

Abstract
Since its reported discovery in 1994, maspin (mammary serine protease inhibitor) has been characterized as a class II tumor suppressor by its ability to promote apoptosis and inhibit cell invasion. Maspin is highly expressed in normal mammary epithelial cells but reduced or absent in aggressive breast carcinomas. However, despite efforts to characterize the mechanism(s) by which maspin functions as a tumor suppressor, its molecular characterization has remained somewhat elusive. Therefore, in an attempt to identify maspin-interacting proteins and thereby gain insight into the functional pathways of maspin, we employed a maspin-baited yeast two-hybrid system and subsequently identified Interferon Regulatory Factor 6 (IRF6) as a maspin-binding protein. IRF6 belongs to the IRF family of transcription factors, which is best known for its regulation of interferon and interferon-inducible genes following a pathogenic stimulus. Although many of the IRF family members have been well characterized, IRF6 remains poorly understood. We report that IRF6 is expressed in normal mammary epithelial cells and that it directly associates with maspin in a yeast two-hybrid system and in vitro. The interaction occurs via the conserved IRF protein association domain and is regulated by phosphorylation of IRF6. We have shown that, similar to maspin, IRF6 expression is inversely correlated with breast cancer invasiveness. We further demonstrated that the transient re-expression of IRF6 in breast cancer cells results in an increase of N-cadherin and a redistribution of vimentin commensurate with changes in cell morphology, suggestive of an epithelial-to-mesenchymal transition event. Concomitantly, we showed that maspin acts as a negative regulator of this process. These findings help to elucidate the molecular mechanisms of maspin and suggest an interactive role between maspin and IRF6 in regulating cellular phenotype, the loss of which can lead to neoplastic transformation.
Maspin2 (mammary serine protease inhibitor, SerpinB5) was first isolated by subtractive hybridization and differential display as a protein that is expressed in normal mammary epithelial cells but reduced or absent in breast carcinomas (1). Since its initial discovery, maspin has been shown to inhibit tumor invasion and metastasis in breast cancer cells (2). Further studies implicate maspin as an angiogenesis inhibitor by its ability to block neovascularization and reduce tumor-associated microvessels and also demonstrate a role for maspin in the induction of apoptosis of tumor cells (3–4). In addition, the overexpression of maspin in transgenic mice disrupts normal mammary gland development by increasing apoptosis and disrupting cell differentiation (5). Despite the characterization of maspin as a tumor suppressor, the molecular mechanisms underlying maspin function are complex and remain predominantly unknown. Therefore, in an effort to decipher the molecular mechanisms of maspin, we employed a yeast two-hybrid system, in which we expressed full-length maspin as bait, to identify maspin-binding proteins. This approach led to the identification of Interferon Regulatory Factor 6 (IRF6) as a possible maspin-binding protein.
IRF6 belongs to the nine-member Interferon Regulatory Factor (IRF) family of transcription factors. IRFs are characterized by two conserved functional domains, namely a distinctive N-terminal DNA binding domain and a protein interaction domain. The protein interaction domain, termed the IRF association domain (IAD), is located near the C-terminal end of the protein. It is thought that protein-protein interactions involving the IAD, including homo- and heterodimerization among other IRF family members, are mediated through phosphorylation of one or multiple serine residues present within the serine-rich region near the C terminus, thus allowing interaction with other proteins (6–9). For several IRFs, this activation step is initiated by various pathogenic stimuli that induce IRF phosphorylation, leading to dimerization and subsequent translocation to the nucleus, where IRFs function to modulate the activation of type I interferons (10–11). Additionally, apart from their role in host defense, IRFs have been linked to the cellular processes of apoptosis, tumor suppression, and embryonic development (12–17). Although sequence alignment suggests that IRF6 possesses the conserved functional domains of the IRF family, there is no evidence to suggest that IRF6 functions in a similar manner to that of other IRFs nor has the initiating signal for IRF6 activation yet been determined. Here, we have demonstrated that IRF6 is expressed in normal mammary epithelial cells and that it interacts with maspin. IRF6 expression, similar to that of maspin, is reduced or lost as cells acquire a neoplastic state and progress toward a metastatic phenotype. We have also provided evidence implicating IRF6 in the regulation of cell differentiation through alterations in N-cadherin and vimentin, important phenotype markers associated with epithelial-to-mesenchymal transition. Importantly, changes induced by the re-expression of IRF6 are mitigated by the presence of maspin, all of which furthers our understanding of the molecular mechanisms of maspin.
MATERIALS AND METHODS
Cell Culture and Adenoviral Transfection
Normal, primary human mammary epithelial cells (HMEC 1331) were purchased from BioWhit-taker, Inc. (Walkersville, MD) and maintained in defined mammary epithelial cell basal medium provided by the company. The 1436N1 cell line, an HPV16E6 immortalized normal mammary epithelial cell line, was a gift from Dr. Shijie Sheng, Wayne State University, Detroit, MI and was maintained in D-complete medium (1:1 α-minimal essential medium and HAM F-12 supplemented with 1% fetal calf serum, 1 ng/ml cholera toxin, 10 mM HEPES, pH 7.7, 50 μM ascorbic acid, and Mito+ with gentamicin. The human breast cancer cell lines MCF-7, T47-D, MDA-MB-231, and HS578T were maintained in RPMI supplemented with 10% fetal calf serum and gentamicin.
IRF6 transfection of MDA-MB-231 and MDA-MB-231 green fluorescent protein-maspin was performed using the pacAd5CMV construct developed at the University of Iowa DNA vector core (supplied at 1011 particles/ml) at varying multiplicities of infection (m.o.i.) ranging from 50 to 250. Cells were treated in RPMI containing 1% serum with no antibiotic for 2 h, following which RPMI complete medium was added to the cells. Transfection efficiency was verified by Western blot analysis of transfected cells. All experiments were performed at 7 days post-transfection.
