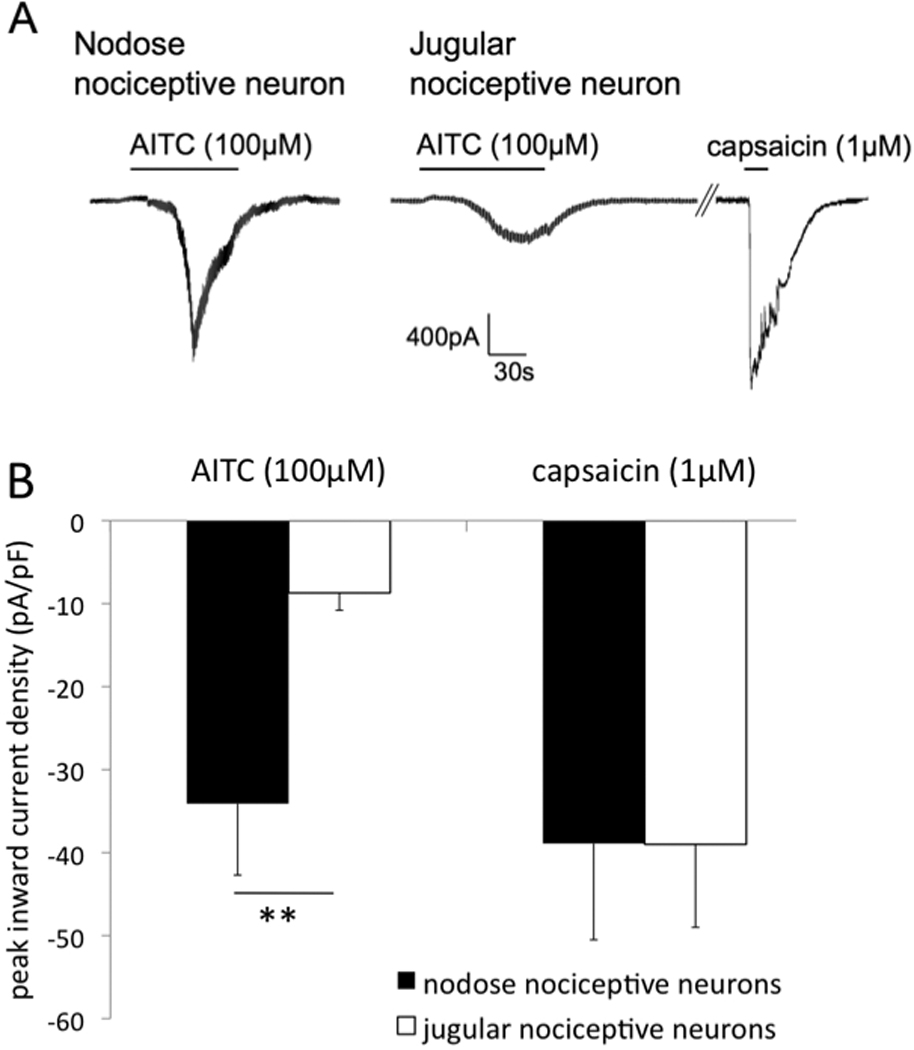
Patch clamp recordings were made from the capsaicin-sensitive vagal afferent neurons retrogradely labeled from the esophagus. (A) Examples of the AITC-induced inward currents in nodose and jugular capsaicin-sensitive neurons. (B) Mean data. 6 of 7 nodose and 8 of 9 jugular esophageal capsaicin-sensitive neurons responded to AITC. Response to maximally-effective concentration of capsaicin (1µM) is shown in 7 nodose and 6 jugular neurons (see methods for details). **p<0.01
Esophageal spinal dorsal root ganglia (DRG) nociceptors
Our data obtained in the vagal afferent system are consistent with the hypothesis that the TRPA1 agonists are substantially more effective activators of the placodes-derived than the neural crest-derived esophageal nociceptors. This hypothesis predicts that the TRPA1 agonists will elicit only a relative weak activation of the spinal DRG nociceptors that are similarly to jugular nociceptors derived from neural crest. In the extracellular recording studies, the spinal DRG nociceptors responded relatively weakly to AITC. AITC (100µM, 20 min) overtly stimulated only 3/11 DRG nociceptors (peak action potential discharge 13, 7 and 5Hz compared to baseline peak 0, 1 and 1Hz, respectively). In the AITC-unresponsive DRG nociceptors the drug access to the nerve terminal was confirmed by the response to capsaicin (1µM). Capsaicin (1µM) induced activation with the peak frequency 11±1Hz (n=11). In their relatively weak responsiveness to AITC the DRG nociceptors are similar to vagal jugular nociceptors (both derived from neural crest), but distinct from the vagal nodose nociceptors derived from epibranchial placodes.
