

Abstract
Much evidence suggests that astrocytes protect neurons against ischemic injury. Although astrocytes are more resistant to some insults than neurons, few studies offer insight into the real time changes of astrocytic protective functions with stress. Mitochondria are one of the primary targets of ischemic injury in astrocytes. We investigated the time course of changes in astrocytic ATP levels, plasma membrane potential, and glutamate uptake, a key protective function, induced by mitochondrial inhibition. Our results show that significant functional change precedes reduction in astrocytic viability with mitochondrial inhibition. Using the mitochondrial inhibitor fluorocitrate (FC, 0.25 mM) that is preferentially taken by astrocytes we found that inhibition of astrocyte mitochondria increased vulnerability of co-cultured neurons to glutamate toxicity. In our studies the rates of FC-induced astrocytic mitochondrial depolarization were accelerated in mixed astrocyte/neuron cultures. We hypothesized that the more rapid mitochondrial depolarization was promoted by an additional energetic demand imposed be the co-cultured neurons. To test this hypothesis we exposed pure astrocytic cultures to 0.01 –1 mM aspartate as a metabolic load. Aspartate application accelerated the rates of FC-induced mitochondrial depolarization, and, at 1 mM, induced astrocytic death, suggesting that strong energetic demands during ischemia can compromise astrocytic function and viability.
Keywords: ischemia, mitochondria, astrocyte, neuron, fluorocitrate
Introduction
Increasing evidence shows that astrocytes play a critical role in neuronal protection during ischemic injury (Chen and Swanson 2003; Dienel and Hertz 2005). Astrocytes contain the principal glycogen store in the brain (Dringen et al. 1993) and provide energy substrates like lactate and pyruvate to neurons (Anderson et al. 2003). Astrocytes spatially buffer ions including K+ and play a crucial role in regulation of the extracellular ionic environment (Walz 1989; Leis et al. 2005). These astrocytic functions are particularly important within ischemic areas where energy depletion and ionic disruption are severe (Lo et al. 2003). During ischemic injury astrocytes also provide neurons with antioxidant substrates, such as glutathione and reduced ascorbate (Anderson et al. 2003; Swanson et al. 2004). Massive release of glutamate occurs during ischemia and plays a major role in ischemia-induced cell death (Benveniste 1991; Choi 1994). Astrocyte glutamate uptake is crucial for limiting the excitotoxic effect of glutamate (Rosenberg and Aizenman 1989; Dugan et al. 1995). Downregulation of EAAT1/GLAST or EAAT2/GLT1, astrocytic glutamate transporters, but not EAAT3/EAAC1, the neuronal subtype, has been shown to increase ischemic injury following focal cerebral ischemia in rat (Watase et al. 1998; Fukamachi et al. 2001; Rao et al. 2001).
Mitochondria are one of the primary targets of ischemic injury in astrocytes (Bambrick et al. 2004). Astrocytes have high rates of oxidative metabolism and derive a substantial amount of ATP from mitochondrial production (Hertz et al. 2006). Multiple astrocytic functions, including high affinity glutamate uptake, are energy demanding (Anderson and Swanson 2000), thus mitochondrial dysfunction can seriously impair astrocytic neuroprotective function.
Astrocytes have been shown to be more resistant than neurons to some forms of ischemic insults in vitro (Goldberg and Choi 1993; Xu et al. 2001), and astrocytic markers are better preserved in animal stroke models (Chen et al. 1993; Lee et al. 2003). However, viability studies provide little information about the time course and extent of functional astrocytic impairment that develops before cell death. In this study we used the mitochondrial inhibitor fluorocitrate (FC) to investigate changes in ATP levels, plasma membrane potential, and glutamate uptake, and correlated them with changes in astrocytic viability. FC blocks the activity of Kreb’s cycle enzyme aconitase acting as a “suicide” substrate (Clarke 1991). FC preferentially inhibits the Kreb’s cycle of astrocytes over neurons, possibly due to its more avid uptake by glial cells, and has been shown, within a certain range of concentrations and exposure times, to produce selective damage to glial cells only (Hassel et al. 1992; Fonnum et al. 1997). Our results indicate that mitochondrial inhibition induced significant functional impairment that occured before astrocytic death. We also used FC to selectively compromise mitochondrial function in astrocytes co-cultured with neurons to assess whether this could affect neuronal vulnerability to glutamate toxicity.
We performed mitochondrial inhibition studies in glucose-free medium, with similarly treated cultures in glucose-free medium without FC serving as a control. We chose these conditions based on our observations and earlier reports (Pauwels et al. 1985; Marrif and Juurlink 1999; Almeida et al. 2001) indicating that astrocytes readily compensate for decreased rates of mitochondrial ATP production by strong upregulation of glycolysis and increased glucose consumption from the medium. Because glucose depletion is characteristic of ischemia, glucose-free medium might also have physiological relevance to ischemic conditions.
A growing body of studies indicates the reciprocal nature of neuron-astrocyte interactions (Magistretti et al. 1993; Reichert et al. 2001; Chen and Swanson 2003), but the mechanisms by which neurons might influence astrocyte response and survival remain practically unknown. In this study we observed that the addition of neurons to astrocyte cultures influenced the rates of astrocytic mitochondrial depolarization, and identified one mechanism that can be partially responsible for this phenomenon.
Methods
Cell culture
Primary cultures of cortical astrocytes and neurons were prepared as previously described (Dugan et al. 1995) using a protocol approved by the Stanford Animal Care and Use Committee. In brief, for astrocyte cultures cerebral cortices from 1–2 day old mouse pups were dissociated in trypsin (0.25%), followed by trituration. The cells were suspended in minimal essential medium (MEM) containing 10% fetal bovine serum (FBS) (HyClone, Logan, UT), 10% equine serum (ES) (HyClone, Logan, UT), 2 mM glutamine and 10 ng/ml EGF (Sigma) and plated at density 2 hemispheres per 24 well plate. The cultures were fed biweekly with astrocyte growth medium (GM) (MEM supplemented with 10% ES and 2 mM glutamine) and used on d.i.v. 22–30. Some astrocytic cultures were exposed upon confluence (days in vitro (DIV) 12–16) to 3 μM cytosine arabinoside for 48 h to inhibit growth of other cell types. Neuronal cultures were prepared by suspending dissociated E14-15 cortexes in MEM with 2mM glutamine, 5% FBS, 5% ES and plating on poly-D-lysine (100 ng/ml) and laminin (4 ng/ml) coated wells at a density of 3 hemispheres/24 well plate. Twenty four hours after plating half of the culture medium was replaced with glial conditioned medium (GCM, collected from confluent astrocyte cultures in flasks that had been fed with MEM containing 5% FBS for at least 3 days) and supplemented with 2% B-27 supplement (Gibco, Grand Island, NY) and 3 μM cytosine arabinoside. Half of the medium was replaced with GCM on DIV 5–7, and the cells were used on DIV 10–12. For mixed astrocytic/neuronal cultures dissociated E14-15 cortexes were plated at 3 hemispheres/plate on a confluent astrocyte (DIV 12–16) monolayer. One day after plating the cells were treated with 3 μM cytosine arabinoside. After 5–6 days half of the medium was exchanged with GM, the cultures were used for experiments 9–11 days after neuronal cell plating.