Semiquantitative PCR and Real-time PCR
Semiquantitative and real-time PCR were performed using standard protocols (18): for IRF6, forward 5′-GAGCGGTCAAGGGAAAGAC-3′ and reverse 5′-TGATGTTCAGGAAGGGGAAG-3′ and for maspin, forward 5′-CCACAGGCTTGGAGAAGATTGA-3′ and reverse 5′–GGTCAGCATTCAATTCATCCTTGT-3′. Primers for GAPDH were purchased from Clontech (Palo Alto, CA). PCR was performed for 27 cycles with a melting temperature of 60 °C. For maspin and GAPDH, primers were reported previously. For real-time PCR, total RNA was reverse transcribed using the Advantage PCR kit according to the manufacturer’s instructions (Clontech). Real-time PCR was performed on a 7500 Real Time PCR System (Applied Biosystems, Foster City, CA) using TaqMan® gene expression human primer/probe sets for the genes IRF6 (Hs00196213_m1) and N-cadherin (Hs00170423_m1) (Applied Biosystems). Briefly, 5 μl of cDNA, 1.25 μl of 20× Gene Expression Assay Mix, and 12.5 μl of 2× TaqMan® Universal PCR Master Mix in a total of 25 μl were amplified with the following thermocycler protocol: 1 cycle at 50 °C for 2 min, 1 cycle at 95 °C for 10 min, and 40 cycles at 95 °C for 15 s/60 °C for 1 min. All data were analyzed with Sequence Detection software (version 1.2.3; Applied Biosystems). The expression of each target gene was normalized to an endogenous control gene (GAPDH, 4333764F).
Cellular Fractionation and Western Blot Analysis
Cytosolic fractionation of cell lysate was performed similar to that previously described (19). Briefly, cells were washed in cold PBS and lysed in Buffer A (10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.1% Nonidet P-40 with 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 2 mM NaF plus protease inhibitor). Cells were scraped, and the slurry was incubated on ice for 30 min. Cells were then centrifuged at 10,000 × g for 10 min at 4 °C. Following the spin, the cytoplasmic fraction was removed and the pellet was washed three times in Buffer A. The remaining pellet was then lysed with Buffer B (20 mM HEPES (pH 7.9), 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol plus protease inhibitors mentioned in Buffer A), incubated on ice for 30 min, and centrifuged at 14,000 × g for 30 min to collect the nuclear fraction. Equal amounts of cellular protein were subjected to SDS-PAGE (10% resolving gel) and Western blot analysis using specific antibody to maspin (1:1000, monoclonal; BD Biosciences) or IRF6 (1:1000). IRF6 polyclonal rabbit antibody was custom ordered from Research Genetics (Huntsville, AL) using the peptides EDELEQSQHHVPIQDTFPF (amino acids 147–165) and SPEASWPK-TEPLEMEV (amino acids 187–102). The antibody was affinity purified, and its specificity was evaluated using the two blocking peptides (Fig. 2a).
FIGURE 2. Validation of IRF6 antibody (a) and IRF6 and maspin (b– d) expression profile in normal primary and cultured mammary epithelial cells.

Post-nuclear cytosolic fractions from 1436N1 cells and Ad5 IRF6-infected MDA-MB-231 cells were evaluated by Western blot (a) before and after preincubation of the antibody with blocking peptides, demonstrating the specificity of the IRF6 antibody. Shown are semiquantitative PCR (b), Western blot analysis (c), and confocal microscopy (d) of IRF6 and maspin in normal primary human mammary epithelial cells (HMEpC) and the immortalized mammary cell line 1436N1. Protein from the cytosolic fraction was evaluated by SDS-PAGE on a 10% gel. Actin was used as a protein loading control. GAPDH was used as an RNA loading control. For confocal microscopy, 1436N1 cells were dual-stained for maspin (green) and IRF6 (red; original magnification ×40).
Immunofluorescence
1436N1 mammary epithelial cells were seeded onto a Falcon 8-well CultureSlide (BD Biosciences) and grown to 70% confluency. Cells were fixed and stained as described previously (19). Briefly, cells were washed 3× in PBS and fixed in 100% ice-cold methanol for 5 min. Following fixation, cells were again washed 3× in PBS and blocked with 10% fetal calf serum in PBS for 1 h. Primary antibody was then added and incubated for 1 h. Following incubation, cells were washed 3× in PBS and incubated with the secondary antibody for 1 h. Cells were then washed 3× in PBS and mounted onto coverslips with gelvatol. Primary antibody for IRF6 was used at 1:750 and for maspin at 1:200. Rhodamine-conjugated goat anti-rabbit secondary antibody was used at 1:200 and Alexa Fluor 488-conjugated anti-mouse secondary antibody was used at 1:200 (Molecular Probes, Eugene, OR). Immunostaining for N-cadherin and vimentin was performed on MDA-MB-231neo and MDA-MB-231maspin cells 7 days post-infection with IRF6-containing adenovirus. Monoclonal antibody to N-cadherin (BD Biosciences) was used at 1:400, and monoclonal vimentin antibody (V-9 conjugated with Texas Red-X) was used at 1:1000 (Dako, Carpinteria, CA).
Phosphatase Treatment
50 μg of total cell lysate from HMEC 1331 cells was treated with 0.4 units of lambda-phosphatase (Sigma) with the supplied 10× buffer and MnCl2 for 1 h at room temperature. The sample was then analyzed using SDS-PAGE and Western blot analysis with antibody against IRF6.