DISCUSSION
We show that the majority of the esophageal placodes- and neural crest-derived vagal nociceptors express the TRPA1 ion channel. While both the placodes- and the neural crest-derived nociceptive subtypes respond to TRPA1 agonists with overt activation (action potential discharge), we found that TRPA1 agonists are substantially more effective in activating the placodes-derived (vagal nodose) nociceptive subtype than the neural crest-derived (spinal DRG and vagal jugular) nociceptive subtypes. We confirmed the higher efficacy of TRPA1 agonists in the activation of placodes-derived nociceptive subtype at the level of isolated vagal neurons indicating that the difference in efficacy is intrinsic to sensory neurons. Our results predict that the presence of endogenous TRPA1 activators in the esopahgeal tissue innervated by both nociceptive subtypes will be preferentially signaled by the placodes-derived nodose nociceptors that are contained exclusively in the vagal pathways.
TRPA1 serves as a broad irritancy receptor for a variety of endogenous reactive molecules such as the products of oxidative and nitrative stress. In the esophagus the major source of oxidative stress is the exposure to refluxate including acid and, in case of inflammation, the infiltration by polymorphonuclear leucocytes (28). Multiple products of oxidative stress including 4-hydroxynonenal, 4-oxo-nonenal, and to some extend hydrogen peroxide directly activate TRPA1 (9, 20, 29). In addition, entero-salivary recirculation of dietary nitrate in combination with acid results in production of relative large quantities of nitric oxide (NO) (11). While NO has a weak activating effect on TRPA1 (8), it combines with the oxidative stress products to form reactive nitrogen species resulting in formation of nitrated fatty acids, such as nitro-oleic acid that is a potent TRPA1 activator (25). In addition, certain arachidonic acid metabolites, such as electrophilic carbon-containing A- and J-series prostaglandins (metabolites of prostaglandins PGE2 and PGD2, respectively) produced by activated immune cells are effective activators of TRPA1(12). Thus, many types of endogenous TRPA1 activators are likely generated in the esophagus in patients with esophageal diseases.
The afferent nerves that can encode noxious stimuli (nociceptors) in the guinea pig esophagus originate from two embryonic sources: the epibranchial placodes that provides vagal nodose nociceptors, and the neural crest that provides vagal jugular and spinal DRG nociceptors. Both placodes-derived and neural crest-derived nociceptive subtypes encode a number of noxious chemical stimuli relevant for the esophagus including the gastroesophageal reflux mediator acid (30), the esophageal chest pain mediator adenosine (19) and inflammatory mediators such as bradykinin (14). However, the placodes- and the neural crest-derived nociceptive subtypes differ in a number of sensory transduction mechanisms. Unlike the neural crest-derived nociceptive subtypes, placodes-derived nociceptors respond to serotonin (via the serotonin 5-HT3 receptor) (18). Similarly, placodes-derived nociceptors, but not neural crest-derived subtypes, respond to purinergic agonists of the P2X receptors for ATP (16, 17). We reported that this difference is attributable to differential expression of the purinergic P2X receptors in the placodes-derived (P2X2/3) and the neural crest-derived (P2X3 only) subtypes (17). The placodes-derived and neural crest-derived nociceptive subtypes are similarly activated by adenosine, but the placodes-derived subtype is activated via the adenosine A1 and/or A2A receptors, while the neural crest-derived subtypes are activated via the A1 receptor only (19). The present study shows that in addition to these qualitative differences, the placodes- and the neural crest-derived subtypes differ also in a quantitative aspect, the substantially more effective activation of the placodes-derived subtype via the TRPA1 receptor.
The spinal DRG nociceptors are thought to be the main pathway for the visceral pain (31). Vagal afferent nerves mediate reflex regulation of visceral organs and may modulate and even contribute to the pain sensations from viscera. The precise consequences of the activation of the nodose and jugular nociceptive subtypes in the esophagus for reflex regulation and sensations have not been elucidated yet. In the neighboring lungs the nodose and jugular nociceptors have opposing effects on respiratory reflexes (32). For example, activation of the nodose subtype increases respiratory rate and inhibits the cough reflex, while activation of the jugular subtype inhibits the respiratory rate and enhances cough. It is therefore likely that the nodose and jugular nociceptive subtypes in the esophagus have different effects on reflex regulation and sensations. Our data predict that the presence of endogenous TRPA1 agonists in the esophageal tissue innervated by both subtypes the overall effect would be dominated by the consequences of activation of the nodose subtype. It is of note that the TRPA1 agonists were as effective as the bona-fide noxious stimuli (capsaicin and noxious distention to 100mmHg) to activate the nodose nociceptors.
