

This study demonstrates a novel corneal endothelial response to genotoxic stress which involves rapid changes in connexin-43 and gap junction intercellular communication.
Abstract
Purpose.
To determine how corneal endothelial (CE) cells respond to acute genotoxic stress through changes in connexin-43 (Cx43) and gap junction intercellular communication (GJIC).
Methods.
Cultured bovine CE cells were exposed to mitomycin C or other DNA-damaging agents. Changes in the levels, stability, binding partners, and trafficking of Cx43 were assessed by Western blot analysis and immunostaining. Live-cell imaging of a Cx43–green fluorescent protein (GFP) fusion protein was used to evaluate internalization of cell surface Cx43. Dye transfer and fluorescent recovery after photobleaching (FRAP) assessed GJIC.
Results.
After genotoxic stress, Cx43 accumulated in large gap junction plaques, had reduced zonula occludens-1 binding, and displayed increased stability. Live-cell imaging of Cx43–GFP plaques in stressed CE cells revealed reduced gap junction internalization and degradation compared to control cells. Mitomycin C enhanced transport of Cx43 from the endoplasmic reticulum to the cell surface and formation of gap junction plaques. Mitomycin C treatment also protected GJIC from disruption after cytokine treatment.
Discussion.
These results show a novel CE cell response to genotoxic stress mediated by marked and rapid changes in Cx43 and GJIC. This stabilization of cell–cell communication may be an important early adaptation to acute stressors encountered by CE.
The corneal endothelium (CE) is a monolayer of neural crest–derived cells that is essential for corneal transparency. Located at the posterior surface of the cornea, the CE separates the cornea from the aqueous humor, relying on cell–cell junctions to control corneal hydration. The CE is a fragile cell layer that is vulnerable to the effects of intraocular surgery, systemic and ocular disease, and topical drugs.1,2 In addition, endogenous oxidative stress appears to play a significant role in the degeneration of CE with age3,4 and in Fuchs' dystrophy.5,6 Human CE does not regenerate in vivo, so existing cells must compensate for cell loss to maintain the pump and barrier functions required for corneal homeostasis. It is clear that surviving cells respond to CE cell loss by spreading to cover the posterior corneal surface. This spreading is associated with thinning of the cell layer,7 but the full range of physiological responses of the CE to cell loss has not been characterized. CE function is dependent on the cell–cell junctions, which maintain the integrity of this monolayer. The goal of the present study was to examine how CE cells mediate these junctions in response to genotoxic stressors.
Gap junctions are intercellular channels composed primarily of the connexin family of proteins. These channels provide rapid intercellular transfer of small signaling molecules, such as nucleotides, inositol trisphosphate (IP3), glutathione, and Ca2+ between connected cells.8 Such gap junction intercellular communications (GJIC) function in the maintenance of tissue homeostasis and influence cellular survival and death in response to oxidative stress,9–11 metabolic stress,9,12 ischemia reperfusion injury,13,14 and genotoxic stress.9,15 Connexins may also mediate cell survival by GJIC-independent mechanisms in addition to functions associated with gap junctions.16
With a half-life of 1.5 to 5 hours, connexin proteins respond rapidly to physiologic changes by altering gap junction coupling between cells. The Cx43 C terminus contains 14 documented phosphorylation sites,17 and different phosphorylated species of Cx43 can be distinguished experimentally by SDS-PAGE. Aggregation of Cx43 into functional gap junction plaques, opening of the junctional pores, and Cx43 degradation have all been linked to site-specific phosphorylation of the Cx43 protein.18 Given the pervasive role of connexins in the maintenance of cell and tissue homeostasis, we hypothesized that exogenous genotoxic stress would alter homeostasis-regulating proteins such as Cx43 in the CE. Previously, we reported DNA damage in goat CE after brief doses of the DNA interstrand cross-linking agent mitomycin C (MMC) during procedures emulating photorefractive keratectomy.19 In this study, we identified specific changes that occur in Cx43 and in GJIC in CE as a result of genotoxic stress induced by exposures such as MMC. Determining how CE cells respond to various stressors may provide opportunities for CE protection and preservation.