Co-immunoprecipitation Assay
500 μg of protein from cytosolic fraction of 1436N1 cell lysate was precleared with 30 μl of protein A-Sepharose (Sigma). The lysate was then incubated with IRF6 polyclonal antibody for 2 h at room temperature. Immune complex was precipitated by the addition of Protein A-Sepharose (1 h at room temperature), washed 3× in PBS, and eluted using 2× SDS reducing buffer (sample buffer) for 5 min at room temperature. Elutions were assessed using Western blot on 10% acrylamide gels. For phosphatase-treated samples, cell lysate was treated with 2 units of lambda-phosphatase for 30 min at room temperature prior to the addition of the primary antibody.
Yeast Two-hybrid Assays
Yeast two-hybrid experiments were performed using the Matchmaker™ Gal4 Two-hybrid System 3 from Clontech. We cloned the full-length maspin coding sequence into pGBKT7 to generate the pGBKT7-maspin bait plasmid in which maspin is fused with the Gal4 DNA binding domain. The human mammary gland Matchmaker™ cDNA library was expressed in the vector pGADT7 as prey. All procedures were performed according to the manufacturer’s protocol. Selection was performed on S.D.:-Leu/-Trp/-Ade/-His medium containing 5-bromo-4-chloro-3-indolyl-α-D-galac-toside as a visual selection marker. Plasmids were isolated from yeast using a standard smash-and-grab protocol with glass beads (425–500 microns, acid-washed; Sigma). Sequencing of the prey plasmids was performed at the University of Iowa DNA Sequencing Facility. For co-transformation of IRF6 deletion mutant constructs, competent AH109 yeast was prepared using a standard lithium acetate protocol. In brief, yeast was suspended in TE/LiAc (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.1 M lithium acetate, pH 7.5). Bait and prey plasmids were added to competent cells in the presence of polyethylene glycol (40%), 10 mM Tris-HCl, 0.1 M lithium acetate, and subjected to heat shock for 15 min at 42 °C. Herring testis DNA was used as a carrier. Successful co-transformation was selected with S.D.:-Leu/-Trp and bait-prey interactions were assayed on S.D.:-Leu/-Trp/-Ade/-His quadruple knockout medium. All medium supplements were obtained from Clontech.
Tissue Specimens and Immunohistochemistry
Archival tissue from patients with no prior drug treatment was obtained from the Department of Pathology at The University of Iowa and the Department of Pathology at Northwestern University, Robert H. Lurie Comprehensive Cancer Center, and were used in compliance with requirements of the Institutional Review Board for the Protection of Human Subjects. Tissue specimens were sectioned at 4 μm, deparaffinized, and subjected to a water bath antigen-recovery protocol using citrate buffer (pH 6.0). Maspin and IRF6 expression were determined on the Dako Autostainer using the Vectastain universal Elite ABC Peroxidase kit (Vector Laboratories, Inc., Burlingame, CA). Antibody to maspin was used at a 1:200 dilution as previously described (20). Antibody to IRF6 was used at a 1:1200 dilution. Expression was determined by semiquantitative assessment of the stained cells and the staining intensity by diaminobenzidine. All the samples were reviewed by a board-certified pathologist (Michael R. Pins) who was blinded to the clinical outcome of these patients until the scoring had been completed. An experimental flow chart outlining the design of the study is available in the supplemental data.
RESULTS
Yeast Two-hybrid and in Vitro Verification of Maspin-IRF6 Interaction
Because the tumor suppressive functions of maspin were first identified in mammary epithelial cells, we utilized a cDNA library derived from normal human mammary epithelial cells in a maspin-baited yeast two-hybrid assay. Evaluation of the resultant clones revealed IRF6 as a putative maspin-binding protein, with the amino acid sequence from residues 139 to 334 spanning almost the entire IRF6 proline-rich region and two-thirds of the IAD domain identified as the interaction site (highlighted in the box, Fig. 1). Following the identification of IRF6 as a possible target for maspin interaction, we confirmed the presence of endogenous IRF6 in human mammary epithelial cells (HMEpC) by semiquantitative PCR and Western blot and verified the interaction between maspin and IRF6 in vitro through co-immunoprecipitation (Fig. 2). Post-nuclear cytosolic fractions from 1436N1 cells and Ad5 IRF6-infected MDA-MB-231 cells were evaluated by Western blot before and after preincubation of the antibody with blocking peptides (Fig. 2a) to demonstrate the specificity of the IRF6 antibody. Both mRNA (Fig. 2b) and protein levels (Fig. 2c) of endogenous IRF6 are clearly demonstrated in HMEpC and the immortalized mammary epithelial cell line 1436N1. Importantly, cell lysates used in the Western blot analysis represent the post-nuclear cytosolic fraction, where both IRF6 and maspin primarily reside. Under the experimental conditions used, no IRF6 was detected in the nuclear fraction (see supplemental Fig. S2). This is supported by confocal microscopy, which demonstrates the cellular localization of these proteins in the cytoplasmic compartment of cultured mammary epithelial cells (Fig. 2d).
FIGURE 1. Amino acid sequence of IRF6.

Shading indicates identity (black) or similarity (gray) with other IRFs (IRF3–9). Inverted triangles (▼) indicate sites for introns with the number representing the following exon. The start codon is at the beginning of exon 3, and the stop codon is in exon 9. The functional domains consist of the DNA binding domain (exons 3– 4), a less conserved proline-rich domain (exons 5– 6), the interferon association domain (exons 7– 8), and the serine-rich region (red bar, exon 9). Indicated are a putative nuclear localization sequence (red), the part of IRF6 encoded by the partial cDNA clone pulled out in the original yeast two-hybrid experiment (black box), pentatryptophan residues of DNA binding domain (*), and potential phosphorylation sites (orange).