Fluorocitrate (FC) and glutamate treatments
FC was purchased as the Ba2+ salt (Sigma). 8 mg of FC Ba2+ salt was dissolved in 100 μL of 1 mM HCL. 600μL of 0.1 mM Na2SO4 was added, and the suspension was centrifuged at 4,000 g for 2 min. The supernatant was mixed with BSS0 to a final volume of 16 mL, and the pH adjusted to 7.4. All treatments, unless indicated, were performed under the condition of glucose deprivation. Balanced salt solution (BSS0: 116 mM NaCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 5.4 mM KCl, 1 mM NaH2PO4,14.7 mM NaHCO3, 10 mM HEPES, pH 7.4) was added to each well to bring the total well volume to 1 mL. This was followed by 3 washes during which 0.8 mL medium was removed and replaced by 0.8 mL BSS0. After the final wash the volume was adjusted to 0.5 mL, and the cells were kept in a humidified 5% incubator for 1 h. After 1 h 400 μL of medium was removed and replaced with BSS0 for control wells or by FC diluted in BSS0 to a final FC concentration of 0.25 mM. We used 0.25 mM FC in our experiments because at that concentration FC induced relatively quick changes in astrocytic physiology but did not increase neuronal death in pure neuronal cultures (see Results). At the end of FC treatment the medium was exchanged for BSS5.5. BSS5.5 is identical to BSS0 but also contains 5.5 mM glucose. For glutamate exposure, cells were additionally treated with 10–1000 μM glutamate for 10 min, as previously described (Dugan et al. 1995), at the end of a 2 h FC treatment.
Measurements of ATP levels
Cellular ATP concentrations were measured using the CellTiter-Glo luminescent ATP assay kit, based on the luciferase/luciferin reaction, from Promega (Madison, WI) according to the manufacturer’s instructions. A Veritas luminescence counter (Turner BioSystems) was used to measure the luminescence signal of the samples in opaque white 96-well plates. ATP standards (Sigma) were used to create an ATP calibration curve for the assay. Protein concentrations were measured using bicinchoninic acid (BCA) protein assay reagent kit from Pierce (Rockford, IL) per manufacturer’s instructions.
Fluorescence measurements of mitochondrial and plasma membrane potential
For assessment of changes in mitochondrial membrane potential, astrocytes were loaded for 30 min prior to measurements with 50 nM tetramethylrhodamine ethyl ester (TMRE). TMRE fluorescence (excitation 535 nm, emission 590 nm) was visualized on an Axiovert 200M fluorescence microscope (Carl Zeiss, Germany). Several fields per dish were selected at random by phase-contrast optics, and the intensity of six 100×150 μm areas of confluent cells (~30 cell/each area) was analyzed to produce the average intensity reading for each well. The average background intensity of a cell-free region was measured in each well and the background signal was subtracted. The plasma membrane sensitive dye DiBAC4(3) (excitation 488 nm, emission 530 nm, 1 μM) was used to investigate changes in astrocyte plasma membrane potential. The imaging protocol was the same as that decribed for TMRE. The presented fluorescence data were normalized by setting the control conditions to a value of 1.0.
Glutamate uptake
Glutamate uptake was measured as described previously (Swanson 1992). Both control and FC treated cultures had 300 μL of 1.67 μCi/mL L-[14C(U)] glutamate plus unlabeled glutamate added to a total glutamate concentration of 100 μM. Uptake was terminated after a 7 min incubation at 37°C by two washes in ice-cold Hank’s balanced salt solution followed immediately by cell lysis in 0.1 M NaOH. Aliquots were taken for scintillation counting and for protein assay using BCA method.
Cell viability assays
Cell death was quantified 24 h after 10 min of glutamate exposure or indicated FC treatments by measuring lactate dehydrogenase (LDH) efflux as previously described (Koh and Choi 1987). LDH values for pure cultures were expressed as a fraction of freeze-thaw LDH release for maximum LDH release. LDH values for mixed cultures were expressed as a fraction of the maximal neuronal LDH released after 18–24 h exposure to 500 μM NMDA or, alternatively, to 10 min acute 1 mM glutamate exposure. In some instances, the cell death was evaluated by microscopic evaluation of Hoechst 33342 (5 μM) and propidium iodide (PI, 5 μM) labeled cells. PI readily penetrates cells with compromised plasma membranes but does not cross the intact membranes of live cells. Hoescht is a cell-permeant nucleic acid stain that labels cells of both live and dead nuclei.
Statistics
Statistical differences between two treatment groups were determined using unpaired two-tailed Student’s t-test with Welch’s correction. Comparisons between multiple groups were performed with ANOVA followed by Bonferroni test for selected groups. Data in all plots are presented as mean±SD.
Results
Mitochondrial inhibition promotes marked physiological and functional changes in astrocytes preceding astrocytic death
Changes in intracellular ATP levels
FC-induced mitochondrial inhibition in glucose-free BSS0 medium resulted in a rapid decrease of astrocytic ATP levels while control astrocytes treated with BSS0 alone did not demonstrate significant changes in ATP levels over the same time course. As shown in Fig. 1A, FC treatment for 2 and 3 h significantly decreased by 53 % and 84 %, respectively, cellular ATP content compared to control. Longer (4 h and 5 h) FC treatments promoted practically complete depletion of cellular ATP levels. In comparison, FC treatment in glucose-replete BSS5.5 medium induced slow and relatively small changes in astrocytic ATP levels (Fig. 1B). Because of the stronger effect of FC mitochondrial inhibition in glucose-free medium, and because glucose-free conditions are relevant to ischemic conditions of substrate deprivation, all subsequent experiments were performed in BSS0 medium.
