

Abstract
A series of structurally simple bibenzyl-diol and stilbene-diol core molecules, structural analogs of the well-known hexestrol and diethylstilbestrol non-steroidal estrogens, were prepared and evaluated as estrogen receptor (ER) subtype-selective ligands. Analysis of their ERα and ERβ binding showed that certain substitution patterns engendered binding affinities that were >100-fold selective for ERβ. When further investigated in cell-based gene transcription assays, some molecules showed similarly high relative transcriptional potency selectivity in favor of ERβ. Interestingly, the most ERβ-selective molecules were those bearing non-polar substituents on one of the internal carbon atoms. These compounds should be useful probes for determining the physiological roles of ERβ, and they might lead to the development of more selective and thus safer pharmaceuticals.
Keywords: Estrogen receptor, estrogen receptor beta, stilbestrol, hexestrol, selective ligand
1. Introduction
The estrogen receptor (ER) is a member of a superfamily of nuclear receptors (NRs) that are ligand-modulated transcription factors mediating the action of steroid hormones and various other bioactive ligands [1]. ER ligands regulate not only the female reproductive system, but also play an important role in other tissues, maintaining bone mineral density, regulating blood-lipid profiles [2,3], and supporting brain function [4]. The positive effects of estrogens are generally the basis for their use in the treatment of postmenopausal osteroporosis [5], atherosclerosis [6], and Alzheimer’s disease [7]. While the stimulation of ER has important benefits in some tissues, it can, however, increase the risk of cancer in the breast and uterus [8,9]. Much interest has therefore been focused on the development of more tissue-selective estrogens, notably the selective estrogen receptor modulators (SERMs), that might be safer and more effective pharmaceuticals [10]. The tissue selective effect of SERMs is thought to result from differential cell and promoter-specific interactions [11].
The newer ER subtype, ERβ was discovered in 1996 and is encoded by a gene different from the one encoding the classical receptor (now referred to as ERα) [12–16]. The overall amino acid identity of the two ER subtypes is 44%, with the DNA-binding domains being nearly identical but the ligand-binding domains (LBDs) being less conserved (58% amino acid sequence identity). The ligand-binding pockets (LBPs) of ERα and ERβ, however, are very similar and differ in only two amino acids, with Met421 and Leu384 in ERα being substituted in ERβ by Ile373 and Met336, respectively. Perhaps more significantly, the internal volume of the ligand-binding pocket of ERβ is somewhat smaller than in ERα [17].
The fact that the two ER subtypes have different patterns of tissue distribution and transcriptional regulation has opened new opportunities for creating SERMs with improved tissue selectivity that would derive from ERα vs. ERβ selective binding affinity and transcriptional potency [16]. Such ER subtype-selective ligands could be effective probes of the respective biological roles of ERα and ERβ. They could also be used to study the conformation of receptor subtype agonist and antagonist complexes. Moreover, new ER subtype-selective SERMs might function as improved tissue-selective estrogens for the treatment of a variety of estrogen-linked diseases [15,18,19].
The isoflavone phytoestrogen, genistein 2, with a ca. 20-fold ERβ relative binding affinity selectivity, was the first compound known to be selective for ERβ (Fig. 1) [16]. Among synthetic estrogens, the isocoumarin 3 and the benzoxazole 4 (also known as ERB-041) are reported to have 40-fold [20] and 200-fold [21] relative binding affinity selectivities for ERβ, respectively. The latter represents the most ERβ-selective ligand known so far and has been in clinical trials. We previously described a series of compounds having a bibenzyl core and bearing polar substituents on the central ethylene unit. The most selective ligand of this series was diarylpropionitrile (DPN) 5, which had a 70-fold ERβ relative binding selectivity (Fig. 2) [22]. Other compounds showing ERβ selectivity and possessing different core structures have been developed by us and others [20,23–28], and a pharmacophore model has evolved from these studies. According to this model, an ERβ-selective ligand has a structurally slender core with hydroxyl groups at both ends and polar or polarizable groups in the interior of the ligand; this pharmacophore (Fig. 3) is illustrated by DPN 5 and compounds 2–4 in Fig. 1. Notably, estradiol (1), the main endogenous estrogen and the typical standard for comparison with other ligands, has modest binding and transcriptional potency preference in favor of ERα.
Fig. 1.

Estradiol and some ER ligands being selective for ERβ
Fig. 2.

ER ligands having either a bibenzyl- or a stilbene-core
Fig. 3.

ERβ pharmacophore model
Recently, we reexamined isobutestrol 6, a compound that has long been known for its low affinity for ERα [29,30], and we were pleased to find that this simple compound had good affinity and an 18-fold relative binding selectivity for ERβ. Interestingly, isobutestrol 6 is rather non-polar at its center, which stands in contrast to the pharmacophore model (Fig. 3). Isobutestrol (6) also has much higher ERβ binding selectivity than its more familiar congeners, hexestrol 7 and diethylstilbestrol (DES) 8 [29] (cf., Table 1), although its absolute affinity is lower. Because of the encouraging ERβ selectivity of isobutestrol 6, we decided to investigate analogs having various, mostly non-polar substitution patterns on the central ethylene group of the bibenzyl core, to see whether we might obtain ligands with even greater ERβ selectivities. In this study, we describe the synthesis and biological evaluation of such bibenzyl-core compounds, and, in some cases, evaluation of their unsaturated stilbene analogs.
Table 1.
ERα and ERβ relative binding affinity (RBA), relative transcriptional potency (RTP), transcriptional activity (EC50 values), and affinity and potency selectivities for bibenzyl-core and related ligands.
| Ligand | Ligand Binding | Transcription Potency | |||||||
|---|---|---|---|---|---|---|---|---|---|
| RBAa(%) | β/α affinity ratiob | RTPc(%) | β/α potency ratiob | EC50d(nM) | β/α potency ratiob | ||||
| ERα | ERβ | ERα | ERβ | ERα | ERβ | ||||
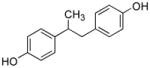
 25 |
0.057 ± 0.017 | 2.73 ± 0.23 | 48 | 0.13 | 5.38 | 41 | 75.1 | 9.3 | 8.1 |
 21 |
0.71 ± 0.08 | 20.5 ± 5.0 | 29 | 1.54 | 41.7 | 27 | 6.5 | 1.2 | 5.4 |
 Isobutestrol, 6 |
2.2 ± 0.4 | 39.5 ± 8.4 | 18 | NDe | ND | ND | NTe | NT | NT |
 22 |
3.2 ± 1.0 | 68 ± 16 | 21 | 5.0 | 22 | 4.4 | 2.0 | 2.3 | 0.89 |
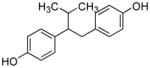 23 |
1.62 ± 0.45 | 7.1 ± 1.8 | 4 | ND | ND | ND | NT | NT | NT |
 24 |
2.10 ± 0.55 | 22.2 ± 3.4 | 11 | ND | ND | ND | NT | NT | NT |
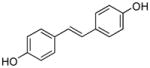 44 |
0.45 | 3.55 | 8 | ND | ND | ND | NT | NT | NT |
 26 |
0.218 ± 0.025 | 12.2 ± 1.2 | 56 | 0.89 | 13.9 | 16 | 11.2 | 3.6 | 3.1 |
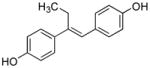 27 |
4.09 ± 0.81 | 76 ± 14 | 19 | 19 | 23 | 1.2 | 0.52 | 2.21 | 0.24 |
 28 |
30.9 ± 6.0 | 21.8 ± 4.0 | 0.7 | ND | ND | ND | NT | NT | NT |
 36b |
0.132 ± 0.002 | 16.5 ± 1.6 | 125 | 0.62 | 45.5 | 73 | 16.2 | 1.1 | 15 |
 37b |
0.32 ± 0.04 | 21.5 ± 4.0 | 66 | 0.93 | 15.6 | 17 | 10.7 | 3.2 | 3.3 |
 38b |
0.77 ± 0.06 | 13.6 ± 3.7 | 18 | 0.50 | 6.0 | 12 | 20.2 | 8.4 | 2.4 |
 39b |
0.85 ± 0.26 | 6.8 ± 1.4 | 8 | ND | ND | ND | NT | NT | NT |
 42b |
0.414 ± 0.021 | 2.81 ± 0.06 | 7 | ND | ND | ND | NT | NT | NT |
 43b |
1.84 ± 0.20 | 2.74 ± 0.70 | 1.5 | ND | ND | ND | NT | NT | NT |
 29b |
< 0.004 | 0.018 ± 0.004 | 4.5 | ND | ND | ND | NT | NT | NT |
 30b |
0.008 ± 0.001 | 0.018 ± 0.001 | 2.3 | ND | ND | ND | NT | NT | NT |
 32b |
0.042 ± 0.006 | 0.018 ± 0.001 | 0.4 | ND | ND | ND | NT | NT | NT |
 DPN, 5f |
0.25 ± 0.15 | 18 ± 2 | 72 | 0.15 | 59 | 390 | 66.00 | 0.85 | 78 |
 Hexestrol, 7 |
277 ± 57 | 697 ± 194 | 2.5 | ND | ND | ND | NT | NT | NT |
 DES, 8 |
372 ± 106 | 278 ± 54 | 0.7 | ND | ND | ND | NT | NT | NT |
Relative binding affinity (RBA) values are determined by competitive radiometric binding assays and are expressed as EC50[estradiol]/EC50[compound] × 100 (RBA, estradiol = 100). All chiral compounds were tested as racemates. In these assays, the Kd for estradiol is 0.2 nM for ERα and 0.5 nM for ERβ. For details, see Experimental Section.
For each value, the β/α ratio is calculated such that the ratio is >1 for compounds having higher affinity or potency on ERβ than on ERα.
Relative transcriptional potency (RTP) values are expressed as EC50(estradiol)/EC50(ligand) × 100 (RTP, estradiol = 100). In these assays, the EC50 for estradiol is 0.1 nM for ERα and 0.5 nM for ERβ.
Transcriptional activity was measured using a cotransfection assay in human endometrial cancer (HEC-1) cells (see Experimental and Fig. 4). All chiral compounds were tested as racemates. Transcriptional potency = EC50.
ND = not determined; NT = not tested.
Data are from the literature [22].