When examined by Western blot, IRF6 appears as a doublet with molecular masses of ~59 and ~63 kDa. Because it is known that phosphorylation of IRFs is important for IRF activation, we tested whether the IRF6 doublet represents different phosphorylation states of IRF6 in HMEpCs. Treatment of HMEpC cell lysate with the nonspecific lambda-phosphatase resulted in the disappearance of the upper band, whereas the intensity of the lower band increased, indicating that the upper band represents a phosphorylated form of IRF6 (Fig. 3a). To further confirm the phosphorylation status of IRF6 and identify the target amino acid, we tested immunoprecipitated IRF6 with commercially available antibodies to phosphoserine, threonine, and tyrosine. We were unable to confirm the phosphorylation status using this approach, and because no phospho-specific antibody to IRF6 is yet available, further work is required to resolve this important point.
FIGURE 3. a, effect of phosphatase treatment on IRF6 and in vitro confirmation (b– c) of maspin-IRF6 interaction.

a, post-nuclear cytosolic fractions from HMEpC cells were treated with lambda-phosphatase (0.4 units) for 1 h at room temperature in the absence of phosphatase inhibitors NaF and sodium orthovanadate. Samples were analyzed using SDS-PAGE and Western blot. b, 500 μg of total protein from the cytosolic fraction of 1436N1 cells was precleared with Protein A-coated Sepharose beads for 30 min and then incubated for 2 h with anti-IRF6 polyclonal antibody at room temperature. Protein A-coated Sepharose beads were added to the lysate and incubated for an additional hour at room temperature. Beads were washed three times in PBS, and then protein was eluted with 2× sample buffer with SDS for 5 min. For phosphatase-treated samples, 500 μg of protein was treated with 2 units of phosphatase for 30 min at room temperature prior to the addition of primary antibody. Lysate was evaluated prior to and following immunoprecipitation and shows the efficient removal of IRF6 from the lysate, whereas only a small portion of maspin was removed. c, full-length IRF6 and eight different IRF6 deletion mutant constructs were expressed as prey and co-transformed with maspin (bait) in the yeast strain AH109. -Leu/-Trp dropout medium was used to select for bait and prey plasmids. -Leu/-Trp/-Ade is a medium stringency dropout medium, and -Leu/-Trp/-Ade/-His is a high stringency dropout medium. The p53-large T antigen interaction was used as a positive control. The graph on the left illustrates the various IRF6 deletion constructs, with arrows depicting the IRF6 fragment used as prey (full-length IRF6 contains exons 3–9). Protein interactions were measured by yeast growth on appropriate selective media.
To verify the maspin-IRF6 interaction in cell culture, we performed a co-immunoprecipitation assay utilizing the cytosolic fraction from 1436N1 cells. As shown in Fig. 3b, immunoprecipitation with anti-IRF6 antibody precipitated endogenous IRF6 and co-precipitated maspin, indicative of an interaction between these two proteins in cultured mammary epithelial cells. It is important to note that the amount of maspin interacting with IRF6 under these conditions represents a small fraction of the total maspin found in the cells, and further work must be done to quantify the stoichiometric ratios of this protein interaction.
To examine the role of IRF6 phosphorylation in regulating the maspin-IRF6 interaction, we performed co-immunoprecipitation with cell lysate pretreated with lambda-phosphatase (Fig. 3b). This led to a significant decrease in the amount of maspin interacting with IRF6, indicating a role for phosphorylation in the regulation of the maspin-IRF6 interaction. These observations were further strengthened by IRF6 protein truncation analysis (Fig. 3c). The IRF family shares at least two common domains, namely an N-terminal DNA binding domain and a protein association domain termed the IRF association domain (IAD). Because the IAD is the primary site of protein interaction among the IRFs, we hypothesized that the IAD would be necessary for an interaction with maspin. To test this, we constructed a series of IRF6 deletion mutant constructs and co-expressed these constructs with the maspin-bait vector in a yeast two-hybrid system to evaluate interaction efficiency. The results demonstrate that the IAD (exons 7–8) is both necessary and sufficient for growth on selective media, verifying the IRF6 IAD as the site of interaction with maspin. Importantly, the inclusion of exon 9 in the IRF6 vector was sufficient to effectively abrogate this growth response, indicating an inefficient maspin-IRF6 interaction when exon 9 is present.
Maspin and IRF6 Expression in Breast Cancer
As we attempted to assess the biological relevance of the maspin-IRF6 interaction, we focused our studies on breast cancer, where the expression of maspin is inversely correlated with the advancement of tumor progression. In HMEpCs, maspin is present in high levels but dramatically reduced in non-aggressive mammary tumors and absent in a majority of highly invasive, metastatic breast carcinomas. Because of maspin’s implications in breast cancer development and progression, we examined IRF6 expression as it relates to maspin expression in a series of neoplastic cell lines. Semiquantitative PCR analysis of the non-aggressive, poorly invasive breast cancer cell lines MCF-7 and T47-D revealed a moderate reduction in IRF6 mRNA compared with HMEpC and 1436N1 (Fig. 4a). No IRF6 mRNA was detected in the highly invasive, metastatic cell lines MDA-MB-231 and HS578T, which mimics the expression pattern of maspin in these same cells. This IRF6 expression pattern was confirmed at the protein level by Western blot analysis, where the loss of IRF6 in both the poorly invasive and highly invasive cell lines is more pronounced (Fig. 4b). These data were confirmed by in situ immunohistochemistry of patient tissues. Immunohistochemical analysis of maspin and IRF6 in tissue samples from normal mammary gland, non-invasive ductal carcinoma in situ and invasive ductal carcinoma revealed an expression pattern in situ similar to that seen in the cell lines tested), where non-invasive neoplastic cells demonstrated a modest reduction of IRF6 and the loss of both maspin and IRF6 coincided with the appearance of invasive cancer cells (Fig. 5 and TABLE ONE).
FIGURE 4. IRF6 and maspin expression profiles in poorly invasive and highly invasive metastatic breast cancer lines.