Fig. 1.

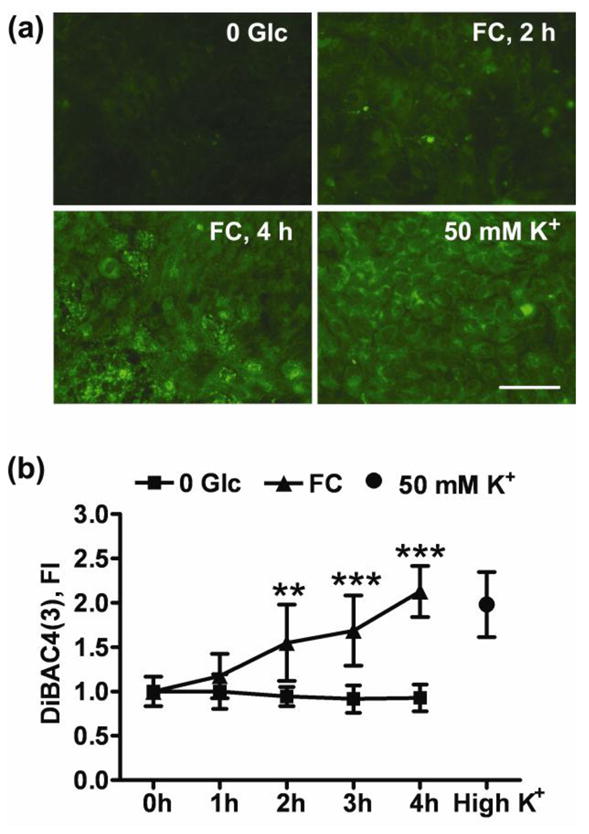
FC-induced plasma membrane depolarization
Because astrocyte plasma membrane potential is maintained by a powerful Na+/K+ ATPase that consumes about 20% of cellular ATP during non-stimulated conditions (Silver and Erecinska 1997), we investigated changes in astrocyte plasma membrane potential accompanying FC-induced changes in ATP levels. Fig. 2 shows that a significant 1.5 fold increase in DiBAC4(3) fluorescence developed after 2h of FC treatment reflecting astrocytic plasma membrane depolarization. A significant increase in DiBAC4(3) signal was also observed after 3 and 4 h of FC treatment. Observations were not extended for more than 4 h because plasma membrane integrity was strongly compromised beyond that time point. Exposure to high K+ extracellular buffer induced plasma membrane depolarization and was used to confirm DiBAC4(3) response (Fig. 2A, B).
Fig. 2.
The effect of FC treatment on mitochondrial membrane potential
We next investigated changes in mitochondrial membrane potential (Ψm) promoted by FC inhibition. As demonstrated in Fig. 3 no changes in Ψm were observed during the first 2 h of FC exposure, with a significant (p=0.003) Ψm decline developing after 3 h of FC treatment. Because the rapid decline in ATP levels in the first 2 h (Fig. 1A) was not accompanied by changes in Ψm, we investigated the possibility that Ψm was maintained by mitochondrial ATPase reversal. In this case Ψm is maintained by hydrolysis of glycolytically produced ATP at mitochondrial complex V. As demonstrated in Fig. 3, application of 1 μM oligomycin, a specific complex V inhibitor at that concentration, promoted quick Ψm depolarization in FC treated astrocytes. This indicates that during mitochondrial inhibition with FC, ATP is consumed rather than produced by astrocyte mitochondria. It has been shown that TMRE staining can be influenced by plasma membrane potential according to the Nernst equation (Ward et al. 2000). In our experiments we observed a mild non-significant increase in TMRE staining at 2 h, despite the significant plasma membrane depolarization at this time point (Fig. 2). Mitochondrial inhibition and subsequent F1F0-ATPase reversal have been shown to lead to mitochondrial hyperpolarization in different cell types including astrocytes (Hortelano et al. 1999; Almeida et al. 2001; Beltran et al. 2002). This phenomenon can be responsible for the slight increase in TMRE staining at 2 h time point notwithstanding the apparent plasma membrane depolarization. We also investigated the effect of high K+- induced plasma membrane depolarization on TMRE signal. In our experiments strong high K+-induced plasma membrane depolarization induced by high K+ exposure did not promote changes in TMRE staining of astrocytes (Fig. 3C).
Fig. 3.

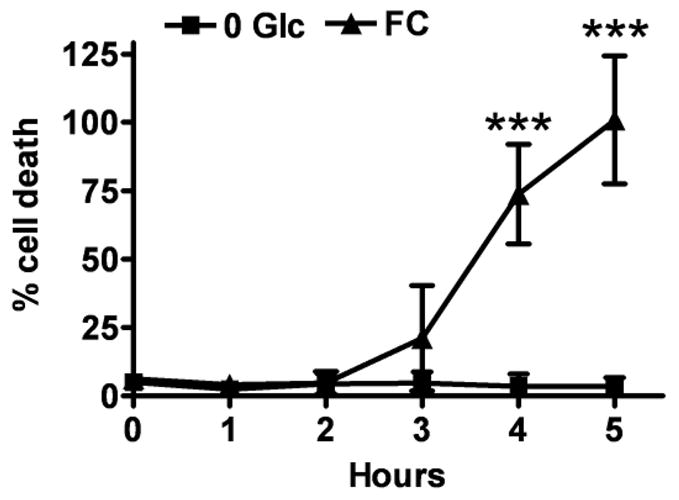
Cell viability
Before proceeding to measure changes in glutamate uptake we assessed whether FC treatment reduced astrocyte viability. As indicated in Fig. 4, control treatment with glucose deprivation alone did not reduce astrocyte viability during the observation time of 5 h. FC treatment induced a trend to increased astrocytic death after 3 h that did not reach significance. A 74% increase in cell death was observed after 4 h. 5 h FC exposure resulted in complete astrocytic death.
Fig. 4.