2. Chemical Synthesis
2.1 Synthesis of Monoalkyl Bibenzyl and Stilbene Diols
Scheme 1 illustrates the syntheses of bibenzyl-core compounds and stilbenes with one of the two carbons of the central ethylene unit bearing one alkyl chain. All syntheses started with the introduction of alkyl substituents into desoxyanisoin 9 by Grignard addition. Steric effects had a strong influence on the reaction yields, which were excellent for small and moderate for large Grignard reagents. To complete the preparation of the bibenzyl-core compounds, the intermediate alcohols 10–14 were dehydroxylated [31] to give molecules 15–19. These conversions took only about 30 min and gave excellent yields. In the final deprotection step, the phenolic hydroxy groups were revealed in convenient overnight reactions at room temperature using BBr3, furnishing the target compounds 6 and 21–25 in very good yields [32].
Scheme 1.

(a) RMgCl, THF; (b) and (d) BBr3, CH2Cl2; (c) Et3SiH, BF3.·OEt2, CH2Cl2
The stilbene derivatives 26–28 were obtained by treating racemic alcohols 10–12 with BBr3 at low temperature, which effected dehydration as well as deprotection of the methoxy groups in a single step [32]. The elimination took place at −78 °C, and while no ether cleavage appeared at that temperature, when the reactions were allowed to warm to 0 °C, deprotection occurred rapidly, and the stilbenes were obtained in moderate yield. The stilbene derivatives proved to be much more sensitive to the acidic reaction conditions than their saturated analogs 6, 21 and 22. Extended reaction time especially at room temperature gave low yields, most possibly due to acid-catalyzed polymerization. However, it is noteworthy that the olefin formation occurred with remarkable regio-and stereoselectivity. In case of 27 and 28, the trans-stilbenes were the major products, with only small amounts of the corresponding cis-isomers and both possible styrene derivatives being evident. In the case of compound 26, the trans-isomer was the only product isolated. In addition, a certain amount of decomposition products were obtained, possibly a result of polymerization of possible styrene intermediates, which have a terminal and thus more reactive double bond than those of the stilbene species.
The geometry of the alkenes was assigned initially by the chemical shift of the olefinic proton. In accordance to literature data on similar molecules [33], we found that the signal of the vinylic proton of the trans-isomers was significantly shifted down field in comparison to the cis-isomers (Δδ ≥ 0.25). This assignment was verified further with NOE studies, exemplified with compounds 26 and 27. Irradiation of the vinylic proton (δ 6.70, 6.54 for 26, 27, respectively) yielded enhancements of the neighboring aromatic protons meta to the phenolic hydroxyl (26: δ 7.39, 13.7%; δ 7.22, 8.3%; 27: δ 7.33, 12.6%, δ 7.19, 8.3%), without exhibiting detectable enhancement of the methylene protons trans to it. These results are in agreement with those obtained with similar molecules that were also investigated by NOE studies [34].
2.2 Synthesis of Geminally Disubstituted Bibenzyl Diols
Scheme 2 shows the synthesis of bibenzyl-core compounds bearing either one or two alkyl chains on one of the carbons of the central ethylene, and in some cases having an additional carbonyl function on the other carbon. Starting with ketone 9, the alkyl chains were introduced by enolate alkylation to obtain the substituted ketones 29a, 30a and 31 within only 30 min at room temperature. In accordance with literature precedent [35], an excess of base and alkylating agent gave the monoalkylated products in good yields, together with much smaller amounts of the corresponding bis-alkylated compounds. Since the enolate is not preformed in that procedure, it is noteworthy that when one equivalent of base and iodoalkane were used, only a single alkyl chain was introduced selectively. In the next step, the second alkyl chain was introduced in the same manner to give the disubstituted ketones 32a and 33–35. Subsequent removal of the carbonyl oxygen [36] in convenient overnight reactions at room temperature furnished the protected products 36a, 37a, 38a and 39a. Employing the BBr3 deprotection method described above gave the final bisphenol products 29b, 30b, 32b, 36b, 37b, 38b and 39b.
Scheme 2.

(a) KOt-Bu, RI, 18-crown-6, THF; (b) Et3SiH, CF3COOH; (c) BBr3, CH2Cl2
2.3 Synthesis of Spirocyclic Bibenzyl Diols
The introduction of spirocycles onto the central ethylene of the bibenzyl-core started with the double alkylation of ketone 9 using α,ω-dibromoalkanes (Scheme 3). In the literature protocol we followed [37], the reaction is performed at room temperature and takes about two days, but we were able to obtain compounds 40 and 41 within a few hours in good yields by refluxing the reaction mixtures. Despite our use of different bases and reaction conditions, we were unable to prepare smaller spirocycles by this method. The subsequent decarbonylation and deprotection reactions were performed as in case of the acyclic analogs described above, to furnish molecules 42a, 43a, and the final compounds 42b, 43b.
Scheme 3.

(a) NaH, Br(CH2)n+3Br, THF; (b) Et3SiH, CF3COOH; (c) BBr3, CH2Cl2
3. Pharmacology
3.1 Estrogen Receptor Binding Assays
The relative binding affinities (RBA) of the compounds we prepared were measured in a competitive radiometric binding assay using purified full-length human ERα and ERβ [38,39]. The binding affinities are listed in Table 1 and are expressed as relative binding affinity (RBA) values, that is, relative to the affinity of estradiol, which is set at 100%. The table includes also the RBA values of isobutestrol 6, DPN 5, hexestrol 7, and DES 8. All chiral compounds were tested as racemates.
Among the 18 target compounds we have prepared, eight (21, 22, 25, 26, 27, 36b, 37b, 38b) were found to have an ERβ selectivity equal to or higher than the reference compound isobutestrol 6, with compounds 25, 26, 37b and in particular 36b being the most selective ones (48-, 56-, 66- and 125-fold, respectively). With the exception of compound 25, all of these compounds had also quite good ERβ binding affinity. The highest affinity was observed with compounds 22 and 27, having RBA values of 68 and 76, respectively, which is almost twice as high as isobutestrol 6 and even four times higher than DPN 5.
The series of monosubstituted bibenzyl compounds having a propyl or shorter substituent R on the central ethylene showed an interesting trend: Compound 25 (R = H) was the most selective one, but it also had the lowest affinity. Increasing the chain length led to higher affinities; however, the affinity for ERα increased faster than for ERβ, resulting overall in lower ERβ selectivity. Thus, compound 22 (R = Pr) had the highest affinity, while still having good selectivity. On the other hand, branching or further increasing the size of the substituent lowered the selectivity as well as the affinity (23, 24). Interestingly, when one of the two carbons of the ethylene unit was di-substituted with small alkyl chains, the ERβ selectivity was higher compared to the corresponding mono-substituted analogs (36b, 37b vs. 21, 6).
It is known that the two ethyl chains of hexestrol 7 and diethylstilbestrol (DES) 8 fit nicely into the 7α and 11β subpockets of the ERs [40], resulting in high affinity but very low selectivity. In comparison, isobutestrol 6 with just one ethyl chain on the two linking carbons, has lower affinity, but increased ERβ selectivity. In this study, we found the same behavior in case of the stilbene analogs: Monoethylstilbestrol 27 had lower affinity than DES, but increased ERβ selectivity. The monomethylstilbestrol 26 had even higher selectivity than monoethylstilbestrol 27, or compound 21, the saturated analog of 26.
In general, increasing the size of substituents (36b, 37b vs 38b, 39b) or constraining them into a spirocycle (38b, 39b vs 42b, 43b) gave lower selectivities and affinities for both ERs. These lower affinities suggest that these compounds are too large or too rigid to fit well into the LBD pockets of the ERs. The decrease in ERβ selectivity is consistent with the larger ligand-binding pocket of ERα, which is being better able to accommodate the large and constrained ligands than ERβ. Another trend noted is that the compounds with a carbonyl function on the central ethylene had much lower affinities and selectivities than their reduced analogs (29b vs. 21; 30b vs. 6; 32b vs. 36b).
3.2 Activity in Gene Transcription
Eight compounds (21, 22, 25, 26, 27, 36b, 37b, and 38b) that showed good selectivity between ERα and ERβ in the binding assay were tested for their agonistic character as regulators of gene transcription. They were assayed in a cotransfection assay in human endometrial cancer (HEC-1) cells, using expression plasmids for either ERα or ERβ and an estrogen-responsive reporter gene [41]. The dose-response curves for these compounds are given in Fig. 4, and the EC50 values are listed in Table 1. This table also includes the EC50 values for our ERβ-selective ligand, DPN 5, for comparison. All chiral compounds were tested as racemates.
Fig. 4.

Transcriptional activation by ERα and ERβ in response to ligands 21, 22, 25, 26, 27, 36b 37b and 38b and estradiol (E2, lowest panel right). Human endometrial cancer (HEC-1) cells were transfected with expression vectors for ERα and ERβ and the estrogen responsive gene 2ERE-pS2-Luc, and were incubated with the indicated concentrations of ligand for 24h. Luciferase and β-galactosidase activity were assayed as described [41]. Estradiol activity at 10 nM is set at 100%. All chiral compounds were tested as racemates.
To facilitate comparisons of the ER subtype transcriptional potencies of our compounds with their ER subtype binding affinities, the EC50 values from the transcription assays were converted to relative transcriptional potency (RTP) values (See Table 1, footnote c). The RTPs provide a measure of transcriptional potency relative to that of estradiol and thus can be better compared to their binding affinities, which are also measured relative to estradiol by the competitive radiometric binding assays. Estradiol has a 2.5-fold preference in favor of ERα in terms of binding (Kd [ERα] = 0.2 nM vs. [ERβ] = 0.5 nM) and a 5-fold preference in terms of transcriptional potency (EC50 [ERα] = 0.1 nM vs. [ERβ] = 0.5 nM).
Of the eight compounds tested, seven showed very good potency selectivity for ERβ (note RTP ratio) that was either very similar to their RBA ratios (compounds 21, 25, 36b, and 38b) or at least of the same magnitude (compounds 22, 26, and 37b); only compound 27 showed no ERβ transcriptional potency preference yet had good ERβ binding preference. Thus, our findings show overall a very good concordance of relative binding affinities and relative transcriptional potencies. This was not always the case, however, because affinity represents binding to the ER, whereas potency can also depend on other factors that can depend on the cell and promoter context: The interactions that ER-agonist ligand complexes have with coregulators and promoter elements can vary, depending on the precise conformations of the ER that are induced by binding to ligands of different structure, irrespective of their binding affinity. Compared to our previously prepared ERβ-selective ligand, DPN 5, some of the compounds we present here have higher ERβ binding affinity (compounds 6, 21, 22, 24, 27, 28, and 37b) and comparable or higher ERβ binding selectivity (compounds 25, 26, 36b, and 37b) and somewhat less to nearly equivalent ERβ transcriptional potency (compounds 21 and 36b).