Methods
Cell Culture and Reagents
Primary bovine CE cells were isolated as previously described19 and cultured in low glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and antibiotics/antimycotics in a humidified 5% CO2, 37°C environment. Passage 1 to 4 cells, split 1:4, were used and grown 2 to 3 days past confluency for every experiment unless otherwise indicated. At least 24 hours before treatment with MMC, cycloheximide, forskolin, and epidermal growth factor (all from Sigma, Saint Louis, MO), the medium was changed to serum-free DMEM containing no antibiotics/antimycotics. MMC was added to cells by changing medium with prewarmed media containing 5 μM freshly diluted MMC. Primary bovine corneal fibroblasts were isolated and grown as previously described.20
Immunofluorescence
Cells were fixed in 3.2% paraformaldehyde (PFA) solution (Electron Microscopy Science, Hatfield, PA) in PBS for 20 minutes at room temperature. Permeabilization with 0.1% Triton X-100 (Thermo Fisher, Pittsburgh, PA) in PBS for 1 minute was followed by blocking with 10% heat-inactivated goat serum in PBS for 1 hour at room temperature. Cells were incubated with a polyclonal rabbit anti-Cx43 antibody to the C terminus (Invitrogen), monoclonal mouse anti-Cx43 to the C terminus (Millipore, Billerica, MA), or monoclonal mouse anti-zonula occludens-1 (ZO-1; Invitrogen) diluted 1:200 in 1% bovine serum albumin (BSA) in PBS solution overnight at 4°C. After multiple washes in PBS, cells were incubated for 1 hour at room temperature with species-appropriate Alexa-conjugated secondary antibodies (Invitrogen) diluted to 0.8 μg/mL in 1% BSA in PBS containing 0.5 μg/mL 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Cells were observed on a laser scanning confocal microscope (Olympus Fluoview FV1000, Olympus, Center Valley, PA) and images captured and processed using Olympus Fluoview software. To quantify plaque size and intensity, ImageJ (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html) was used to manually trace Cx43 plaques in three separate high magnification confocal images. Area and intensity values were calculated and compared with the Mann–Whitney U test.
Immunoblotting
Cells were lysed directly in 1X SDS sample buffer [1.6% sodium dodecyl sulfate, 0.06M Tris, 5.5% glycerol, and 0.002% bromophenol blue], scraped into tubes, heated at 95°C for 5 minutes, then sonicated until solubilized. For alkaline phosphatase treatment of cell lysates, cells were lysed in RIPA buffer (Thermo Fisher) [25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS] containing protease inhibitor cocktail (Sigma), sonicated, and incubated with 0.1 U/μL cell lysate of calf alkaline phosphatase (Sigma) for 1 hour at 37°C. Protein concentration was determined by Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA) and then 2-mercaptoethanol was added to a final concentration of 1% to the lysates and heated at 70°C for 20 minutes. An equal amount of protein was added to precast 4–20% gradient or 10% polyacrylamide gels (Bio-Rad) and electrophoresis was performed for 1 hour at 200 V. Protein was transferred to PVDF membrane (Millipore) and blocked for 1 hour at room temperature in 0.2 M Tris, 0.15 M NaCl, 0.01% thimeresol, 0.2% Tween-20, pH 7.4 (TTTBS) for ECL detection or with fluorescent blocker (Millipore) for Odyssey infrared imaging (LI-COR, Lincoln, NE). Membranes were incubated with the following primary antibodies as indicated: mouse anti-Cx43NT1, which recognizes the N-terminal region of Cx43 (Fred Hutchinson Cancer Center, Seattle, WA); rabbit anti-Cx43, which recognizes the C-terminal region of Cx43 (Invitrogen); mouse anti-ZO-1 (Invitrogen); and mouse anti-α-tubulin (Sigma), all diluted in 1% BSA in TTTBS or fluorescent blocker. Secondary antibodies included horseradish peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) for enhanced chemiluminescence (ECL) detection and IRDye800 and IRDye680 labeled secondary antibodies (LI-COR) for infrared imaging. Where indicated, blots were stripped for 15 minutes at room temperature (Restore Western Blot Stripping Buffer, Thermo Fisher Scientific, Rockford, IL). Chemiluminescent signal was visualized with ECL substrate (Millipore) followed by detection and capture of 16-bit images with a Bio-Rad FX imager (Bio-Rad), and fluorescent signal was detected with Odyssey infrared imaging system (LI-COR). Densitometry was performed using digital analysis software for ECL (Bio-Rad Quantity One) and fluorescent detection (LI-COR) followed by appropriate statistical analysis for comparisons with GraphPad (GraphPad Software Inc., La Jolla, CA). All forms of Cx43 (phosphorylated and nonphosphorylated) were quantified in densitometry.
Immunoprecipitation
Cells were lysed directly in cold coimmunoprecipitation buffer [0.1% Nonidet P40, 25 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA] containing HALT protease and phosphatase inhibitors (Thermo Fisher) and briefly sonicated until solubilized. Lysates were precleared with magnetic protein G beads (Millipore) for 1 hour at room temperature to remove any nonspecific binding of cell proteins to beads. The beads were discarded and then cleared lysates were incubated with 1 μg antibody per 500 μg of lysate using a rabbit polyclonal anti-Cx43 antibody (Invitrogen) overnight at 4°C. Beads were then washed multiple times with lysis buffer and heated at 95°C for 5 minutes in 1X SDS sample buffer with 2-mercaptoethanol. A magnetic rack was used to separate the beads from lysate before loading for SDS-PAGE separation. Mouse anti-ZO-1 (Invitrogen) and rabbit anti-Cx43 (Invitrogen) antibodies were used for immunoblotting.