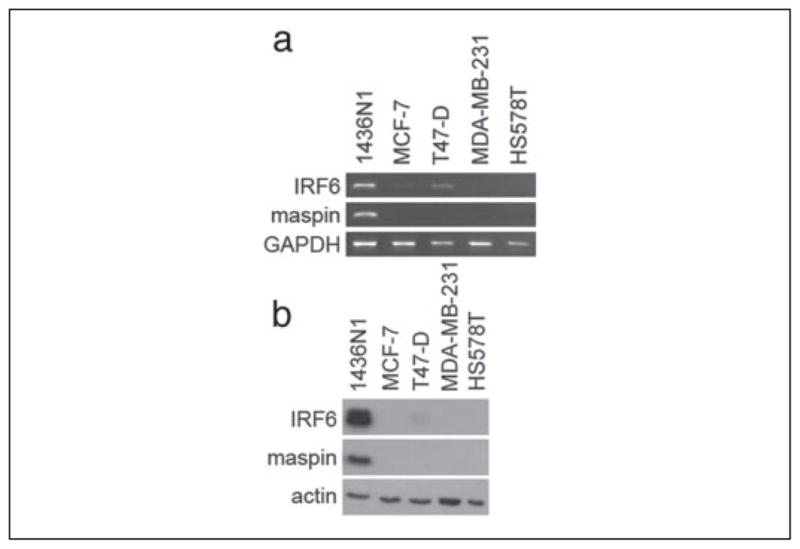
Semiquantitative PCR (a) and Western blot analysis (b) of IRF6 and maspin in the poorly invasive neoplastic breast cell lines MCF-7 and T47-D and the highly invasive metastatic cell lines MDA-MB-231 and HS578T. The post-nuclear cytosolic fraction was analyzed. GAPDH was used as an RNA loading control. Actin was used as a protein loading control.
FIGURE 5. Immunohistochemical analysis of IRF6 and maspin in normal and neoplastic tissue sections.
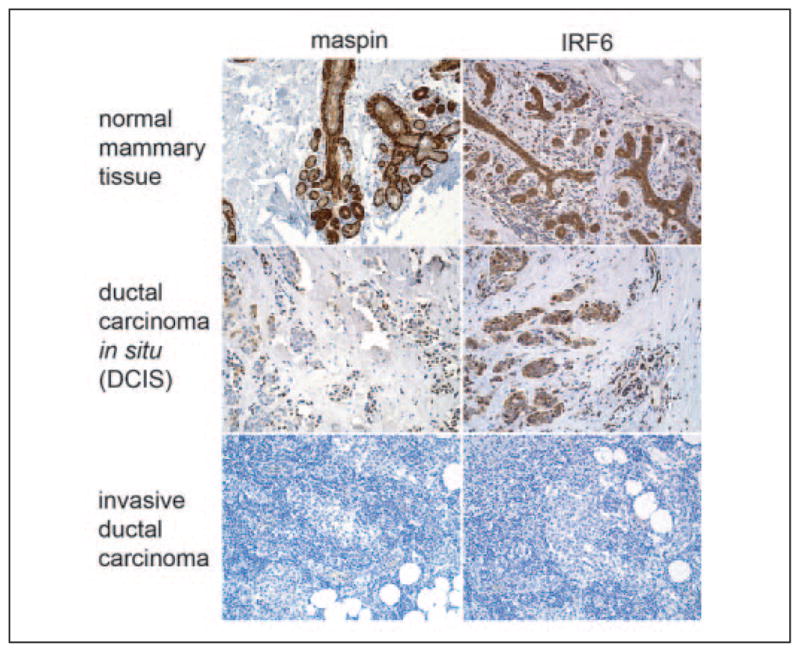
A comparative analysis of maspin and IRF6 immunoreactivity in archival tissue samples from normal mammary and breast cancer patient tissues with ductal carcinoma in situ (DCIS) and invasive ductal carcinoma. Diaminobenzidine was used as the chromogen.
TABLE I. Summary of in vivo analysis of maspin and IRF6 expression.
4-μm sections were deparafinized and stained for maspin and IRF6 as described under “Materials and Methods.” The intensity and degree of staining was evaluated by a board-certified pathologist (MRP) in a blinded fashion; the evaluator did not have prior knowledge of the stage of the patient or the grade of the tumor. Results were scored based upon the greatest degree of staining in the tumor cells constituting at least 10% of the tumor, where a score of zero represented no staining, 1+ represented faint staining significantly greater than the internal and external negative controls, 3+ represented staining equal to or greater than the positive control tissue, and 2+ represented staining intermediate between 1 and 3+. Tissue samples from cases 4 and 5 were matched primary tumor and lymph node metastases from the same patient.
| Patient age | Tumor type | Tumor grade | Maspin | IRF6 | |
|---|---|---|---|---|---|
| Case 1 | 51 | Infiltrating ductal carcinoma | Grade 3 | – | – |
| Case 2 | 77 | Infiltrating ductal carcinoma | Grade 3 | – | – |
| Case 3 | 50 | DCIS/mix | Grade 1 | – | 1–2+ in DCIS |
| Case 4 | 47 | ||||
| Primary tumor | Infiltrating ductal carcinoma | Grade 2 | – | 1+ (focal) | |
| Lymph node | – | 1+ (10% 2+) | |||
| Case 5 | 42 | ||||
| Primary tumor | Infiltrating ductal carcinoma | Grade 3 | 3+ | – | |
| Lymph node | heterogeneous | 3+ (15%) | 2+ |
In an attempt to decipher a possible role for IRF6 in mammary epithelial cells and the consequences resulting from the loss of both IRF6 and maspin, we transiently infected MDA-MB-231 neo (MB-231neo) and MDA-MB-231 green fluorescent protein-maspin-transfected (MB-231maspin) cells with an adenovirus containing wild-type IRF6 (Ad5 IRF6) at different m.o.i., ranging from 50 to 250. At 7 days post-infection, an altered phenotype was noticeable in MB-231neo cells. However, no changes were seen in MB-231maspin cells following Ad5 IRF6 infection (Fig. 6, A–D). The presence of maspin and IRF6 was confirmed by immunofluorescence microscopy of the cells and real-time PCR, respectively (Figs. 6B and 7a).