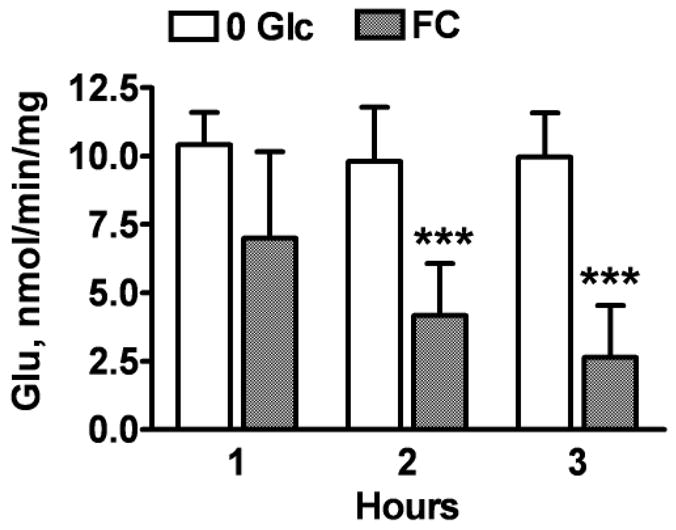
Glutamate uptake
Because astrocyte glutamate uptake is energy demanding, and because the cell plasma membrane potential contributes to the driving force for glutamate uptake we investigated changes in glutamate uptake with FC treatment. As shown in Fig. 5, the time course of changes in glutamate uptake correlated with that of ATP decline (Fig. 1A). There was a significant 58% and 74% reduction in the levels of glutamate uptake after 2 and 3 h of FC exposure, respectively. The measurements were not extended beyond 3 h when significant changes in astrocytic viability were observed (Fig. 4). The results indicate that the important function of glutamate uptake was markedly compromised before any significant increase in astrocyte death.
Fig. 5.
Astrocyte mitochondrial dysfunction increases glutamate toxicity for co-cultured neurons
Earlier studies demonstrated that astrocytes protect neurons against glutamate toxicity in mixed astrocyte/neuron cultures (Rosenberg and Aizenman 1989; Dugan et al. 1995). Because astrocytic glutamate uptake was significantly decreased after 2 h of 0.25 mM FC treatment (Fig. 5), we investigated whether 2 h FC treatment would impair the ability of astrocytes to protect neurons against glutamate toxicity. Astrocytic/neuronal co-cultures were exposed to different concentrations of glutamate at the end of 2 h FC treatment. As shown in Fig. 6A, FC pretreatment potentiated glutamate-induced neuronal death. FC pretreatment and subsequent 10 and 100 μM glutamate exposure resulted in 27% and 70% neuronal death, respectively, compared to 5% and 20% neuronal death in control cultures not treated with FC. To exclude a possible direct effect of FC treatment on neuronal survival, we studied the effect of 2 h FC pretreatment in pure neuronal cultures. As shown in Fig. 6B, FC-pretreated neuronal cultures demonstrated similar levels of neuronal death compared to control cultures exposed to the same conditions of glucose deprivation and subsequent glutamate treatment. Thus FC-induced astrocyte mitochondrial inhibition compromised neuronal survival during glutamate challenge. Morphological observations showed that the cell death in mixed cell cultures was limited to neurons at all glutamate concentrations tested. The top panels of Fig. 6C show representative images of control and FC-exposed mixed cultures that were exposed to 100 μM glutamate. The phase contrast images of the same areas on the bottom panels of Fig. 6C demonstrate that glutamate-induced death is limited to neurons.
Fig. 6.

Energetic challenge promotes faster mitochondrial depolarization in FC-treated astrocytes
Though co-culturing with neurons did not change astrocyte viability, we observed faster rates of astrocytic mitochondrial depolarization in mixed cultures. As shown in Fig. 3, 2 h FC treatment didn’t affect TMRE fluorescence of pure astrocytic cultures. In contrast, a significant decline in TMRE intensity in astrocytes was commonly observed after 2 h FC treatment in neuron/astrocyte co-cultures (Fig 7). While different mechanisms could be responsible for the change in astrocyte behavior, we hypothesized that co-culturing with neurons imposed additional energetic demands on astrocytes. An energetic load would induce a decline in cellular ATP levels that might otherwise be used to maintain mitochondrial Ψm (by reversal of the mitochondrial ATPase). In order to test our assumption we exposed astrocytes in pure cultures to different concentrations of aspartate during 0.25 mM FC treatment. Aspartate is taken up by the same transporters, with a similar Kd and energy requirements, as glutamate (Drejer et al. 1983), but, unlike glutamate, cannot be used as a mitochondrial fuel during FC inhibition in glucose-free medium. As shown in Fig. 8, co-exposure of astrocytes to different concentrations of aspartate and 0.25 mM FC for 2 h resulted in significant mitochondrial depolarization compared to control cells or astrocytes treated with FC alone (Fig. 3). Treatment with 100 μM and 1 mM aspartate resulted in a 69% and 77% decrease in TMRE fluorescence, respectively. We also investigated whether a decrease in plasma membrane potential contributed to the decrease in TMRE fluorescence induced by FC and aspartate co-treatment. As shown in Fig. 8C, FC+100 μM aspartate treatment caused no significant change in plasma membrane potential compared to FC treatment alone, thus indicating that the decrease in TMRE staining was not due to plasma membrane depolarization in those experiments. Fig 8D demonstrates the effects of the increased metabolic stress induced by exposure to different aspartate concentrations on the rates of ATP depletion in astrocyte cultures (as compared to Fig. 1A).
Fig. 7.