4. Conclusion
By making various analogs of the bibenzyl non-steroidal estrogen isobutestrol and the related stilbenes, we have obtained a number of structurally simple compounds that have high ERβ binding affinity and selectivity, and good ERβ transcriptional potency and selectivity. In terms of ERβ binding affinity, the best bibenzyl is compound 22 and the best stilbene is 27; in terms of ERβ binding selectivity, the best in each series is 36b and 26. In terms of ERβ transcriptional potency and selectivity, 36b is the best bibenzyl, while among the stilbenes, 27 has the highest potency and 26 is the most selective. Although some of these compounds match the ERβ binding selectivity of DPN (5), none matched or exceeded the ERβ transcriptional potency selectivity of this compound. Nevertheless, these compounds represent new structures of remarkable simplicity that have high ERβ activities, and they should thus serve as good probes to further investigate the biological functions of ERβ in vivo, as well as to study ligand-induced conformations of ERβ agonist complexes.
It is also remarkable that many of the ERβ-selective ligands in this study are ones having non-polar substituents on the central ethylene unit, while previous studies by us and others have validated a pharmacophore model for non-steroidal estrogens having rather more polar substituents on the interior of the ligand. Despite differences in the polarity of the ligand interior, the ERβ-selective bibenzyl systems we have studied share with other ERβ-selective ligands an interior that is generally more “slender” and/or less “symmetrical” than typical for ERα-selective ligands, such as hexestrol (7) and DES (8). These ERβ-selective slender ligands appear to have a better fit with the somewhat smaller ligand binding pocket of ERβ compared to ERα [17].
Further structural studies will be needed to elucidate this size/shape preference further, but the results we have obtained here indicate again that the estrogen receptors—both ERα and ERβ—have a rather eclectic taste for the refined features of ligand structure, so that even ligands of simple structure can discriminate between the two ER subtypes. Nevertheless, despite our lack of understanding of the detailed features of this discrimination, our findings open new strategies for designing ligands having high selectivity for ERβ.
5. Experimental Section
5.1 Materials and Methods
Reagents and solvents were purchased from Aldrich and Fisher Scientific; compound 44 was obtained from Aldrich. THF and dichloromethane were dried using a solvent-dispensing system (SDS) (neutral alumina columns) built by J.C. Meyer on the basis of a design developed by Pangborn et al. [42]. Glassware was oven- or flame-dried, assembled while hot, and cooled under nitrogen atmosphere. All reactions were performed in anhydrous solvents and under nitrogen atmosphere, unless stated otherwise. Reaction progress was monitored by thin-layer chromatography (TLC) using 0.25 mm Merck silica gel 60 glass plates containing F254 UV-Indicator. The plates were visualized by either UV light (254 nm), or by dipping in a solution of potassium permanganate followed by heating. Column chromatography was performed using Woelm 32–63 μm silica gel packing. 1H and 13C NMR were recorded on Varian UNITY 400 or 500 MHz spectrometers. NOE experiments were performed with a Varian UNITY INOVIA 500NB instrument. Chemical shifts are reported in ppm and referenced from solvent references. NMR coupling constants are reported in Hertz. Mass spectra were obtained either on a Micromass 70-VSE spectrometer (EI, 70 eV; CI, methane), or a Fisons VG Quattro instrument (ESI, cone voltage 25 V). Melting point (mp) determinations were carried out on Thomas Hoover Unimelt capillary apparatus and are uncorrected. Elemental analyses were performed by the Microanalysis Service Laboratory of the University of Illinois. Analyses indicated by the symbols of the elements or functions were within ± 0.4 % of the theoretical values.
5.2 Chemical Synthesis
General procedure for the Grignard addition to desoxyanisoin (9)
Desoxyanisoin 9 (400 mg, 1.56 mmol) was dissolved in 10 mL THF and the Grignard reagent (4.68 mmol) was added. The mixture was refluxed for 6 h (unless stated otherwise), then cooled to 0 °C, quenched with water (10 mL) and extracted with EtOAc (3 × 15 mL), The organic extracts were dried over Na2SO4 and the solvent was removed under vacuum.
1,2-Bis-(4-methoxy-phenyl)-propan-2-ol (10)
Use of methyl magnesium chloride (1.56 mL of a 3M solution in THF) gave 10 as a white solid (389 mg, 92% yield, mp 68 °C) after purification by flash chromatography (33% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.8, 2H), 6.89 (d, J = 8.6, 2H), 6.86 (d, J = 8.8, 2H), 6.76 (d, J = 8.6, 2H), 3.81 (s, 3H), 3.77 (s, 3H), 3.05 (d, J = 13.5, 1H), 2.94 (d, J = 13.5, 1H), 1.53 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 158.38, 158.23, 139.80, 131.55, 128.81, 126.22, 113.45, 113.28, 74.19, 55.23, 55.14, 49.65, 29.38; MS (ESI) m/z 255 (M+-17, 100).
1,2-Bis-(4-methoxy-phenyl)-butan-2-ol (11)
Use of ethyl magnesium bromide (4.68 mL of a 1M solution in THF) gave 11 as a white solid (344 mg, 77% yield, mp 63 °C) after purification by flash chromatography (33% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.22 (d, J = 8.8, 2H), 6.87-6.84 (m, 4H), 6.73 (d, J = 8.6, 2H), 3.81 (s, 3H), 3.75 (s, 3H), 3.07 (d, J = 13.5, 1H), 2.96 (d, J = 13.5, 1H), 1.92 (dq, J = 14.7, 7.4, 1H), 1.79 (dq, J = 14.7, 7.4, 1H), 0.76 (t, J = 7.4, 3H); 13C NMR (500 MHz, CDCl3) δ 158.32, 158.01, 137.65, 131.61, 128.45, 126.76, 113.42, 113.18, 76.66, 55.15, 55.11, 48.47, 34.42, 7.83; MS (ESI) m/z 269 (M+-17, 100).
1,2-Bis-(4-methoxy-phenyl)-pentan-2-ol (12)
Use of propyl magnesium chloride (2.34 mL of a 2 M solution in diethyl ether) gave 12 as yellow oil (349 mg, 75% yield) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.22 (d, J = 8.8, 2H), 6.87-6.83 (m, 4H), 6.74 (d, J = 8.8, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 3.07 (d, J = 13.5, 1H), 2.96 (d, J = 13.5, 1H), 1.88 (d, J = 13.7, 4.5, 1H), 1.85 (d, J = 13.7, 4.5, 1H), 1.37-1.26 (m, 1H) 1.13-1.00 (m, 1H), 0.84 (t, J = 7.4, 3H); 13C NMR (500 MHz, CDCl3) δ 158.45, 158.11, 138.19, 131.75, 128.52, 126.77, 113.58, 113.31, 76.64, 55.31, 55.27, 48.92, 44.50, 17.00, 14.54; MS (ESI) m/z 283 (M+-17, 100).
1,2-Bis-(4-methoxy-phenyl)-3-methyl-butan-2-ol (13)
Use of isopropyl magnesium chloride (2.34 mL of a 2 M solution in THF) gave 13 as pale yellow oil (215 mg, 46% yield) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.21 (d, J = 8.8, 2H), 6.83 (d, J = 8.8, 2H), 6.80 (d, J = 8.8, 2H), 6.68 (d, J = 8.8, 2H), 3.80 (s, 3H), 3.73 (s, 3H), 3.16 (d, J = 13.5, 1H), 3.08 (d, J = 13.5, 1H), 2.11-2.06 (m, 1H), 1.00 (d, J = 6.8, 3H), 0.81 (d, J = 6.8, 3H); 13C NMR (500 MHz, CDCl3) δ 158.40, 158.12, 136.93, 131.81, 128.69, 127.61, 113.59, 112.99, 78.68, 55.29, 55.24, 44.87, 37.71, 18.01, 17.07; MS (CI) m/z 283 (M+-17, 19).
1,2-Bis-(4-methoxy-phenyl)-hexan-2-ol (14)
Use of butyl magnesium chloride (2.34 mL of a 2 M solution in diethyl ether) and a reflux time of 4.5 h gave 14 as colorless oil (286 mg, 58% yield) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.22 (d, J = 9.0, 2H), 6.87-6.83 (m, 4H), 6.74 (d, J = 8.6, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 3.07 (d, J = 13.5, 1H), 2.96 (d, J = 13.5, 1H),1.93-1.86 (m, 1H), 1.78-1.72 (m, 1H), 1.33-1.19 (m, 3H), 1.07-0.98 (m, 1H), 0.83 (t, J = 7.2, 3H); 13C NMR (500 MHz, CDCl3) δ 158.32, 157.98, 138.06, 131.62, 128.40, 126.64, 113.42, 113.17, 76.44, 55.13, 55.09, 48.80, 41.80, 25.68, 23.02, 14.02; MS (ESI) m/z 297 (M+-17, 100).
General procedure for the dehydroxylation of 1,2-Bis-(4-methoxy-phenyl)-alkan-2-ols
The starting material was dissolved in CH2Cl2 and the mixture was brought to 0 °C. Et3SiH was added and after 2 min BF3·OEt was added dropwise. The reaction was stirred for 30 min at 0 °C, quenched with water and extracted three times with EtOAc. The organic extracts were dried over Na2SO4 and the solvent was removed under vacuum.
1,2-Bis-(4-methoxy-phenyl)-propane (15)
Use of compound 10 (337 mg, 1.24 mmol), Et3SiH (394 μL, 2.48 mmol) and BF3·OEt (314 μL, 2.48 mmol) in 5 mL CH2Cl2 gave 15 as a white solid (313 mg, quant. yield, mp 68 °C) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.09 (d, J = 8.6, 2H), 6.98 (d, J = 8.8, 2H), 6.83 (d, J = 8.8, 2H), 6.78 (d, J = 8.6, 2H), 3.79 (s, 3H), 3.78 (s, 3H), 2.95-2.88 (m, 1H), 2.84 (dd, J = 13.5, 6.6, 1H), 2.69 (dd, J = 13.5, 6.6, 1H), 1.21 (d, J = 6.9, 3H); 13C NMR (500 MHz, CDCl3) δ 157.74, 157.71, 139.15, 133.00, 130.04, 127.92, 113.59, 113.42, 55.18, 44.30, 41.11, 21.26; MS (EI) m/z 256 (M+, 9).