FIGURE 6. Phenotypic analysis of MDA-MB-231 neo and MDA-MB-231 green fluorescent protein-maspin cells following transient infection with Ad5 IRF6.

Representative phase-contrast micrographs of MDA-MB-231 neo (MB-231neo) cells untransfected for IRF6 expression (A) and Ad5 IRF6 infected (B) at an m.o.i. of 100 were taken 7 days post-infection. The MDA-MB-231 green fluorescent protein-maspin-transfected cells (MB-231maspin) were either negative for IRF6 expression (C, untransfected) or infected with Ad5 IRF6 (D). Representative samples were also fixed for immunohistochemistry and stained for vimentin (red) and N-cadherin (green) as shown under the representative phase-contrast micrographs. The insert depicts immunofluorescence of MB-231maspin cells. Original magnification was ×20 for all pictures.
FIGURE 7. Real-time PCR analysis demonstrating changes in N-cadherin expression following Ad5 IRF6 infection of MDA-MB-231 cells.

A, verification of IRF6 expression in MB-231neo and MB-231maspin cells following infection with various concentrations of Ad5 IRF6 (m.o.i. 50, 100, 250). B, changes in N-cadherin expression in the same cells infected with Ad5 IRF6 (m.o.i. 50, 100, 250).
We next sought to define the altered phenotype induced by IRF6 re-expression by examining the expression of the known phenotypic markers vimentin and N-cadherin. Real-time PCR analysis of transfected and non-transfected MB-231neo cells indicated an increase in the expression of N-cadherin (Fig. 7b.). This was further confirmed at the protein level by immunohistochemical analysis of the fixed cells (Fig. 6, A and B). For vimentin, there was no significant change at the mRNA level, but at the protein level the presence of IRF6 exerted a change in the distribution of vimentin (Fig. 6, A and B). Notably, the alterations in N-cadherin and vimentin were not evident in the MB-231maspin cells infected with Ad5 IRF6 (Fig. 6, C and D).
DISCUSSION
This study presents new findings from a yeast two-hybrid system in which we identified a novel interaction between the tumor suppressor maspin and IRF6, a member of the IRF transcription factor family. Our studies indicate that IRF6 is expressed in normal human mammary epithelial cells, and this interaction with maspin has been verified by co-immunoprecipitation. IRF6 is the least understood of the nine-member IRF family. Functions for this family of transcription factors have generally been associated with the induction and regulation of type I interferons and interferon-inducible genes following pathogenic insult but have also been linked to the cellular processes of growth control, apoptosis, tumor suppression, and embryonic development (12–17, 21–25). Notably, recent studies indicate a crucial role for IRF6 in palatal development (26–27). Palatal fusion requires the dispersion and elimination of the medial edge epithelium lining opposing palatal shelves, which occurs through a combination of apoptosis, epithelial-to-mesenchymal transition, and cell migration (28–30). IRF6 is highly expressed in the medial edge epithelium immediately prior to palatal fusion, indicating a possible role for IRF6 in one or more of these tissue-remodeling processes.
Our studies show for the first time that IRF6 is expressed in mammary epithelial cells where, under our experimental conditions, it resides primarily in the cytoplasm. SDS-PAGE of the cytosolic fraction demonstrated that IRF6 appears as two distinct bands with molecular masses of ~59 and ~63 kDa. Our data indicate that the upper ~63-kDa band represents a phosphorylated form of IRF6, as treatment with lambda-phosphatase abolished the upper band. Phosphorylation is a central step in the activation of many IRFs and acts to facilitate protein interactions involving the IAD. Phosphorylation normally occurs in the serine-rich region of the C terminus, which, in a non-phosphorylated state, exerts an auto-inhibitory effect by blocking protein-protein interactions (31–33). Through detailed IRF6 truncation analysis, we have demonstrated a similar auto-inhibitory effect caused by the IRF6 C-terminal serine-rich region. We have shown that the IRF6 IAD is both necessary and sufficient for an interaction with maspin and that the inclusion of the C-terminal serine-rich region abrogates this interaction. Furthermore, our co-immunoprecipitation data indicate that IRF6 phosphorylation plays a significant role in the maspin-IRF6 interaction, because treatment of cell lysate with lambda-phosphatase prior to co-immunoprecipitation resulted in a significant decrease in the amount of maspin interacting with IRF6. Taken together, we postulate that phosphorylation of IRF6 facilitates its interaction with maspin by relieving the auto-inhibitory effect of the C terminus in a manner similar to other IRFs.
Co-immunoprecipitation studies also suggest that in normal mammary epithelial cells only a small portion of maspin is interacting with IRF6. Because it appears that the interaction between maspin and IRF6 occurs primarily in the cytoplasmic compartment, it is possible that the interaction with maspin serves to regulate IRF6 by controlling its translocation to the nucleus. This type of negative regulation has been reported with other IRFs, such as the cytoplasmic retention of IRF9 by its binding partner Stat2 (34). Indeed, this is supported by our transfection studies that demonstrated the ability of maspin to protect against the effects of IRF6 re-expression.