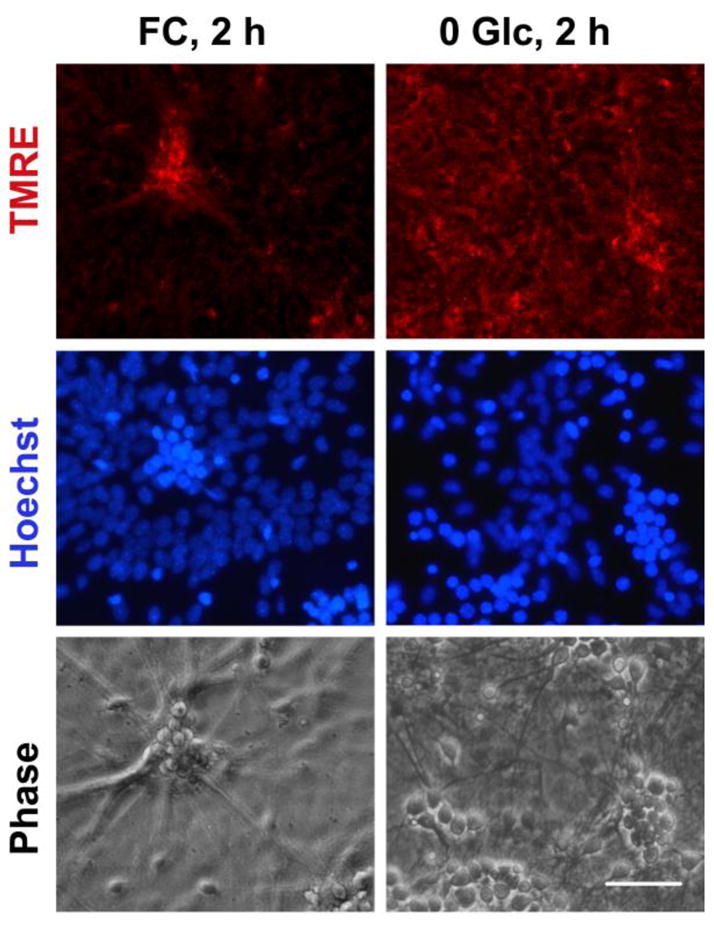
Fig. 8.

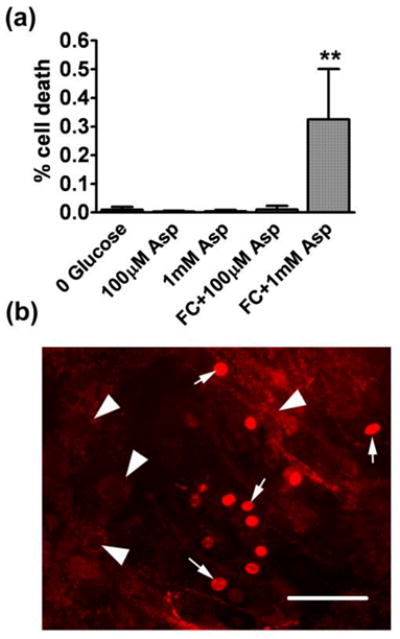
Because mitochondrial depolarization is known to be associated with astrocyte cell death (Dienel and Hertz 2005) we investigated the changes in astrocyte viability induced by co-exposure to FC and aspartate. Fig 9A shows that 1 mM aspartate applied during FC inhibition induced a significant 30% decrease in astrocyte viability. In contrast, 100 μM aspartate + FC treatment was not accompanied by significant astrocyte death, despite the profound loss of mitochondrial membrane potential. This is in striking contrast with the commonly observed coincidence of mitochondrial membrane depolarization and loss of astrocytic viability ((Juurlink and Hertz 1993) and Fig 9B).
Fig. 9.
Discussion
In this paper we investigated the time course of changes in astrocyte physiology promoted by mitochondrial inhibition under the conditions of glucose deprivation. Several interrelated factors contribute to mitochondrial impairment during ischemia. An increase in cytoplasmic free Ca2+ is characteristic of the pathogenesis of brain ischemia (Silver and Erecinska 1992). Ca2+ sequestration in mitochondria (Dux et al. 1987; Zaidan and Sims 1994) triggers mitochondrial dysfunction (Morley et al. 1994; Siesjo et al. 1995). Following arterial occlusion in brain, production of reactive oxygen species (ROS) is increased in ischemic areas during early reperfusion (Chan 1996; Piantadosi and Zhang 1996; Kuroda and Siesjo 1997). Exposure to elevated ROS levels results in oxidation of mitochondrial lipids, sulfhydryl groups and iron sulfur complexes of mitochondrial respiratory enzymes (Wagner et al. 1990; Gilboe et al. 1991; Nakahara et al. 1992; Liu et al. 1993) leading to impairment of mitochondrial oxidative phosphorylation. Different isoforms of nitric oxide synthase are activated during cerebral ischemia (Endoh et al. 1994; Nakashima et al. 1995; Bolanos et al. 1997; Wiencken and Casagrande 1999) leading to excessive production of nitric oxide (NO). NO is a potent competitive inhibitor of mitochondrial cytochrome oxidase (complex IV) (Brown 1995; Giuffre et al. 1996). In addition, both extracellular and intracellular pH drops significantly in ischemic tissue (Siesjo 1988; Kintner et al. 2000). Because mitochondrial respiration is sensitive to inhibition by acidosis (Hillered et al. 1984), systemic pH decrease during ischemia has been associated with mitochondrial impairment (Rehncrona et al. 1979; Kim et al. 1996).
Recent in vivo studies indicated that astrocytic oxidative metabolism could be significantly impaired in parts of the postischemic brain apparently without astrocytic loss (Thoren et al. 2005). Our studies showed that mitochondrial inhibition in glucose-free medium induced a rapid decrease in cellular ATP levels, depolarized plasma membrane, and decreased the rates of glutamate uptake before inducing significant loss of astrocyte viability. In our experiments astrocytes were able to maintain Ψm during mitochondrial inhibition by F1F0-ATPase hydrolysis of cellular ATP (Akerman and Jarvisalo 1980; Buchet and Godinot 1998). This hydrolysis is apparently important for astrocytic viability because Ψm collapse is associated with release of proapoptotic factors such as cytochrome c, apoptosis-inducing factors, and some procaspases (Liu et al. 1996; Susin et al. 1999). On the other hand, the reversal of the mitochondrial ATPase promotes higher rates of cellular ATP depletion and thus faster deterioration of energy requiring astrocytic protective functions. During the pathological conditions, such as ischemia, astrocytes have the potential to ameliorate damage to neurons, depending on the extent to which key functions are preserved. Using astrocyte/neuron co-cultures we confirmed that specific inhibition of astrocytic mitochondria impaired the well-known ability of astrocytes to protect co-cultured neurons against glutamate toxicity (Rosenberg and Aizenman 1989; Dugan et al. 1995). Our results suggest that astrocytic mitochondrial dysfunction, though still not extensively studied, may be an important early event in the development of ischemic injury.