1,2-Bis-(4-methoxy-phenyl)-butane (16)
Use of compound 11 (330 mg, 1.05 mmol), Et3SiH (320 μL, 2.01 mmol) and BF3·OEt (253 μL, 2.01 mmol) in 5 mL CH2Cl2 gave 16 as a colorless oil (249 mg, 88%) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.8, 2H), 6.94 (d, J = 8.6, 2H), 6.82 (d, J = 8.8, 2H), 6.77 (d, J = 8.6, 2H), 3.79 (s, 3H), 3.77 (s, 3H), 2.82-2.74 (m, 2H), 2.67-2.58 (m, 1H), 1.76-1.65 (m, 1H), 1.61-1.50 (m, 1H), 0.76 (t, J = 7.1, 3H); 13C NMR (400 MHz, CDCl3) δ 157.68, 157.56, 137.11, 133.04, 130.02, 128.64, 113.45, 113.33, 55.14, 49.08, 42.72, 28.36, 12.11; (EI) m/z 270 (M+, 11).
1,2-Bis-(4-methoxy-phenyl)-pentane (17)
Use of compound 12 (306 mg, 1.02 mmol), Et3SiH (325 μL, 2.04 mmol) and BF3·OEt (259 μL, 2.04 mmol) in 5 mL CH2Cl2 gave 17 as a white solid (233 mg, 88%, mp 34 °C) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.8, 2H), 6.94 (d, J = 8.6, 2H), 6.81 (d, J = 8.8, 2H), 6.76 (d, J = 8.6, 2H), 3.79 (s, 3H), 3.77 (s, 3H), 2.84-2.69 (m, 3H), 1.67-1.50 (m, 2H), 1.22-1.10 (m, 2H), 0.83 (t, J = 7.3, 3H); 13C NMR (400 MHz, CDCl3) δ 157.64, 157.55, 137.37, 133.00, 130.00, 128.56, 113.44, 113.31, 55.11, 47.04, 43.07, 37.84, 20.62, 14.06; MS (EI) m/z 284 (M+, 12).
1,2-Bis-(4-methoxy-phenyl)-3-methyl-butane (18)
Use of compound 13 (215 mg, 0.72 mmol), Et3SiH (228 μL, 1.43 mmol) and BF3·OEt (181 μL, 1.43 mmol) in 4.5 mL CH2Cl2 gave 18 as a colorless oil (168 mg, 82%) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 6.96 (d, J = 8.8, 2H), 6.88 (d, J = 8.8, 2H), 6.77 (d, J = 8.8, 2H), 6.70 (d, J = 8.8, 2H), 3.77 (s, 3H), 3.74 (s, 3H), 3.04 (dd, J = 13.6, 5.4, 1H), 2.74 (dd, J = 13.6, 9.5, 1H), 2.55-2.51 (m, 1H), 1.87 (qq, J = 6.7, 6.7, 1H), 0.98 (d, J = 6.7, 3H), 0.77 (d, J = 6.7, 3H); 13C NMR (500 MHz, CDCl3) δ 157.79, 157.59, 135.59, 133.61, 130.04, 129.79, 113.46, 113.24, 55.39, 54.31, 38.90, 32.40, 21.53, 19.91; MS (EI) m/z 284 (M+, 7).
1,2-Bis-(4-methoxy-phenyl)-hexane (19)
Use of compound 14 (235 mg, 0.75 mmol), Et3SiH (239 μL, 1.50 mmol) and BF3·OEt (190 μL, 1.50 mmol) in 4.5 mL CH2Cl2 gave 19 as a colorless oil (207 mg, 93%) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 8.6, 2H), 6.92 (d, J = 8.8, 2H), 6.81 (d, J = 8.6, 2H), 6.75 (d, J = 8.8, 2H), 3.79 (s, 3H), 3.76 (s, 3H), 2.80 (dd, J = 13.4, 7.2, 1H), 2.78-2.67 (m, 2H), 1.68-1.61 (m, 1H), 1.59-1.51 (m, 1H), 1.31-1.08 (m, 4H), 0.81 (t, J = 7.3, 3H); 13C NMR (500 MHz, CDCl3) δ 157.68, 157.59, 137.45, 133.04, 130.02, 128.59, 113.47, 113.33, 55.13, 55.11, 47.28, 43.09, 35.33, 29.73, 22.69, 13.99; MS (EI) m/z 298 (M+, 9).
General procedure for the deprotection of methoxy groups with BBr3
The starting material was dissolved in CH2Cl2 and brought to 0 °C. BBr3 was added dropwise and the mixture was brought to rt and stirred for 17 h. The reaction was brought to 0 °C, quenched with water and extracted three times with EtOAc. The organic extracts were dried over Na2SO4 and the solvent was removed under vacuum.
1,2-Bis-(4-hydroxy-phenyl)-propane (21)
Use of compound 15 (286 mg, 1.12 mmol) and BBr3 (3.35 mL of a 1M solution in CH2Cl2) in 11 mL CH2Cl2 gave 21 as a white solid (209 mg, 82%, mp 173–174 °C) after purification by flash chromatography (50% EtOAc/hexanes). 1H NMR (400 MHz, acetone-d6) δ 8.02 (s, OH), 7.01 (d, J = 8.5, 2H), 6.91 (d, J = 8.5, 2H), 6.72 (d, J =8.5, 2H), 6.68 (d, J = 8.5, 2H), 2.91-2.82 (m, 1H), 2.76 (dd, J = 13.3, 6.7, 1H), 2.65 (dd, J = 13.3, 8.0, 1H), 1.14 (d, J = 6.8, 3H); 13C NMR (400 MHz, acetone-d6) δ 156.24, 138.74, 132.60, 130.80, 128.68, 115.74, 115.59, 44.95, 41.97, 21.79; MS (EI) m/z 228 (M+,7). HRMS (EI) calcd for C15H16O2: 228.1150, found 228.1145. Anal. C15H16O2 (C, H).
1,2-Bis-(4-hydroxy-phenyl)-butane (6)
Use of compound 16 (250 mg, 0.93 mmol) and BBr3 (2.79 mL of a 1M solution in CH2Cl2) in 10 mL CH2Cl2 gave 6 as a white solid (209 mg, quant. yield,) after purification by flash chromatography (50% EtOAc/hexanes). The analytical data of compound 6 was consistent with the data published previously [30].
1,2-Bis-(4-hydroxy-phenyl)-pentane (22)
Use of compound 17 (203 mg, 0.71 mmol) and BBr3 (2.14 mL of a 1M solution in CH2Cl2) in 7 mL CH2Cl2 gave 22 as a white solid (157 mg, 86%, mp 99–100 °C) after purification by flash chromatography (50% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.00 (s, OH), 6.95 (d, J = 8.6, 2H), 6.86 (d, J = 8.4, 2H), 6.71 (d, J =8.4, 2H), 6.65 (d, J = 8.6, 2H), 2.81-2.66 (m, 3H), 1.64-1.49 (m, 2H), 1.21-1.09 (m, 2H), 0.78 (d, J = 7.3, 3H); 13C NMR (500 MHz, acetone-d6) δ 156.31, 156.19, 136.96, 132.63, 130.83, 129.42, 115.74, 115.57, 47.86, 43.77, 38.76, 21.26, 14.29; MS (ESI) m/z 255 (M+−-1, 81). HRMS (ESI) calcd for C17H19O2: 255.1385, found 255.1389 Anal. C17H20O2 (C, H).
1,2-Bis-(4-hydroxy-phenyl)-3-methyl-butane (23)
Use of compound 18 (139 mg, 0.49 mmol) and BBr3 (1.48 mL of a 1M solution in CH2Cl2) in 5 mL CH2Cl2 gave 23 as a white solid (106 mg, 85%, mp 101 °C) after purification by flash chromatography (50% EtOAc/hexanes) and re-crystallization from toluene. 1H NMR (400 MHz, CDCl3) δ 6.89 (d, J = 8.6, 2H), 6.81 (d, J = 8.6, 2H), 6.68 (d, J =8.6, 2H), 6.61 (d, J = 8.6, 2H), 5.00 (bs, OH), 3.03 (dd, J = 13.6, 5.1, 1H), 2.69 (dd, J = 13.6, 9.9, 1H), 2.48 (ddd, J = 12.3, 7.1, 5.3, 1H), 1.86 (qq, J = 6.8, 6.8, 1H), 0.98 (d, J = 6.8, 3H), 0.76 (d, J = 6.8, 3H); 13C NMR (500 MHz, CDCl3) δ 153.30, 153.10, 135.67, 135.61, 130.08, 129.79, 114.79, 114.65, 54.34, 38.83, 32.32, 21.33, 19.95; MS (EI) m/z 256 (M+, 7). HRMS (EI) calcd for C17H20O2: 256.1463, found 256.1457. Anal. C17H20O2 (C, H).
1,2-Bis-(4-hydroxy-phenyl)-hexane (24)
Use of compound 19 (184 mg, 0.62 mmol) and BBr3 (1.85 mL of a 1M solution in CH2Cl2) in 6 mL CH2Cl2 gave 24 as a white solid (148 mg, 88%, mp 81 °C) after purification by flash chromatography (50% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.01 (s, OH), 7.98 (s, OH), 6.95 (d, J = 8.6, 2H), 6.86 (d, J = 8.6, 2H), 6.71 (d, J = 8.6, 2H), 6.65 (d, J = 8.6, 2H), 2.78-2.65 (m, 3H), 1.66-1.60 (m, 1H), 1.56-1.49 (m, 1H), 1.29-1.06 (m, 4H), 0.77 (t, J = 7.3, 3H); 13C NMR (500 MHz, acetone-d6) δ 156.30, 156.18, 137.00, 132.64, 130.82, 129.42, 115.74, 115.56, 48.08, 43.80, 36.17, 30.49, 23.29, 14.24; MS (EI) m/z 270 (M+, 8). HRMS (EI) calcd for C18H22O2: 270.1620, found 270.1623. Anal. C18H22O2 (C, H).