We compared the esophageal nociceptive subtypes by using suprathreshold concentrations of TRPA1 agonists. However, exposing the jugular nociceptors to higher concentrations of TRPA1 agonists did not reveal more robust response. Our data also indicate that higher efficacy of TRPA1 agonists to activate the nodose nociceptive subtype is not due to higher excitability of their nerve terminals. Firstly, the nodose and jugular nociceptors responded similarly to capsaicin in both the extracellular studies suggesting that the excitability of their nerve terminals is not substantially different. Secondly, when evaluated in the voltage clamp on isolated neurons and thus not influenced by the excitability, the response to the TRPA1 agonist AITC was also larger in the nodose nociceptotors. Taken together our results are consistent with the conclusion that the difference in responsiveness to TRPA1 agonists is intrinsic to the nociceptive nerves but not caused by different excitability of their nerve terminals.
A recent study in a similar guinea pig preparation (14) reported smaller response of nodose nociceptors to high concentration of AITC (1000µM, relative to EC50 of AITC on TRPA1 reported 10–30µM in most isolated systems (33, 34)) than those observed in our study with 10-times lower concentration of AITC (100µM). The reason for this difference is unknown. We speculate that in a large concentration AITC causes concomitant desensitization and/or inhibition of TRPA1(35). These effects may mask the difference in response between nodose and jugular nociceptors in the referenced study (14).
The mechanisms underlying the quantitative difference of the TRPA1-mediated response between the nodose and jugular nociceptors are unknown. In the guinea pig the normalized amount of TRPA1 protein in the whole jugular ganglia is similar to nodose ganglia (quantitative analysis of the experiments in (36) - S. Yu, personal communication). While it cannot be excluded that the nodose nociceptive (TRPV1-positive) neurons express higher levels of TRPA1 than the jugular nociceptive neurons, this would be difficult to address directly, since the nodose esophageal neurons consist of both TRPV1-positive nociceptive fibers and the TRPV1-negative low threshold (tension) mechanosensors. Published data also suggests that the overall expression of TRPA1 in not higher in the nodose neurons compared to DRG neurons. The qRT-PCR analysis in the mouse showed that the expression of TRPA1 in the DRG tends to be higher than in the nodose ganglia (that are in the mouse mostly placodes-derived (37)) while the percentage of the TRPA1-expressing cells is comparable (13). Similarly, the microarray hybridization studies in the mouse reported higher expression of TRPA1 in the neural crest-derived DRG than in the mostly placodes-derived nodose visceral neurons (38).
Alternatively, the TRPA1 channels are in a more active state in the nodose nociceptors. Multiple intracellular mechanisms, notably including inreacellular calcium, polyphosphates and interaction with TRPV1 regulate the activity of TRPA1 in afferent neurons (23, 26, 39–41). Our experiments indicate that the extracellular calcium (implicated in TRPA1 desensitization) is not critical determinant of the relatively weak response of jugular nerve fibers to TRPA1 agonists. Further studies are required to determine mechanisms underlying differential responsiveness of the nodose and jugular neurons to the TRPA1 agonists. Interestingly, there is a precedent for different function of TRPA1 in the placodes- and neural crest-derived neurons. The TRPA1 receptor mediates the response to noxious cold in nodose neurons, but does not contribute to cold-sensing in the neural crest-derived DRG neurons in mice (42, 43). The mechanisms underlying this difference are unknown.
Acknowledgments
ACKNOWLEDGMENTS AND DISCLOSURES
M.K. is supported by NIH DK74480
M.B. is supported by Center of Excellence for Research in Personalized Therapy (CEVYPET) funded by EU.
L.S. is supported by UK/155/2010
M.B. - performed extracellular recording studies and data analysis
F.R. - performed patch clamp
L.S. - performed single cell RT-PCR
L.M. - performed extracellular recording studies
T.T-C. - contributed essential expertise, performed data analysis, co-wrote the paper
M.K. - designed the study, wrote the paper
Footnotes
COMPETING INTERESTS
The authors have no competing interests.