While the ability of astrocytes to influence neuronal responses to ischemic injury is well known (Swanson et al. 2004), few studies have investigated the effect of neurons on astrocyte vulnerability. In vivo studies demonstrate that therapeutic agents that are beneficial for neuronal survival also decrease the extent of astrocytic death in ischemia (Chen and Swanson 2003). In our study the astrocytes were able to maintain mitochondrial membrane potential during FC inhibition for 2 h by ATPase reversal in pure cultures but not in the presence of co-cultured neurons (Fig 2 and 6). This is in agreement with previous studies that report the failure of astrocytes to use the compensatory mechanism of ATPase reversal in mixed neuron/astrocyte co-cultures (Reichert et al. 2001).
In order to investigate whether energetic requirements imposed by neurons were partially responsible for astrocyte Ψm depolarization, we initially stimulated FC-treated astrocytes with different concentrations of glutamate. We observed that an initial decline in mitochondrial potential upon glutamate addition was commonly followed by complete restoration of Ψm in a fraction of astrocytes (data not shown). This could be attributed to the intracellular conversion of glutamate to α-ketoglutarate that can enter the Kreb’s cycle downstream of FC inhibition of aconitase. We therefore conducted the additional experiments described above using aspartate. While aspartate is taken up by the same glutamate transporters (Drejer et al. 1983) unlike glutamate, it cannot be metabolized and enter the Kreb’s cycle downstream of FC inhibition. Because glutamate is metabolized oxidatively, it also cannot be used as a fuel under ischemic conditions of oxygen and glucose deprivation. For these reasons aspartate exposure in our experiments provided an adequate model of glutamate-induced energetic load during ischemia. Our results showed that FC-treated astrocytes exposed to 100 μM and 1 mM aspartate for 2h demonstrated significantly decreased TMRE staining (Fig. 8). These results suggest that the additional energetic load imposed by co-cultured neurons releasing glutamate is a potential mechanism that increases astrocyte mitochondrial depolarization during FC treatment. In our studies the addition of aspartate to FC-treated astrocytes did not promote any changes in plasma membrane potential, in a sharp contrast to FC-only treatment that demonstrated no changes in mitochondrial membrane potential but significant plasma membrane depolarization at 2 h time point (Fig. 2,3). The regulatory mechanisms that control preservation of either mitochondrial or plasma membrane potential under the conditions of energy deprivation remain to be investigated. Of note, failure of astrocytes to maintain mitochondrial membrane potential by F1F0-ATPase reversal when co-cultured with neurons, without changes in astrocytic plasma membrane potential, was also reported by another group in a previous study (Reichert et al. 2001).
Our studies also showed that the profound loss of mitochondrial potential induced by 2 h co-exposure to 100 μM aspartate + FC in glucose-free medium was not accompanied by astrocytic death. The ability of astrocytes to recover from a profound loss of mitochondrial potential without loss of viability, is in sharp contrast with commonly observed astrocyte behavior (Fig. 9) (Dienel and Hertz 2005), though it has been observed previously (Reichert et al. 2001). Studies of this phenomenon are still very limited.
Exposure of astrocytes to higher aspartate concentration of 1 mM in the presence of mitochondrial inhibition promoted significant increase in astrocytic death. As described above, the energetic requirements of aspartate uptake are equivalent to those for glutamate uptake. During ischemic episodes astrocytes in vivo can be exposed to glutamate concentrations in the millimolar range. Cellular glutamate ranges from 1 to 10 mM in different brain cell types (Erecinska and Silver 1990). Ischemic injury can result in release of intracellular glutamate into the extracellular space. Because the extracellular space comprises only 15–20% of brain volume in vivo (Dienel and Hertz 2005), this would result in local increases of glutamate concentrations in the millimolar range, which have been observed (Benveniste 1991). While astrocytes in primary cultures are relatively resistant to fuel and oxygen restriction, astrocyte death is strongly amplified by acidosis (Goldman et al. 1989; Giffard et al. 1990; Swanson et al. 1997). Extracellular ion shifts, combined with low pH, promote very rapid loss of astrocyte viability under ischemic conditions (Bondarenko and Chesler 2001). Our results indicate that high energetic demands encountered by astrocytes in the ischemic brain, but not commonly present in cell culture experiments, can be an additional source of astrocyte vulnerability in vivo. Indeed, several in vivo studies indicate that astrocytes can be equally or even more rapidly injured than neurons (Liu et al. 1999; Lukaszevicz et al. 2002; Zhao et al. 2003). Because the ischemic infarct area is defined by pan-necrosis of both glial and neuronal cells, the extent of astrocyte death can influence the amount of brain that suffers infarction. The results of our and previous studies indicate that, in addition to severe substrate and oxygen restriction, multiple environmental factors can influence the extent of astrocyte death and thus infarct size.
In summary, we demonstrated that inhibition of astrocyte mitochondria promoted functional changes in astrocytes and compromised their ability to protect neurons against excitotoxicity. Neurons, in turn, influenced the rates of astrocyte mitochondrial depolarization promoted by FC inhibition. In our studies an energy-demanding process such as aspartate uptake at the plasma membrane compromised astrocytes’ ability to maintain mitochondrial membrane potential during mitochondrial inhibition, and ultimately induced astrocyte death. Our results emphasize dynamic and highly complex interactions between neurons and astrocytes during stress that affect the ability of astrocytes to maintain mitochondrial membrane potential and survive energy deprivation as well as maintain brain protective functions during ischemic injury. The strategies aimed at the protection of astrocytic mitochondria, both by supply of mitochondrial metabolites or preservation of mitochondrial membrane integrity, might be useful in developing therapies to attenuate ischemic injury.
Acknowledgments
This work was supported in part by NIH grants GM49831, NS014543 and NS37520.