1,2-Bis-(4-hydroxy-phenyl)-ethane (25)
Use of commercially available 1,2-Bis-(4-methoxy-phenyl)-ethane 20 (32 mg, 0.13 mmol) and BBr3 (0.5 mL of a 1M solution in CH2Cl2) in 1 mL CH2Cl2 gave 25 as a white solid (27 mg, quant. yield, mp 196 °C) after purification by flash chromatography (50% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.06 (s, OH), 7.01 (d, J = 8.6, 4H), 6.72 (d, J = 8.6, 4H), 2.74 (s, 4H); 13C NMR (500 MHz, acetone-d6) δ 156.31, 133.57, 130.18, 115.80, 38.17; MS (EI) m/z 214 (M+, 11). HRMS (EI) calcd for C14H14O2: 214.0994, found 214.0993. Anal. C14H14O2 (C, H).
1,2-Bis-(4-hydroxy-phenyl)-prop-1-ene (26)
The starting material (500 mg, 1.84 mmol) 10 was dissolved in 20 mL CH2Cl2 and brought to −78 °C. BBr3 (7.4 mL of a 1M solution in CH2Cl2) was added dropwise and the reaction was stirred for 90 min. The mixture was brought to 0 °C and stirred for another 2 h. The reaction was quenched with water and extracted three times with EtOAc. The organic extracts were dried over Na2SO4 and the solvent was removed under vacuum. After purification by flash chromatography (25% EtOAc/hexanes) and re-crystallization from EtOAc/hexanes, 26 was obtained as a white solid (212 mg, 51%, mp 176 °C). 1H NMR (500 MHz, acetone-d6) δ 8.37 (s, OH), 8.35 (s, OH), 7.39 (d, J = 8.6, 2H), 7.22 (d, J = 8.6, 2H), 6.86 - 6.81 (m, 4H), 6.70 (s, 1H), 2.20 (s, 3H); 13C NMR (500 MHz, acetone-d6) δ 157.53, 156.80, 136.29, 135.44, 131.22, 130.90, 127.74, 126.13, 115.89, 115.85, 17.53; MS (CI) m/z 227 (M+ +1, 100). HRMS (CI) calcd for C15H15O2: 227.1072, found 227.1070. Anal. C15H14O2: Calcd. C, 79.62; H, 6.24. Found C, 79.19; H, 6.17.
1,2-Bis-(4-hydroxy-phenyl)-but-1-ene (27)
The reaction was performed as described above for 26. Use of 11 (400 mg, 1.40 mmol) and BBr3 (5.59 mL of a 1M solution in CH2Cl2) in 16 mL CH2Cl2 gave 27 as a white solid (156 mg, 46%, mp 109 °C) after purification by flash chromatography (25% EtOAc/hexanes) and re-crystallization from diethyl ether/hexanes. 1H NMR (500 MHz, acetone-d6) δ 8.35 (s, OH), 7.33 (d, J = 8.8, 2H), 7.19 (d, J = 8.8, 2H), 6.86-6.81 (m, 4H), 6.54 (s, 1H), 2.70 (quart., J = 7.5, 2H), 1.03 (t, J = 7.5, 3H); 13C NMR (500 MHz, acetone-d6) δ 157.52, 156.89, 142.66, 134.80, 130.79, 130.73, 128.33, 126.22, 115.96, 115.92, 23.46, 13.86; MS (CI) m/z 241 (M+ +1, 100). HRMS (CI) calcd for C16H17O2: 241.1229, found 241.1230. Anal. C16H16O2: Calcd. C, 79.97; H, 6.72. Found C, 79.42; H, 6.70.
1,2-Bis-(4-hydroxy-phenyl)-pent-1-ene (28)
The reaction was performed as described above for 26. Use of 12 (500 mg, 1.67 mmol) and BBr3 (6.65 mL of a 1M solution in CH2Cl2) in 19 mL CH2Cl2 gave 28 as a white solid (224 mg, 53%, mp 117 °C) after purification by flash chromatography (25% EtOAc/hexanes) and re-crystallization from EtOAc/hexanes. 1H NMR (500 MHz, acetone-d6) δ 8.36 (s, OH), 8.33 (s, OH), 7.33 (d, J = 8.8, 2H), 7.18 (d, J = 8.8, 2H), 6.86-6.81 (m, 4H), 6.56 (s, 1H), 2.65 (t, J = 7.9, 2H), 1.48-1.40 (m, 2H), 0.89 (t, J = 7.4, 3H); 13C NMR (500 MHz, acetone-d6) δ 157.49, 156.87, 141.45, 135.29, 130.82, 130.81, 128.33, 126.92, 115.94, 115.92, 32.57, 22.71, 14.27; MS (CI) m/z 255 (M+ +1, 100). HRMS (CI) calcd for C17H19O2: 255.1385, found 255.1379. Anal. C17H18O2: Calcd. C, 80.28; H, 7.13. Found C, 79.76; H, 7.15.
General procedure for the alkylation of desoxyanisoin (9) and 1,2-Bis-(4-methoxy-phenyl)-alkan-1-ones
KOtBu and 18-crown-6 were added to THF at rt and the suspension was stirred for 15 min. A solution of the alkyl iodide and starting material were added at rt and stirring was continued for 30 min. The mixture was filtered and the filter cake was washed with EtOAc. The filtrate was absorbed on silica gel and the solvent was removed under vacuum. The silica gel was charged on a column and flash chromatography was performed as usual.
1,2-Bis-(4-methoxy-phenyl)-propan-1-one (29a)
Use of KOtBu (524 mg, 4.68 mmol) and 18-crown-6 (41 mg, 0.156 mmol) in 13 mL THF, as well as 9 (400 mg, 1.56 mmol) and methyl iodide (390 μL, 6.24 mmol) in 3 mL THF gave 29a as a colorless oil (233 mg, 55%) after purification by flash chromatography (20% EtOAc/hexanes). In addition, 129 mg (29%) of the dimethylated product 32a were isolated. 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 8.8, 2H), 7.20 (d, J = 8.8, 2H), 6.85 (d, J = 8.8, 2H), 6.82 (d, J = 8.8, 2H), 4.60 (quart., J = 6.9, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 1.49 (d, J = 6.9, 3H); 13C NMR (400 MHz, CDCl3) δ 199.03, 163.08, 158.32, 133.90, 130.97, 129.36, 128.63, 114.25, 113.57, 55.33, 55.13, 46.52, 19.52; MS (EI) m/z 270 (M+, 6).
1,2-Bis-(4-hydroxy-phenyl)-propan-1-one (29b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 29a (73 mg, 0.27 mmol) and BBr3 (1.62 mL of a 1M solution in CH2Cl2) in 2 mL CH2Cl2 gave 29b as a white solid (38 mg, 58%, mp 151 °C) after purification by flash chromatography (20% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 7.92 (d, J = 8.8, 2H), 7.14 (d, J =8.6, 2H), 6.83 (d, J = 8.8, 2H), 6.74 (d, J = 8.6, 2H), 4.70 (quart., J = 6.9, 1H), 1.37 (d, J = 6.9, 3H); 13C NMR (500 MHz, acetone-d6) δ 199.06, 162.37, 157.00, 134.03, 132.01, 129.58, 116.38, 115.86, 46.54, 19.89; MS (EI) m/z 242 (M+, 4). HRMS (EI) calcd for C15H14O3: 242.0943, found 242.0946. Anal. C15H14O3 • H2O: Calcd. C, 69.22; H, 6.20. Found C, 69.68; H, 5.70.
1,2-Bis-(4-methoxy-phenyl)-butan-1-one (30a)
Use of KOtBu (2.63 mg, 23.45 mmol) and 18-crown-6 (410 mg, 1.55 mmol) in 130 mL THF, as well as 9 (4.00 g, 15.63 mmol) and ethyl iodide (1.52 mL, 18.76 mmol) in 30 mL THF gave 30a as a colorless oil (27 mg, 92%) after purification by flash chromatography (20% EtOAc/hexanes.) 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 8.8, 2H), 7.21 (d, J = 8.6, 2H), 6.86 (d, J = 8.8, 2H), 6.81 (d, J = 8.6, 2H), 4.34 (t, J = 7.3, 1H), 3.82 (s, 3H), 3.75 (s, 3H), 2.19-2.11 (m, 1H), 1.85-1.77 (m, 1H), 0.88 (t, J = 7.3, 3H); 13C NMR (400 MHz, CDCl3) δ 198.82, 163.12, 158.40, 132.06, 130.87, 129.93, 129.14, 114.13, 113.58, 55.37, 55.14, 54.08, 27.05, 12.27; MS (CI) m/z 285 (M+ +1, 100).
1,2-Bis-(4-hydroxy-phenyl)-butan-1-one (30b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 30a (225 mg, 0.79 mmol) and BBr3 (3.17 mL of a 1M solution in CH2Cl2) in 8 mL CH2Cl2 gave 30b as a white solid (100 mg, 49%, mp 117 °C) after purification by flash chromatography (40% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 7.95 (d, J = 8.6, 2H), 7.17 (d, J = 8.6, 2H), 6.85 (d, J = 8.6, 2H), 6.75 (d, J = 8.6, 2H), 4.48 (t, J = 7.2, 1H), 2.12-2.03 (m, 1H), 1.77-1.68 (m, 1H), 0.84 (t, J = 7.4, 3H); 13C NMR (500 MHz, acetone-d6) δ 198.89, 162.41, 157.07, 132.15, 131.92, 130.10, 130.05, 116.27, 115.89, 54.07, 27.78, 12.44; MS (EI) m/z 256 (M+, 6). HRMS (EI) calcd for C16H16O3: 256.1099, found 256.1102. Anal. C16H16O3 (C, H).
1,2-Bis-(4-methoxy-phenyl)-pentan-1-one (31)
Use of KOtBu (1.96 g, 17.54 mmol) and 18-crown-6 (154 mg, 0.58 mmol) in 50 mL THF, as well as 9 (1.50 g, 5.86 mmol) and propyl iodide (2.28 mL, 23.4 mmol) in 10 mL THF gave 31 as a colorless oil (1.13 g, 65%) after purification by flash chromatography (10% EtOAc/hexanes). In addition, 259 mg (13%) of dipropylated product 35 were isolated. 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 8.8, 2H), 7.22 (d, J = 8.8, 2H), 6.87 (d, J = 8.8, 2H), 6.82 (d, J = 8.8, 2H), 4.34 (t, J = 7.3, 1H), 3.81 (s, 3H), 3.74 (s, 3H) 2.15-2.07 (m, 1H), 1.81-1.74 (m, 1H), 1.36-1.20 (m, 2H), 0.91 (t, J = 7.4, 3H); 13C NMR (500 MHz, CDCl3) δ 198.87, 163.14, 158.40, 132.24, 130.88, 129.91, 129.11, 114.13, 113.60, 55.34, 55.11, 52.00, 36.10, 20.78, 14.03; MS (CI) m/z 299 (M+ +1, 100).