REFERENCES
- 1.Fass R, Sifrim D. Management of heartburn not responding to proton pump inhibitors. Gut. 2009;58:295–309. doi: 10.1136/gut.2007.145581. [DOI] [PubMed] [Google Scholar]
- 2.Galmiche JP, Clouse RE, Balint A, et al. Functional esophageal disorders. Gastroenterology. 2006;130:1459–1465. doi: 10.1053/j.gastro.2005.08.060. [DOI] [PubMed] [Google Scholar]
- 3.Rohof WO, Hirsch DP, Boeckxstaens GE. Pathophysiology and management of gastroesophageal reflux disease. Minerva Gastroenterol Dietol. 2009;55:289–300. [PubMed] [Google Scholar]
- 4.Page AJ, Blackshaw LA. Roles of gastro-oesophageal afferents in the mechanisms and symptoms of reflux disease. Handb Exp Pharmacol. 2009:227–257. doi: 10.1007/978-3-540-79090-7_7. [DOI] [PubMed] [Google Scholar]
- 5.Patapoutian A, Tate S, Woolf CJ. Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov. 2009;8:55–68. doi: 10.1038/nrd2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackshaw LA, Brierley SM, Hughes PA. TRP channels: new targets for visceral pain. Gut. 2010;59:126–135. doi: 10.1136/gut.2009.179523. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi N, Mizuno Y, Kozai D, et al. Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin) 2008;2:287–298. doi: 10.4161/chan.2.4.6745. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto T, Dubin AE, Petrus MJ, Patapoutian A. TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS One. 2009;4:e7596. doi: 10.1371/journal.pone.0007596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersson DA, Gentry C, Moss S, Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28:2485–2494. doi: 10.1523/JNEUROSCI.5369-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harnett KM, Rieder F, Behar J, Biancani P. Viewpoints on Acid-induced inflammatory mediators in esophageal mucosa. J Neurogastroenterol Motil. 2010;16:374–388. doi: 10.5056/jnm.2010.16.4.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iijima K, Henry E, Moriya A, Wirz A, Kelman AW, McColl KE. Dietary nitrate generates potentially mutagenic concentrations of nitric oxide at the gastroesophageal junction. Gastroenterology. 2002;122:1248–1257. doi: 10.1053/gast.2002.32963. [DOI] [PubMed] [Google Scholar]
- 12.Taylor-Clark TE, Undem BJ, Macglashan DW, Jr, Ghatta S, Carr MJ, McAlexander MA. Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1) Mol Pharmacol. 2008;73:274–281. doi: 10.1124/mol.107.040832. [DOI] [PubMed] [Google Scholar]
- 13.Brierley SM, Hughes PA, Page AJ, et al. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology. 2009;137:2084–2095. e2083. doi: 10.1053/j.gastro.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu S, Ouyang A. TRPA1 in bradykinin-induced mechanical hypersensitivity of vagal C fibers in guinea pig esophagus. Am J Physiol Gastrointest Liver Physiol. 2009;296:G255–G265. doi: 10.1152/ajpgi.90530.2008. [DOI] [PubMed] [Google Scholar]
- 15.Kollarik M, Ru F, Brozmanova M. Vagal afferent nerves with the properties of nociceptors. Auton Neurosci. 2010;153:12–20. doi: 10.1016/j.autneu.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu S, Undem BJ, Kollarik M. Vagal afferent nerves with nociceptive properties in guinea-pig oesophagus. J Physiol. 2005;563:831–842. doi: 10.1113/jphysiol.2004.079574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwong K, Kollarik M, Nassenstein C, Ru F, Undem BJ. P2X2 receptors differentiate placodal vs. neural crest C-fiber phenotypes innervating guinea pig lungs and esophagus. Am J Physiol Lung Cell Mol Physiol. 2008;295:L858–L865. doi: 10.1152/ajplung.90360.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu S, Ru F, Ouyang A, Kollarik M. 5-Hydroxytryptamine selectively activates the vagal nodose C-fibre subtype in the guinea-pig oesophagus. Neurogastroenterol Motil. 2008;20:1042–1050. doi: 10.1111/j.1365-2982.2008.01136.x. [DOI] [PubMed] [Google Scholar]
- 19.Ru F, Surdenikova L, Brozmanova M, Kollarik M. Adenosine-induced activation of esophageal nociceptors. Am J Physiol Gastrointest Liver Physiol. 2011;300:G485–G493. doi: 10.1152/ajpgi.00361.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor-Clark TE, McAlexander MA, Nassenstein C, et al. Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J Physiol. 2008;586:3447–3459. doi: 10.1113/jphysiol.2008.153585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nassenstein C, Kwong K, Taylor-Clark T, et al. Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. J Physiol. 2008;586:1595–1604. doi: 10.1113/jphysiol.2007.148379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Q, Tang Z, Surdenikova L, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139:1353–1365. doi: 10.1016/j.cell.