References
- Akerman KE, Jarvisalo JO. Effects of ionophores and metabolic inhibitors on the mitochondrial membrane potential within isolated hepatocytes as measured with the safranine method. Biochem J. 1980;192:183–190. doi: 10.1042/bj1920183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Anderson MF, Blomstrand F, Blomstrand C, Eriksson PS, Nilsson M. Astrocytes and stroke: networking for survival? Neurochem Res. 2003;28:293–305. doi: 10.1023/a:1022385402197. [DOI] [PubMed] [Google Scholar]
- Bambrick L, Kristian T, Fiskum G. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res. 2004;29:601–608. doi: 10.1023/b:nere.0000014830.06376.e6. [DOI] [PubMed] [Google Scholar]
- Beltran B, Quintero M, Garcia-Zaragoza E, O’Connor E, Esplugues JV, Moncada S. Inhibition of mitochondrial respiration by endogenous nitric oxide: a critical step in Fas signaling. Proc Natl Acad Sci U S A. 2002;99:8892–8897. doi: 10.1073/pnas.092259799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste H. The excitotoxin hypothesis in relation to cerebral ischemia. Cerebrovasc Brain Metab Rev. 1991;3:213–245. [PubMed] [Google Scholar]
- Bolanos JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heales SJ. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem. 1997;68:2227–2240. doi: 10.1046/j.1471-4159.1997.68062227.x. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001;34:134–142. doi: 10.1002/glia.1048. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- Buchet K, Godinot C. Functional F1-ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA-depleted rho degrees cells. J Biol Chem. 1998;273:22983–22989. doi: 10.1074/jbc.273.36.22983. [DOI] [PubMed] [Google Scholar]
- Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- Chen H, Chopp M, Schultz L, Bodzin G, Garcia JH. Sequential neuronal and astrocytic changes after transient middle cerebral artery occlusion in the rat. J Neurol Sci. 1993;118:109–106. doi: 10.1016/0022-510x(93)90099-k. [DOI] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Clarke DD. Fluoroacetate and fluorocitrate: mechanism of action. Neurochem Res. 1991;16:1055–1058. doi: 10.1007/BF00965850. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50:362–388. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- Drejer J, Larsson OM, Schousboe A. Characterization of uptake and release processes for D- and L-aspartate in primary cultures of astrocytes and cerebellar granule cells. Neurochem Res. 1983;8:231–243. doi: 10.1007/BF00963923. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 1993;623:208–214. doi: 10.1016/0006-8993(93)91429-v. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Bruno VM, Amagasu SM, Giffard RG. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dux E, Mies G, Hossmann KA, Siklos L. Calcium in the mitochondria following brief ischemia of gerbil brain. Neurosci Lett. 1987;78:295–300. doi: 10.1016/0304-3940(87)90376-4. [DOI] [PubMed] [Google Scholar]
- Endoh M, Maiese K, Wagner J. Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res. 1994;651:92–100. doi: 10.1016/0006-8993(94)90683-1. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Silver IA. Metabolism and role of glutamate in mammalian brain. Prog Neurobiol. 1990;35:245–296. doi: 10.1016/0301-0082(90)90013-7. [DOI] [PubMed] [Google Scholar]
- Fonnum F, Johnsen A, Hassel B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia. 1997;21:106–113. [PubMed] [Google Scholar]
- Fukamachi S, Furuta A, Ikeda T, Ikenoue T, Kaneoka T, Rothstein JD, Iwaki T. Altered expressions of glutamate transporter subtypes in rat model of neonatal cerebral hypoxia-ischemia. Brain Res Dev Brain Res. 2001;132:131–139. doi: 10.1016/s0165-3806(01)00303-0. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Monyer H, Choi DW. Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res. 1990;530:138–141. doi: 10.1016/0006-8993(90)90670-7. [DOI] [PubMed] [Google Scholar]
- Gilboe DD, Kintner D, Fitzpatrick JH, Emoto SE, Esanu A, Braquet PG, Bazan NG. Recovery of postischemic brain metabolism and function following treatment with a free radical scavenger and platelet-activating factor antagonists. J Neurochem. 1991;56:311–319. doi: 10.1111/j.1471-4159.1991.tb02597.x. [DOI] [PubMed] [Google Scholar]
- Giuffre A, Sarti P, D’Itri E, Buse G, Soulimane T, Brunori M. On the mechanism of inhibition of cytochrome c oxidase by nitric oxide. J Biol Chem. 1996;271:33404–33408. doi: 10.1074/jbc.271.52.33404. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Pulsinelli WA, Clarke WY, Kraig RP, Plum F. The effects of extracellular acidosis on neurons and glia in vitro. J Cereb Blood Flow Metab. 1989;9:471–477. doi: 10.1038/jcbfm.1989.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Paulsen RE, Johnsen A, Fonnum F. Selective inhibition of glial cell metabolism in vivo by fluorocitrate. Brain Res. 1992;576:120–124. doi: 10.1016/0006-8993(92)90616-h. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2006 doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Hillered L, Ernster L, Siesjo BK. Influence of in vitro lactic acidosis and hypercapnia on respiratory activity of isolated rat brain mitochondria. J Cereb Blood Flow Metab. 1984;4:430–437. doi: 10.1038/jcbfm.1984.62. [DOI] [PubMed] [Google Scholar]
- Hortelano S, Alvarez AM, Bosca L. Nitric oxide induces tyrosine nitration and release of cytochrome c preceding an increase of mitochondrial transmembrane potential in macrophages. Faseb J. 1999;13:2311–2317. doi: 10.1096/fasebj.13.15.2311. [DOI] [PubMed] [Google Scholar]
- Juurlink BH, Hertz L. Ischemia-induced death of astrocytes and neurons in primary culture: pitfalls in quantifying neuronal cell death. Brain Res Dev Brain Res. 1993;71:239–246. doi: 10.1016/0165-3806(93)90175-a. [DOI] [PubMed] [Google Scholar]
- Kim H, Koehler RC, Hurn PD, Hall ED, Traystman RJ. Amelioration of impaired cerebral metabolism after severe acidotic ischemia by tirilazad posttreatment in dogs. Stroke. 1996;27:114–121. doi: 10.1161/01.str.27.1.114. [DOI] [PubMed] [Google Scholar]
- Kintner DB, Anderson MK, Fitzpatrick JH, Jr, Sailor KA, Gilboe DD. 31P-MRS-based determination of brain intracellular and interstitial pH: its application to in vivo H+ compartmentation and cellular regulation during hypoxic/ischemic conditions. Neurochem Res. 2000;25:1385–1396. doi: 10.1023/a:1007664700661. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Siesjo BK. Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. Clin Neurosci. 1997;4:199–212. [PubMed] [Google Scholar]