1,2-Bis-(4-methoxy-phenyl)-2-methyl-propan-1-one (32a)
Use of KOtBu (417 mg, 3.72 mmol) and 18-crown-6 (2.3 mg, 0.009 mmol) in 8 mL THF, as well as 29a (250 mg, 0.93 mmol) and methyl iodide (232 μL, 3.72 mmol) in 2 mL THF gave 32a as a white solid (186 mg, 70%, mp 60 °C) after purification by flash chromatography (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 9.0, 2H), 7.21 (d, J = 8.8, 2H), 6.88 (d, J = 8.8, 2H), 6.71 (d, J = 9.0, 2H), 3.79 (s, 3H), 3.76 (s, 3H), 1.57 (s, 6H); 13C NMR (500 MHz, CDCl3) δ 202.27, 162.15, 158.20, 137.95, 132.22, 128.65, 126.68, 114.25, 113.06, 55.22, 55.16, 50.46, 28.14; MS (EI) m/z 285 (M++1, 34)
1,2-Bis-(4-hydroxy-phenyl)-2-methyl-propan-1-one (32b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 32a (100 mg, 0.35 mmol) and BBr3 (1.06 mL of a 1M solution in CH2Cl2) in 3.5 mL CH2Cl2 gave 32b as a white solid (24 mg, 27%, mp 140 °C) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 7.50 (d, J = 9.0, 2H), 7.13 (d, J = 8.8, 2H), 6.83 (d, J = 8.8, 2H), 6.70 (d, J = 9.0, 2H), 1.51 (s, 6H); 13C NMR (500 MHz, acetone-d6) δ 201.99, 163.18, 156.95, 137.53, 132.87, 129.53, 127.53, 116.54, 113.86, 55.66, 50.94; MS (EI) m/z 256 (M+, 4). HRMS (EI) calcd for C16H16O3: 256.1099, found 256.1106. Anal. C16H16O3 (C, H).
1,2-Bis-(4-methoxy-phenyl)-2-methyl-butan-1-one (33)
Use of KOtBu (3.56 g, 31.8 mmol) and 18-crown-6 (280 mg, 1.06 mmol) in 90 mL THF, as well as 30a (3.00 g, 10.6 mmol) and methyl iodide (2.63 mL, 42.3 mmol) in 20 mL THF gave 33 as a colorless oil (1.55 g, 50%) after purification by flash chromatography (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 9.0, 2H), 7.18 (d, J = 8.8, 2H), 6.87 (d, J = 8.8, 2H), 6.70 (d, J = 9.0, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 2.17-2.10 (m, 1H), 2.06-1.98 (m, 1H), 1.51 (s, 3H), 0.73 (t, J = 7.5, 3H); 13C NMR (500 MHz, CDCl3) δ 202.33, 162.08, 158.20, 137.04, 131.95, 129.37, 127.26, 114.16, 113.05, 55.24, 55.18, 54.02, 32.25, 24.21, 8.61; MS (CI) m/z 299 (M+ +1, 49).
1,2-Bis-(4-methoxy-phenyl)-2-methyl-pentan-1-one (34)
Use of KOtBu (676 mg, 6.04 mmol) and 18-crown-6 (53 mg, 0.2 mmol) in 17 mL THF, as well as 31 (600 mg, 2.01 mmol) and methyl iodide (501 μL, 8.05 mmol) in 4 mL THF gave 34 as a colorless oil (334 mg, 53%) after purification by flash chromatography (10% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 9.0, 2H), 7.18 (d, J = 9.0, 2H), 6.87 (d, J = 9.0, 2H), 6.70 (d, J = 9.0, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 2.09-1.90 (m, 2H), 1.53 (s, 3H), 1.19-1.01 (m, 2H), 0.84 (t, J = 7.2, 3H); 13C NMR (400 MHz, CDCl3) δ 202.23, 162.02, 158.12, 137.28, 131.93, 129.28, 127.15, 114.12, 113.02, 55.24, 55.18, 53.80, 42.07, 25.00, 17.50, 14.75; MS (CI) m/z 340 (M+ +1, 25).
1,2-Bis-(4-methoxy-phenyl)-2-propyl-pentan-1-one (35)
Use of KOtBu (752 mg, 6.71 mmol) and 18-crown-6 (177 mg, 0.67 mmol) in 14 mL THF, as well as 31 (500 mg, 1.68 mmol) and propyl iodide (654 μL, 6.71 mmol) in 3 mL THF gave 35 as a colorless oil (257 mg, 45%) after purification by flash chromatography (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 9.0, 2H), 7.16 (d, J = 8.8, 2H), 6.86 (d, J = 9.0, 2H), 6.68 (d, J = 8.8, 2H), 3.80 (s, 3H), 3.76 (s, 3H), 2.06-1.93 (m, 4H), 1.09-0.90 (m, 4H), 0.80 (t, J = 7.2, 3H); 13C NMR (500 MHz, CDCl3) δ 202.46, 161.92, 158.13, 136.06, 131.68, 129.75, 127.76, 113.96, 112.97, 57.11, 55.17, 55.11, 37.31, 16.75, 14.69; MS (EI) m/z 341 (M+ +1, 16).
General procedure for the deoxygenation with Et3SiH/TFA
The starting material was dissolved in TFA, and Et3SiH was added dropwise at rt. The reaction was stirred for 24 h, quenched with water and extracted three times with diethyl ether. The organic extracts were dried over Na2SO4 and the solvent was removed under vacuum.
1,2-Bis-(4-methoxy-phenyl)-2-methyl-propane (36a)
Use of 32a (100 mg, 0.35 mmol) and Et3SiH (140 μL, 0.88 mmol) in 2.70 mL TFA gave 36a as a white solid (90 mg, quant. yield, mp 50 °C) after purification by flash chromatography (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.22 (d, J = 9.0, 2H), 6.86 (d, J = 9.0, 2H), 6.75-6.70 (m, 4H), 3.83 (s, 3H), 3.77 (s, 3H), 2.81 (s, 2H), 1.31 (s, 6H); 13C NMR (500 MHz, CDCl3) δ 157.78, 157.38, 141.12, 131.26, 131.07, 127.19, 113.11, 112.79, 55.13, 55.04, 50.30, 38.15, 28.29; MS (EI) m/z 270 (M+, 3).
1,2-Bis-(4-hydroxy-phenyl)-2-methyl-propane (36b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 36a (70 mg, 0.26 mmol) and BBr3 (780 μL of a 1M solution in CH2Cl2) in 2.5 mL CH2Cl2 gave 36b as a white solid (50 mg, 79%, mp 113 °C) after purification by flash chromatography (20% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.07 (s, OH), 8.02 (s, OH), 7.12 (d, J = 8.8, 2H), 6.74 (d, J = 8.8, 2H), 6.66 (d, J = 8.6, 2H), 6.59 (d, J = 8.6, 2H), 2.73 (s, 2H), 1.23 (s, 6H); 13C NMR (500 MHz, acetone-d6) δ 156.39, 155.94, 140.71, 132.08, 130.66, 127.95, 115.35, 115.00, 50.81, 38.65, 28.72; MS (EI) m/z 242 (M+, 3). HRMS (EI) calcd for C16H18O2: 242.1307, found 242.1307. Anal. C16H18O2: Calcd. C, 79.31; H, 7.49. Found C, 78.90; H, 7.51.
1,2-Bis-(4-methoxy-phenyl)-2-methyl-butane (37a)
Use of 33 (1.00 g, 3.36 mmol) and Et3SiH (1.33 mL, 8.4 mmol) in 2.57 mL TFA gave 37a as a colorless oil (679 mg, 70%) after purification by flash chromatography (5% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.12 (d, J = 8.9, 2H), 6.83 (d, J = 8.9, 2H), 6.70-6.65 (m, 4H), 3.81 (s, 3H), 3.75 (s, 3H), 2.85 (d, J = 13.3, 1H), 2.72 (d, J = 13.3, 1H), 1.94-1.86 (m, 1H), 1.59-1.52 (m, 1H), 1.19 (s, 3H), 0.70 (t, J = 7.4, 3H); 13C NMR (500 MHz, CDCl3) δ 157.71, 157.25, 138.88, 131.37, 130.85, 127.91, 113.09, 112.73, 55.11, 55.06, 49.82, 41.71, 34.37, 22.80, 8.71; MS (CI) m/z 283 (M+- 1, 3).
1,2-Bis-(4-hydroxy-phenyl)-2-methyl-butane (37b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 37a (630 mg, 2.22 mmol) and BBr3 (8.87 mL of a 1M solution in CH2Cl2) in 20 mL CH2Cl2 gave 37b as a white solid (486 mg, 86%, mp 106 °C) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.08 (s, OH), 8.02 (s, OH), 7.05 (d, J = 8.9, 2H), 6.74 (d, J = 8.9, 2H), 6.63-6.56 (m, 4H), 2.81 (d, J = 13.2, 1H), 2.66 (d, J = 13.2, 1H), 1.92-1.85 (m, 1H), 1.55-1.47 (m, 1H), 1.14 (s, 3H), 0.64 (t, J = 7.4, 3H); 13C NMR (500 MHz, acetone-d6) δ 156.35, 155.90, 138.30, 132.19, 130.38, 128.71, 115.39, 114.94, 50.41, 42.29, 35.19, 23.03, 9.00; MS (CI) m/z 255 (M+ -1, 3). HRMS (EI) calcd for C17H19O2: 255.1385, found 255.1381. Anal. C17H20O2 (C, H).