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM. Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J Physiol. 2007;583:175–193. doi: 10.1113/jphysiol.2007.133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrus M, Peier AM, Bandell M, et al. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain. 2007;3:40. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor-Clark TE, Ghatta S, Bettner W, Undem BJ. Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol Pharmacol. 2009;75:820–829. doi: 10.1124/mol.108.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang YY, Chang RB, Waters HN, McKemy DD, Liman ER. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J Biol Chem. 2008;283:32691–32703. doi: 10.1074/jbc.M803568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Undem BJ, Oh EJ, Lancaster E, Weinreich D. Effect of extracellular calcium on excitability of guinea pig airway vagal afferent nerves. J Neurophysiol. 2003;89:1196–1204. doi: 10.1152/jn.00553.2002. [DOI] [PubMed] [Google Scholar]
- 28.Farhadi A, Fields J, Banan A, Keshavarzian A. Reactive oxygen species: are they involved in the pathogenesis of GERD, Barrett's esophagus, and the latter's progression toward esophageal cancer? Am J Gastroenterol. 2002;97:22–26. doi: 10.1111/j.1572-0241.2002.05444.x. [DOI] [PubMed] [Google Scholar]
- 29.Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest. 2008;118:1899–1910. doi: 10.1172/JCI34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kollarik M, Ru F, Undem BJ. Acid-sensitive vagal sensory pathways and cough. Pulm Pharmacol Ther. 2007;20:402–411. doi: 10.1016/j.pupt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grundy D. Neuroanatomy of visceral nociception: vagal and splanchnic afferent. Gut. 2002;51 Suppl 1:i2–i5. doi: 10.1136/gut.51.suppl_1.i2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canning BJ, Chou YL. Cough sensors. I. Physiological and pharmacological properties of the afferent nerves regulating cough. Handb Exp Pharmacol. 2009:23–47. doi: 10.1007/978-3-540-79842-2_2. [DOI] [PubMed] [Google Scholar]
- 33.Savidge J, Davis C, Shah K, et al. Cloning and functional characterization of the guinea pig vanilloid receptor 1. Neuropharmacology. 2002;43:450–456. doi: 10.1016/s0028-3908(02)00122-3. [DOI] [PubMed] [Google Scholar]
- 34.Bandell M, Story GM, Hwang SW, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 35.Everaerts W, Gees M, Alpizar YA, et al. The capsaicin receptor TRPV1 is a crucial mediator of the noxious effects of mustard oil. Curr Biol. 2011;21:316–321. doi: 10.1016/j.cub.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 36.Yu S, Gao G, Peterson BZ, Ouyang A. TRPA1 in mast cell activation-induced long-lasting mechanical hypersensitivity of vagal afferent C-fibers in guinea pig esophagus. Am J Physiol Gastrointest Liver Physiol. 2009;297:G34–G42. doi: 10.1152/ajpgi.00068.2009. [DOI] [PubMed] [Google Scholar]
- 37.Nassenstein C, Taylor-Clark TE, Myers AC, et al. Phenotypic distinctions between neural crest and placodal derived vagal C-fibres in mouse lungs. J Physiol. 2010;588:4769–4783. doi: 10.1113/jphysiol.2010.195339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peeters PJ, Aerssens J, de Hoogt R, et al. Molecular profiling of murine sensory neurons in the nodose and dorsal root ganglia labeled from the peritoneal cavity. Physiol Genomics. 2006;24:252–263. doi: 10.1152/physiolgenomics.00169.2005. [DOI] [PubMed] [Google Scholar]
- 39.Kim D, Cavanaugh EJ. Requirement of a soluble intracellular factor for activation of transient receptor potential A1 by pungent chemicals: role of inorganic polyphosphates. J Neurosci. 2007;27:6500–6509. doi: 10.1523/JNEUROSCI.0623-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai Y, Wang S, Tominaga M, et al. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest. 2007;117:1979–1987. doi: 10.1172/JCI30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Dai Y, Fukuoka T, et al. Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain. 2008;131:1241–1251. doi: 10.1093/brain/awn060. [DOI] [PubMed] [Google Scholar]
- 42.Fajardo O, Meseguer V, Belmonte C, Viana F. TRPA1 channels mediate cold temperature sensing in mammalian vagal sensory neurons: pharmacological and genetic evidence. J Neurosci. 2008;28:7863–7875. doi: 10.1523/JNEUROSCI.1696-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.del Camino D, Murphy S, Heiry M, et al. TRPA1 contributes to cold hypersensitivity. J Neurosci. 2010;30:15165–15174. doi: 10.1523/JNEUROSCI.2580-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]