- Lee DR, Helps SC, Gibbins IL, Nilsson M, Sims NR. Losses of NG2 and NeuN immunoreactivity but not astrocytic markers during early reperfusion following severe focal cerebral ischemia. Brain Res. 2003;989:221–230. doi: 10.1016/s0006-8993(03)03373-0. [DOI] [PubMed] [Google Scholar]
- Leis JA, Bekar LK, Walz W. Potassium homeostasis in the ischemic brain. Glia. 2005;50:407–416. doi: 10.1002/glia.20145. [DOI] [PubMed] [Google Scholar]
- Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, Simpson IA. Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Brain Res Mol Brain Res. 1999;68:29–41. doi: 10.1016/s0169-328x(99)00063-7. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rosenthal RE, Starke-Reed P, Fiskum G. Inhibition of postcardiac arrest brain protein oxidation by acetyl-L-carnitine. Free Radic Biol Med. 1993;15:667–670. doi: 10.1016/0891-5849(93)90171-p. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Lukaszevicz AC, Sampaio N, Guegan C, Benchoua A, Couriaud C, Chevalier E, Sola B, Lacombe P, Onteniente B. High sensitivity of protoplasmic cortical astroglia to focal ischemia. J Cereb Blood Flow Metab. 2002;22:289–298. doi: 10.1097/00004647-200203000-00006. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev Neurosci. 1993;15:306–312. doi: 10.1159/000111349. [DOI] [PubMed] [Google Scholar]
- Marrif H, Juurlink BH. Astrocytes respond to hypoxia by increasing glycolytic capacity. J Neurosci Res. 1999;57:255–260. doi: 10.1002/(SICI)1097-4547(19990715)57:2<255::AID-JNR11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Morley P, Hogan MJ, Hakim AM. Calcium-mediated mechanisms of ischemic injury and protection. Brain Pathol. 1994;4:37–47. doi: 10.1111/j.1750-3639.1994.tb00809.x. [DOI] [PubMed] [Google Scholar]
- Nakahara I, Kikuchi H, Taki W, Nishi S, Kito M, Yonekawa Y, Goto Y, Ogata N. Changes in major phospholipids of mitochondria during postischemic reperfusion in rat brain. J Neurosurg. 1992;76:244–250. doi: 10.3171/jns.1992.76.2.0244. [DOI] [PubMed] [Google Scholar]
- Nakashima MN, Yamashita K, Kataoka Y, Yamashita YS, Niwa M. Time course of nitric oxide synthase activity in neuronal, glial, and endothelial cells of rat striatum following focal cerebral ischemia. Cell Mol Neurobiol. 1995;15:341–349. doi: 10.1007/BF02089944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels PJ, Opperdoes FR, Trouet A. Effects of antimycin, glucose deprivation, and serum on cultures of neurons, astrocytes, and neuroblastoma cells. J Neurochem. 1985;44:143–148. doi: 10.1111/j.1471-4159.1985.tb07123.x. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. discussion 332. [DOI] [PubMed] [Google Scholar]
- Rao VL, Dogan A, Todd KG, Bowen KK, Kim BT, Rothstein JD, Dempsey RJ. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci. 2001;21:1876–1883. doi: 10.1523/JNEUROSCI.21-06-01876.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehncrona S, Mela L, Siesjo BK. Recovery of brain mitochondrial function in the rat after complete and incomplete cerebral ischemia. Stroke. 1979;10:437–446. doi: 10.1161/01.str.10.4.437. [DOI] [PubMed] [Google Scholar]
- Reichert SA, Kim-Han JS, Dugan LL. The mitochondrial permeability transition pore and nitric oxide synthase mediate early mitochondrial depolarization in astrocytes during oxygen-glucose deprivation. J Neurosci. 2001;21:6608–6616. doi: 10.1523/JNEUROSCI.21-17-06608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Acidosis and ischemic brain damage. Neurochem Pathol. 1988;9:31–88. doi: 10.1007/BF03160355. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Katsura K, Zhao Q, Folbergrova J, Pahlmark K, Siesjo P, Smith ML. Mechanisms of secondary brain damage in global and focal ischemia: a speculative synthesis. J Neurotrauma. 1995;12:943–956. doi: 10.1089/neu.1995.12.943. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Ion homeostasis in rat brain in vivo: intra- and extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. J Cereb Blood Flow Metab. 1992;12:759–772. doi: 10.1038/jcbfm.1992.107. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Energetic demands of the Na+/K+ ATPase in mammalian astrocytes. Glia. 1997;21:35–45. doi: 10.1002/(sici)1098-1136(199709)21:1<35::aid-glia4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci Lett. 1992;147:143–146. doi: 10.1016/0304-3940(92)90580-z. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Farrell K, Stein BA. Astrocyte energetics, function, and death under conditions of incomplete ischemia: a mechanism of glial death in the penumbra. Glia. 1997;21:142–153. doi: 10.1002/(sici)1098-1136(199709)21:1<142::aid-glia16>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Thoren AE, Helps SC, Nilsson M, Sims NR. Astrocytic function assessed from 1-14C-acetate metabolism after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2005;25:440–450. doi: 10.1038/sj.jcbfm.9600035. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Kleinholz M, Myers RE. Delayed decreases in specific brain mitochondrial electron transfer complex activities and cytochrome concentrations following anoxia/ischemia. J Neurol Sci. 1990;100:142–151. doi: 10.1016/0022-510x(90)90025-i. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of glial cells in the regulation of the brain ion microenvironment. Prog Neurobiol. 1989;33:309–333. doi: 10.1016/0301-0082(89)90005-1. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Wiencken AE, Casagrande VA. Endothelial nitric oxide synthetase (eNOS) in astrocytes: another source of nitric oxide in neocortex. Glia. 1999;26:280–290. [PubMed] [Google Scholar]
- Xu L, Sapolsky RM, Giffard RG. Differential sensitivity of murine astrocytes and neurons from different brain regions to injury. Exp Neurol. 2001;169:416–424. doi: 10.1006/exnr.2001.7678. [DOI] [PubMed] [Google Scholar]
- Zaidan E, Sims NR. The calcium content of mitochondria from brain subregions following short-term forebrain ischemia and recirculation in the rat. J Neurochem. 1994;63:1812–1819. doi: 10.1046/j.1471-4159.1994.63051812.x. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG. Early loss of astrocytes after experimental traumatic brain injury. Glia. 2003;44:140–152. doi: 10.1002/glia.10283. [DOI] [PubMed] [Google Scholar]