Because of the role of maspin in tumor suppression, we evaluated the expression of IRF6 in both poorly invasive and highly invasive breast cancer cell lines. A comparison of maspin and IRF6 expression by semi-quantitative PCR demonstrated a moderate decrease in IRF6 mRNA levels during progression from a normal to a neoplastic, poorly invasive phenotype, whereas IRF6 expression was absent in the highly invasive and metastatic cells. This differs from maspin expression, which is highly reduced or completely absent in the poorly invasive cells. Studies have shown that methylation of the maspin promoter is involved in maspin silencing and is an early event in breast cancer development (35). In contrast, the moderate down-regulation of IRF6 in poorly invasive cells suggests an altered regulatory pathway rather than epigenetic gene silencing. The reduction of IRF6 is more pronounced at the level of protein expression, where Western blot analysis revealed only minimal amounts of IRF6 in the poorly invasive cell lines. Importantly, the stable re-expression of maspin in MDA-MB-231 cells did not result in any changes in IRF6 expression nor did the re-expression of IRF6 lead to changes in maspin expression, suggesting that neither of these genes directly regulates the expression of the other. We postulate that the stepwise decline of IRF6 could be indicative of a role for IRF6 in regulating cellular differentiation, because the poorly invasive cells still maintain some epithelial characteristics, whereas the aggressive, metastatic cells have adopted a mesenchymal phenotype. Studies indicate that silencing of the maspin gene is one of the initial steps in tumor progression (35). It is plausible to speculate that this could result in deregulated IRF6, leading to transcriptional activation of genes involved in epithelial-to-mesenchymal transition. This is supported by our IRF6 transfection approach, which indicates that in the absence of maspin IRF6 leads to increased expression of epithelial-to-mesenchymal transition-associated genes such as N-cadherin. It is also possible that IRF6 is temporally required for gene regulation (as is the case in palatal fusion (26)) and the presence of maspin regulates this process. In the absence of maspin, continual activation of IRF6 might lead to its silencing, which might explain the loss of IRF6 in highly invasive breast cancer cells. The increase in N-cadherin (and decrease in E-cadherin) in cancer cells with epithelial origin has been reported to facilitate the infiltration of cancer cells into the host basement membrane. Indeed, studies have identified the up-regulation of N-cadherin as characteristic of a more invasive and less differentiated breast cancer cell phenotype (36–38).
Our in vitro findings are further supported by immunohistochemical analysis of breast tissue. These studies indicate that both maspin and IRF6 are moderately expressed in both the ductal and glandular epithelium of normal tissue and reduced or absent in neoplastic lesions. Interestingly, in normal tissue the myoepithelial cell layer, which surrounds the gland, expresses a high level of maspin but is devoid of IRF6. This lack of IRF6 expression in myoepithelial cells could be the result of an alternate differentiation pathway because both the myoepithelial and luminal epithelial cells are derived from a common mammary progenitor cell, further supporting a role for IRF6 and maspin in regulating the mammary epithelial cell phenotype (39).
In conclusion, we have shown that the transcription factor IRF6 is expressed in normal mammary epithelium and that it interacts with the tumor suppressor maspin. We further demonstrated that IRF6 expression is lost in invasive breast cancer cells and hypothesize that the maspin-IRF6 interaction is important in maintaining a normal mammary epithelial cell phenotype. We further postulate that the maspin-IRF6 interaction plays an important role in tissue remodeling by regulating cell differentiation, an important aspect of mammary gland morphogenesis. This work helps elucidate the tumor suppressive mechanisms of maspin and supports a model in which maspin realizes its function in part through the regulation of IRF6. Understanding the molecular mechanisms of maspin and its interaction with IRF6 will allow us to translate the potential of maspin as a possible therapeutic agent and prognostic indicator in breast cancer.
Acknowledgments
We thank Dr. Dawn Kirschmann, Dr. Lynne-Marie Postovit, and Dr. Zoe Demou and Margaret Malik, Nicholas Rorick, and Elisabeth Seftor for helpful scientific discussions and research contributions.
Footnotes
This work was supported by National Institutes of Health Grant CA 75681 (to M. J. C. H.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The abbreviations used are: maspin, mammary serine protease inhibitor; IRF6, interferon regulatory factor 6; IAD, IRF association domain; m.o.i., multiplicity of infection; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PBS, phosphate-buffered saline; HMEpC, human mammary epithelial cell.