1,2-Bis-(4-methoxy-phenyl)-2-methyl-pentane (38a)
Use of 34 (285 mg, 1.07 mmol) and Et3SiH (425 μL, 2.68 mmol) in 820 μL TFA gave 38a as a colorless oil (212 mg, 66%) after purification by flash chromatography (5% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.15 (d, J = 8.8, 2H), 6.85 (d, J = 8.8, 2H), 6.72-6.68 (m, 4H), 3.83 (s, 3H), 3.77 (s, 3H), 2.88 (d, J = 13.2, 1H), 2.74 (d, J = 13.2, 1H), 1.83 (dd, J = 13.0, 4.0, 1H), 1.52 (dd, J = 13.0, 4.5, 1H), 1.28-1.18 (m, 4H), 1.08-0.96 (m, 1H), 0.87 (t, J = 7.4, 3H); 13C NMR (500 MHz, CDCl3) δ 157.67, 157.22, 139.29, 131.37, 130.75, 127.76, 127.70, 113.07, 113.03, 112.70, 55.08, 55.03, 50.00, 44.59, 41.47, 23.42, 17.48, 14.70; MS (EI) m/z 298 (M+, 3).
1,2-Bis-(4-hydroxy-phenyl)-2-methyl-pentane (38b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 38a (190 mg, 0.64 mmol) and BBr3 (1.91 mL of a 1M solution in CH2Cl2) in 6.5 mL CH2Cl2 gave 38b as a white solid (270 mg, quant. yield, mp 55 °C) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.07 (s, OH), 8.01 (s, OH), 7.06 (d, J = 8.7, 2H), 6.74 (d, J = 8.7, 2H), 6.63-6.56 (m, 4H), 2.81 (d, J = 13.2, 1H), 2.67 (d, J = 13.2, 1H), 1.80 (dd, J = 13.0, 4.0, 1H), 1.46 (dd, J = 13.0, 4.7, 1H), 1.22-1.11 (m, 4H), 1.02-0.90 (m, 1H), 0.80 (t, J = 7.3, 3H); 13C NMR (500 MHz, acetone-d6) δ 156.36, 155.87, 138.79, 132.20, 130.33, 128.54, 115.38, 114.97, 50.59, 45.52, 42.04, 23.77, 18.16, 15.04; MS (EI) m/z 270 (M+, 2). HRMS (EI) calcd for C18H22O2: 270.1620, found 270.1616.
1,2-Bis-(4-methoxy-phenyl)-2-propyl-pentane (39a)
Use of 35 (180 mg, 0.53 mmol) and Et3SiH (210 μL, 1.32 mmol) in 407 μL TFA gave 39a as a colorless oil (36 mg, 21%) after purification by flash chromatography (5% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 9.0, 2H), 6.82 (d, J = 9.0, 2H), 6.67-6.57 (m, 4H), 3.81 (s, 3H), 3.74 (s, 3H), 2.81 (s, 2H), 1.64-1.49 (m, 4H), 1.31-1.15 (m, 4H), 0.88 (t, J = 7.3, 6H); 13C NMR (400 MHz, CDCl3) δ 157.59, 157.09, 139.10, 131.06, 130.71, 127.86, 113.00, 112.75, 55.10, 55.04, 44.45, 44.14, 38.70 16.93, 14.71; MS (CI) m/z 325 (M+ -1, 7).
1,2-Bis-(4-hyroxy-phenyl)-2-propyl-pentane (39b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 39a (40 mg, 0.12 mmol) and BBr3 (370 μL of a 1M solution in CH2Cl2) in 1.2 mL CH2Cl2 gave 39b as a white solid (35 mg, quant. yield, mp 137 °C) after purification by flash chromatography (20% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.07 (s, OH), 8.01 (s, OH), 7.06 (d, J = 8.8, 2H), 6.75 (d, J = 8.8, 2H), 6.58-6.52 (m, 4H), 2.79 (s, 2H), 1.61-1.48 (m, 4H), 1.28-1.13 (m, 4H), 0.85 (t, J = 7.3, 6H); 13C NMR (500 MHz, acetone-d6) δ 156.31, 155.80, 138.57, 131.88, 130.22, 128.65, 115.40, 115.03, 44.75, 44.66, 39.60 17.54, 14.95; MS (CI) m/z 297 (M+-1, 2). HRMS (CI) calcd for C20H25O2: 297.1855, found 297.1858. Anal. C20H26O2: Calcd. C, 80.50; H, 8.78. Found C, 79.61; H, 8.89.
(4-methoxy-phenyl)- [1-(4-methoxy-phenyl)-cyclopentyl]-methanone (40)
NaH (312 mg of a 60% dispersion in oil, 7.80 mmol) was dissolved in 20 mL THF at rt and stirred for 20 min. 9 (500 mg, 1.95 mmol) was added as a solid at 0 °C and the reaction was continued for 4 h at rt. 1,4-dibromo-butane (344 μL, 2.93 mmol) was added at 0 °C and the mixture was refluxed for 3.5 h. After cooling to rt, the reaction was quenched with NH4Cl (sat. aqueous solution) and extracted three times with EtOAc. The organic extracts were dried over Na2SO4 and the solvent was removed under vacuum. After purification by flash chromatography (10% EtOAc/hexanes), 40 was obtained as a colorless oil (390 mg, 65%). 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 8.8, 2H), 7.20 (d, J = 8.8, 2H), 6.84 (d, J = 8.8, 2H), 6.73 (d, J = 8.8, 2H), 3.77 (s, 6H), 2.47 (ddd, J = 11.9, 6.4, 6.4, 2H), 2.05 (ddd, J = 11.9, 5.9, 5.9, 2H), 1.78-1.64 (m, 4H); 13C NMR (500 MHz, CDCl3) δ 200.72, 162.21, 158.01, 137.12, 132.30, 128.70, 126.96, 114.13, 113.06, 62.38, 55.25, 55.15, 37.60, 24.54; MS (CI) m/z 311 (M+ +1, 11).
(4-methoxy-phenyl)- [1-(4-methoxy-phenyl)-cyclohexyl]-methanone (41)
The reaction was performed as described above for 40. Use of NaH (312 mg of a 60% dispersion in oil, 7.80 mmol), 9 (500 mg, 1.95 mmol), and 1,4-dibromo-pentane (400 μL, 2.93 mmol) in 20 mL THF gave 41 as a colorless oil (371 mg, 59%) after purification by flash chromatography (10% EtOAc/hexanes). 1 H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.6, 2H), 7.31 (d, J = 8.6, 2H), 6.90 (d, J = 8.6, 2H), 6.71 (d, J = 8.6, 2H), 3.80 (s, 3H), 3.76 (s, 3H), 2.49 (d, J = 13.3, 2H), 1.74 (dt, J = 13.3, 3.4, 2H), 1.67-1.57 (m, 3H), 1.49-1.38 (m, 2H), 1.31-1.20 (m, 1H); 13C NMR (500 MHz, CDCl3) δ 203.24, 161.75, 158.30, 136.82, 131.17, 130.65, 127.13, 114.27, 112.95, 55.19, 55.14, 54.45, 36.39, 25.84, 23.27; MS (CI) m/z 325 (M+ +1, 21).
(4-methoxy-phenyl)- [1-(4-methoxy-phenyl)-methyl]-cyclopentane (42a)
The reaction was performed according to the general procedure for the deoxygenation with Et3SiH/TFA. Use of 40 (360 mg, 1.16 mmol) and Et3SiH (921 μL, 5.80 mmol) in 895 μL TFA gave 42a as a colorless oil (286 mg, 83%) after purification by flash chromatography (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 6.97 (d, J = 8.6, 2H), 6.78 (d, J = 8.6, 2H), 6.64 (d, J = 8.6, 2H), 6.52 (d, J = 8.6, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 2.74 (s, 2H), 1.96-1.66 (m, 8H); 13C NMR (500 MHz, CDCl3) δ 157.67, 157.32, 140.47, 131.27, 131.10, 128.37, 112.89, 112.66, 55.14, 55.06, 51.69, 46.53, 36.83, 22.88; MS (CI) m/z 295 (M+ - 1, 4).
(4-hydroxy-phenyl)- [1-(4-hydroxy-phenyl)-methyl]-cyclopentane (42b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 42a (260 mg, 0.88 mmol) and BBr3 (2.64 mL of a 1M solution in CH2Cl2) in 9 mL CH2Cl2 gave 42b as a white solid (205 mg, 87%, mp 121 °C) after purification by flash chromatography (25% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.04 (s, OH), 7.98 (s, OH), 6.89 (d, J = 8.8, 2H), 6.68 (d, J = 8.8, 2H), 6.54 (d, J = 8.6, 2H), 6.45 (d, J = 8.6, 2H), 2.70 (s, 2H), 1.92-1.62 (m, 6H); 13C NMR (500 MHz, acetone-d6) δ 156.31, 155.94, 139.85, 131.86, 130.83, 129.05, 115.17, 114.91, 52.43, 47.22, 37.55, 23.42; MS (CI) m/z 267 (M+ -1, 6). HRMS (CI) calcd for C18H19O2: 267.1385, found 267.1389. Anal. C18H20O2 (C, H).
(4-methoxy-phenyl)- [1-(4-methoxy-phenyl)-methyl]-cyclohexane (43a)
The reaction was performed according to the general procedure for the deoxygenation with Et3SiH/TFA. Use of 41 (250 mg, 0.77 mmol) and Et3SiH (613 μL, 3.86 mmol) in 600 μL TFA gave 43a as a colorless oil (178 mg, 78%) after purification by flash chromatography (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.03 (d, J = 8.6, 2H), 6.81 (d, J = 8.6, 2H), 6.63 (d, J = 8.6, 2H), 6.51 (d, J = 8.6, 2H), 3.81 (s, 3H), 3.74 (s, 3H), 2.67 (s, 2H), 2.09 (d, J = 13.3, 2H), 1.59-1.45 (m, 5H), 1.38-1.24 (m, 3H); 13C NMR (500 MHz, CDCl3) δ 157.66, 157.15, 131.42, 130.19, 128.43, 113.15, 112.54, 55.11, 55.06, 42.08, 35.87, 26.58, 22.30; MS (CI) m/z 309 (M+ -1, 6).