References
- 1.Zou Z, Anisowicz A, Hendrix MJC, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 2.Sheng S, Carey J, Seftor E, Dias L, Hendrix MJC, Sager R. Proc Natl Acad Sci U S A. 1996;93:11669–11674. doi: 10.1073/pnas.93.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang M, Volpert O, Shi YH, Bouck N. Nat Med. 2000;6:196–199. doi: 10.1038/72303. [DOI] [PubMed] [Google Scholar]
- 4.Jiang N, Meng Y, Zhang S, Mensah-Osman E, Sheng S. Oncogene. 2002;21:4089–4098. doi: 10.1038/sj.onc.1205507. [DOI] [PubMed] [Google Scholar]
- 5.Zhang M, Magit D, Botteri F, Shi HY, He K, Li M, Furth P, Sager R. Dev Biol. 1999;215:278–287. doi: 10.1006/dbio.1999.9442. [DOI] [PubMed] [Google Scholar]
- 6.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. J Biol Chem. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 7.Lin R, Mamane Y, Hiscott J. J Biol Chem. 2000;275:34320–34327. doi: 10.1074/jbc.M002814200. [DOI] [PubMed] [Google Scholar]
- 8.Meraro D, Hashmueli S, Koren B, Azriel A, Oumard A, Kirchhoff S, Hauser H, Nagulapalli S, Atchison ML, Levi B. J Immunol. 1999;163:6468–6478. [PubMed] [Google Scholar]
- 9.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. Nat Struct Biol. 2003;10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 10.Barnes BJ, Kellum MJ, Field AE, Pitha PM. Mol Cell Biol. 2002;22:5721–5740. doi: 10.1128/MCB.22.16.5721-5740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin R, Heylbroeck C, Pith PM, Hiscott J. Mol Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM. Cancer Res. 2003;63:6424–6431. [PubMed] [Google Scholar]
- 13.Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T. Cell. 1994;77:829–839. doi: 10.1016/0092-8674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 14.Duguay D, Mercier F, Stagg J, Martineau D, Branson J, Servant M, Lin R, Galipeau J, Hiscott J. Cancer Res. 2002;62:5148–5152. [PubMed] [Google Scholar]
- 15.Moriyama Y, Nishiguchi S, Tamori A, Koh N, Yano Y, Kubo S, Hirohashi K, Otani S. Clin Cancer Res. 2001;7:1293–1298. [PubMed] [Google Scholar]
- 16.Hatada S, Kinoshita M, Takahashi S, Nishihara R, Sakumoto H, Fukui A, Noda M, Asashima M. Gene (Amst) 1997;203:183–188. doi: 10.1016/s0378-1119(97)00512-x. [DOI] [PubMed] [Google Scholar]
- 17.De Creus A, Van Beneden K, Stevenaert F, Debacker V, Plum J, Leclercq G. J Immunol. 2002;168:6486–6493. doi: 10.4049/jimmunol.168.12.6486. [DOI] [PubMed] [Google Scholar]
- 18.Kirschmann DA, Seftor EA, Fong SFT, Nieva DRC, Sullivan CM, Edwards EM, Sommer P, Csiszar K, Hendrix MJC. Cancer Res. 2002;62:4478–4483. [PubMed] [Google Scholar]
- 19.Khalkhali-Ellis Z, Christian AL, Kirschmann DA, Edwards EM, Rezaie-Thompson M, Vasef MA, Gruman LM, Seftor RE, Norwood LE, Hendrix MJ. Clin Cancer Res. 2004;10:449–454. doi: 10.1158/1078-0432.ccr-1002-03. [DOI] [PubMed] [Google Scholar]
- 20.Hendrix MJC, Seftor EA, Seftor REB, Trevor KT. Am J Pathol. 1997;150:483–493. [PMC free article] [PubMed] [Google Scholar]
- 21.Civas A, Island ML, Genin P, Morin P, Navarro S. Biochimie (Paris) 2002;84:643–654. doi: 10.1016/s0300-9084(02)01431-1. [DOI] [PubMed] [Google Scholar]
- 22.Williams BR, Sen GC. Science. 2003;300:1100–1101. doi: 10.1126/science.1085277. [DOI] [PubMed] [Google Scholar]
- 23.Fulda S, Debatin KM. Oncogene. 2002;21:2295–2308. doi: 10.1038/sj.onc.1205255. [DOI] [PubMed] [Google Scholar]
- 24.Kim TY, Lee KH, Chang S, Chung C, Lee HW, Yim J, Kim TK. J Biol Chem. 2003;278:15272–15278. doi: 10.1074/jbc.M205792200. [DOI] [PubMed] [Google Scholar]
- 25.Masumi A, Yamakawa Y, Fukazawa H, Ozato K, Komuro K. J Biol Chem. 2003;278:25401–25407. doi: 10.1074/jbc.M213037200. [DOI] [PubMed] [Google Scholar]
- 26.Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, Natsume N, Yoshiura K, Vieira AR, Orioli IM, Castilla EE, Moreno L, Arcos-Burgos M, Lidral AC, Field LL, Liu YE, Ray A, Goldstein TH, Schultz RE, Shi M, Johnson MK, Kondo S, Schutte BC, Marazita ML, Murray JC. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Alvarez C, Tudela C, Perez-Miguelsanz J, O’Kane S, Puerta J, Ferguson MW. Dev Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- 29.Fitchett JE, Hay ED. Dev Biol. 1989;131:455–474. doi: 10.1016/s0012-1606(89)80017-x. [DOI] [PubMed] [Google Scholar]
- 30.Cuervo R, Valencia C, Chandraratna RA, Covarrubias L. Dev Biol. 2002;245:145–156. doi: 10.1006/dbio.2002.0620. [DOI] [PubMed] [Google Scholar]
- 31.Reich NC. J Interferon Cytokine Res. 2002;22:103–109. doi: 10.1089/107999002753452719. [DOI] [PubMed] [Google Scholar]
- 32.Marié I, Smith E, Prakash A, Levy DE. Mol Cell Biol. 2000;20:8803–8814. doi: 10.1128/mcb.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eroshkin A, Mushegian A. J Mol Med. 1999;77:403–405. doi: 10.1007/s001090050369. [DOI] [PubMed] [Google Scholar]
- 34.Lau JF, Parisien J, Horvath CM. Proc Natl Acad Sci U S A. 2000;97:7278–7283. doi: 10.1073/pnas.97.13.7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domann FE, Rice JC, Hendrix MJ, Futscher BW. Int J Cancer. 2000;85:805–810. doi: 10.1002/(sici)1097-0215(20000315)85:6<805::aid-ijc12>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Knudsen KA, Wheelock MJ. J Cell Biochem. 2005;95:488–496. doi: 10.1002/jcb.20419. [DOI] [PubMed] [Google Scholar]
- 37.Strizzi L, Bianco C, Normanno N, Seno M, Wechselberger C, Wallace-Jones B, Khan NI, Hirota M, Sun Y, Sanicola M, Salomon DS. J Cell Physiol. 2004;201:266–276. doi: 10.1002/jcp.20062. [DOI] [PubMed] [Google Scholar]
- 38.Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boecker W, Buerger H. Cell Prolif. 2003;36(Suppl 1):73–84. doi: 10.1046/j.1365-2184.36.s.1.7.x. [DOI] [PMC free article] [PubMed] [Google Scholar]