(4-hydroxy-phenyl)- [1-(4-hydroxy-phenyl)-methyl]-cyclohexane (43b)
The reaction was performed according to the general procedure for the deprotection of methoxy groups. Use of 43a (180 mg, 0.58 mmol) and BBr3 (1.74 mL of a 1M solution in CH2Cl2) in 6 mL CH2Cl2 gave 43b as a white solid (130 mg, 79%, mp 122 °C) after purification by flash chromatography (33% EtOAc/hexanes). 1H NMR (500 MHz, acetone-d6) δ 8.06 (s, OH), 7.97 (s, OH), 6.96 (d, J = 8.8, 2H), 6.72 (d, J = 8.8, 2H), 6.53 (d, J = 8.6, 2H), 6.45 (d, J = 8.6, 2H), 2.62 (s, 2H), 2.07 (d, J = 13.3, 2H), 1.56-1.42 (m, 5H), 1.35-1.25 (m, 3H); 13C NMR (500 MHz, acetone-d6) δ 156.25, 155.74, 132.19, 129.65, 129.14, 115.48, 114.78, 42.65, 36.49, 27.29, 23.02; MS (CI) m/z 281 (M+ -1, 6). HRMS (CI) calcd for C19H21O2: 281.1542, found 281.1549. Anal. C19H22O2 (C, H).
5.3 Estrogen Receptor Binding Affinity Assays
Relative binding affinities were determined by a competitive radiometric binding assay as previously described [38,39], using 10 nM [3H]estradiol as tracer (GE Healthcare, Piscataway, NJ), and purified full-length human ERα and ERβ (PanVera/InVitrogen, Carlsbad, CA). Incubations were for 18–24 h at 0 °C, then the receptor-ligand complexes were absorbed onto hydroxyapatite (BioRad, Hercules, CA) and unbound ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values, with the RBA of estradiol set to 100. The values given are the average ± range or SD of two or more independent determinations. All chiral compounds were tested as racemates. Estradiol binds to ERα with a Kd of 0.2 nM and to ERβ with a Kd of 0.5 nM.
5.4 Cell Culture and Transient Transfections
Human endometrial cancer (HEC-1) cells were maintained in Minimum Essential Medium (MEM) plus phenol-red supplemented with 5% calf serum and were changed to phenol-red-free Improved MEM and 5% charcoal dextran-treated calf serum (CDCS) for 3–4 days before use in experiments. Transfection assays were performed in 24-well plates using a mixture of 0.35 mL of serum-free improved MEM medium and 0.15 mL of Hank’s balanced salt solution containing 5 μg of lipofectin (Life Technologies, Inc., Gaithersburg, MD), 20 μg of transferrin (Sigma, St. Louis, MO), 0.2 μg of pCMV β-galactosidase as internal control, 0.5 μg of 2ERE-pS2-Luc, and 50 ng of ER expression vector per well. The cells were incubated at 37 °C in a 5% CO2-containing incubator for 6 h. The medium was then replaced with fresh Improved MEM supplemented with 5% CDCS plus the desired concentrations of ligands. Cells were harvested 24 h later. Luciferase and β-galactosidase activity were assayed as described [41]. All chiral compounds were tested as racemates.
Acknowledgments
This work was supported through grants from the National Institutes of Health (R37DK15556 and P01AG024387 to J.A.K., and R01CA18119 to B.S.K.).
Abbreviations
- DPN
diarylpropionitrile
- ER
estrogen receptor
- LBD
ligand-binding
- NR
nuclear receptor
- RBA
relative binding affinity
- RTP
relative transcriptional potency
- SERM
selective estrogen receptor modulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katzenellenbogen JA, Katzenellenbogen BS. Chemistry and Biology. 1996;3:529–536. doi: 10.1016/s1074-5521(96)90143-x. [DOI] [PubMed] [Google Scholar]
- 2.Gennari L, De Paola V, Merlotti D, Martini G, Nuti R. Expert Opinion on Pharmacotherapy. 2007;8:537–553. doi: 10.1517/14656566.8.5.537. [DOI] [PubMed] [Google Scholar]
- 3.Sammartino A, Cirillo D, Mandato VD, Di Carlo C, Nappi C. Journal of Endocrinological Investigation. 2005;28:80–84. [PubMed] [Google Scholar]
- 4.Markou A, Duka T, Prelevic GM. Hormones (Athens) 2005;4:9–17. doi: 10.14310/horm.2002.11138. [DOI] [PubMed] [Google Scholar]
- 5.Barret-Connor E, Cox DA, Anderson PW. Trends Endocrinol Metab. 1999;10:320–325. doi: 10.1016/s1043-2760(99)00182-4. [DOI] [PubMed] [Google Scholar]
- 6.Arnal JF, Gourdy P, Elhage R, Garmy-Susini B, Delmas E, Brouchet L, Castano C, Barreira Y, Couloumiers JC, Prats H, Prats AC, Bayard F. European Journal of Endocrinology. 2004;150:113–117. doi: 10.1530/eje.0.1500113. [DOI] [PubMed] [Google Scholar]
- 7.Yaffe K, Sawaya G, Lieberburg I, Grady D. JAMA, the Journal of the American Medical Association. 1998;279:688–695. doi: 10.1001/jama.279.9.688. [DOI] [PubMed] [Google Scholar]
- 8.Beresford SA, Weiss NS, Voigt LF, McKnight B. Lancet. 1997;349:458–461. doi: 10.1016/S0140-6736(96)07365-5. [DOI] [PubMed] [Google Scholar]
- 9.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 10.Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, Norton L, Nickelsen T, Bjarnason NH, Morrow M, Lippman ME, Black D, Glusman JE, Costa A, Jordan VC. JAMA. 1999;281:2189–2197. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- 11.McDonnell DP. Trends in Endocrinology and Metabolism. 1999;10:301–311. doi: 10.1016/s1043-2760(99)00177-0. [DOI] [PubMed] [Google Scholar]
- 12.Katzenellenbogen BS, Katzenellenbogen JA. Breast Cancer Research. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katzenellenbogen JA, O’Malley BW, Katzenellenbogen BS. Molecular Endocrinology. 1996;10:119–131. doi: 10.1210/mend.10.2.8825552. [DOI] [PubMed] [Google Scholar]
- 14.Mosselman S, Polman J, Dijkema R. FEBS Letters. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 15.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 16.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. EMBO Journal. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris HA. Ernst Schering Found Symp Proc. 2006;1:149–161. doi: 10.1007/2789_2006_021. [DOI] [PubMed] [Google Scholar]
- 19.Harris HA. Mol Endocrinol. 2007;21:1–13. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 20.De Angelis M, Stossi F, Waibel M, Katzenellenbogen BS, Katzenellenbogen JA. Bioorganic & Medicinal Chemistry. 2005;13:6529–6542. doi: 10.1016/j.bmc.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC., Jr Endocrinology. 2003;144:4241–4249. doi: 10.1210/en.2003-0550. [DOI] [PubMed] [Google Scholar]
- 22.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Journal of Medicinal Chemistry. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 23.De Angelis M, Stossi F, Carlson KA, Katzenellenbogen BS, Katzenellenbogen JA. Journal of the American Chemical Society. 2005;48:1132–1144. doi: 10.1021/jm049223g. [DOI] [PubMed] [Google Scholar]
- 24.Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, Collini MD, Miller CP, Dinh T, Henderson RA, Keith JC, Jr, Harris HA. Journal of Medicinal Chemistry. 2004;47:5021–5040. doi: 10.1021/jm049719y. [DOI] [PubMed] [Google Scholar]
- 25.Norman BH, Dodge JA, Richardson TI, Borromeo PS, Lugar CW, Jones SA, Chen K, Wang Y, Durst GL, Barr RJ, Montrose-Rafizadeh C, Osborne HE, Amos RM, Guo S, Boodhoo A, Krishnan V. Journal of Medicinal Chemistry. 2006;49:6155–6157. doi: 10.1021/jm060491j. [DOI] [PubMed] [Google Scholar]
- 26.Richardson TI, Norman BH, Lugar CW, Jones SA, Wang Y, Durbin JD, Krishnan V, Dodge JA. Bioorganic & Medicinal Chemistry. 2007;17:3570–3574. doi: 10.1016/j.bmcl.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 27.Schopfer U, Schoeffter P, Bischoff SF, Nozulak J, Feuerbach D, Floersheim P. Journal of Medicinal Chemistry. 2002;45:1399–1401. doi: 10.1021/jm015577l. [DOI] [PubMed] [Google Scholar]
- 28.Vu AT, Campbell AN, Harris HA, Unwalla RJ, Manas ES, Mewshaw RE. Bioorganic & Medicinal Chemistry Letters. 2007;17:4053–4056. doi: 10.1016/j.bmcl.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 29.De Angelis M, Katzenellenbogen JA. Bioorganic & Medicinal Chemistry Letters. 2004;14:5835–5839. doi: 10.1016/j.bmcl.2004.09.048. [DOI] [PubMed] [Google Scholar]
- 30.Kilbourn MR, Arduengo AJ, Park JT, Katzenellenbogen JA. Molecular Pharmacology. 1981;19:388–398. [PubMed] [Google Scholar]
- 31.Adlington MG, Orfanopoulos M, Fry JL. Tetrahedron Letters. 1976:2955–2958. [Google Scholar]
- 32.Felix AM. Journal of Organic Chemistry. 1974;39:1427–1429. [Google Scholar]
- 33.Krohn K, Kulikowski K, Leclercq G. Journal of Medicinal Chemistry. 1989;32:1532–1538. doi: 10.1021/jm00127a022. [DOI] [PubMed] [Google Scholar]
- 34.Tingoli M, Tiecco M, Testaferri L, Temperini A, Pelizzi G, Bacchi A. Tetrahedron. 1995;51:4691–4700. [Google Scholar]
- 35.Kamada A, Sasaki A, Kitazawa N, Okabe T, Nara K, Hamaoka S, Araki S, Hagiwara H. Chemical & Pharmaceutical Bulletin. 2004;52:79–88. doi: 10.1248/cpb.52.79. [DOI] [PubMed] [Google Scholar]
- 36.West CT, Donnelly SJ, Kooistra DA, Doyle MP. Journal of Organic Chemistry. 1973;38:2675–2681. [Google Scholar]
- 37.Scribner AW, Haroutounian SA, Carlson KE, Katzenellenbogen JA. Journal of Organic Chemistry. 1997;62:1043–1057. [Google Scholar]
- 38.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 39.Katzenellenbogen JA, Johnson HJ, Jr, Myers HN. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 40.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 41.McInerney EM, Tsai MJ, O’Malley BW, Katzenellenbogen BS. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10069–10073. doi: 10.1073/pnas.93.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]