Triton X-100 Insolubility
Cells were lysed in cell lysis buffer containing 1% Triton X-100 and 1 mM EDTA with protease inhibitors (Sigma) and then chilled on ice for 30 minutes with mixing. Cell lysates were spun at 10,000 g for 30 minutes at 4°C. The soluble fraction was removed and the Triton X-100 insoluble pellet was further dissolved in 1X SDS buffer followed by sonication. Both the insoluble and soluble fractions were used for immunoblotting.
Live-Cell Imaging
CE cells grown on Bioptech dishes (Bioptechs Inc., Butler, PA) to 70% to 80% confluency were transfected with 1 μg of Cx43–GFP plasmid (gift of Professor Matthias Falk, Lehigh University) using Lipofectamine 2000 (Invitrogen). This construct was previously described to have GFP at the most C-terminal end of Cx43,21 and 24 hours after transfection, cells expressing the Cx43–GFP fusion protein were imaged with the Nikon Eclipse TE200-E microscope (Nikon Instruments Inc., Melville, NY) on a heated stage with 5% CO2. Images were captured using software (Metamorph, Molecular Devices, Sunnyvale, CA) every 2 minutes over a 4- to 6-hour period after treatment with MMC or medium alone. To determine internalization and degradation frequency, Cx43–GFP assembled into cell surface plaques were identified and followed frame-by-frame exactly 24 hours after transfection. Plaques were counted if they were internalized and/or degraded by reduced fluorescent signal over a 3-hour timeframe. Comparison of the frequency of internalization/degradation events between medium and MMC-treated cells was performed using the Fisher exact test. A 2 × 2 contingency table was analyzed with GraphPad, with P < 0.05 being considered statistically significant.
GJIC Assays
Lucifer yellow (LY) dye (Sigma) scrape-load assays were performed by scraping confluent monolayers of CE cells with a sterile pipette tip in the presence of 1 mg/mL LY in warm PBS. After 2 minutes, cells were rinsed to remove LY dye solution and cells were immediately fixed in 3.2% PFA without permeabilization. Images were captured using a Nikon epifluorescent microscope. Dye transfer images were analyzed using ImageJ software by measuring the distance of dye transfer from the scrape wound at 10 different sites per image. Average distances were then analyzed with ANOVA and a post-hoc Tukey's test for multiple comparisons. P < 0.05 was considered statistically significant. For fluorescent recovery after photobleaching (FRAP), cells grown on 35-mm coverslip-bottom dishes were loaded with 1 μM of Calcein-AM (Invitrogen) for 30 minutes at 37°C. Cells were allowed to recover for 1 to 2 hours then treated with 5 μM MMC for 15 minutes. Individual cells were photobleached using a laser scanning confocal microscope (Olympus) and images of dye recovery were captured. ImageJ software was used to measure dye intensity in image stacks. Photobleached cells were normalized to control unbleached cells. Data were exported into GraphPad for exponential curve fitting and calculation of rate constants (k). Half-life of dye recovery (t1/2) was calculated based on the formula: t1/2=ln(2)/k. The Student's t test was used for statistical comparison of t1/2.
Results
Genotoxic Stress to CE Generates Rapid Changes in the Cx43 Abundance and Localization
We previously reported the rapid induction of DNA lesions after brief exposure to low levels of MMC in CE cells.19 Within a similar timeframe, low dose MMC exposure induced an increase in total cellular Cx43 levels as detected by immunoblotting (Fig. 1A). Increases occurred in each of the three characteristic protein bands representative of differentially phosphorylated Cx43 species present in most eukaryotic cells. The phenomenon was highly repeatable, with total cellular Cx43 increasing somewhat more than twofold 60 to 90 minutes after treatment. These results were confirmed using two Cx43 antibodies targeting different regions of Cx43, the C-terminal (Fig. 1A, top) and the N-terminal (Fig. 1A, bottom). The use of antibodies targeting different regions of the protein reduces the possibility that the observed quantitative changes result from epitope masking. Alkaline phosphatase (AP) treatment of whole cell lysates before electrophoresis dephosphorylates Cx43 collapsing the characteristic multiple bands into a single faster moving band (Fig. 1B). The rapid and transient increase in Cx43 was maintained after AP treatment, indicating that total levels of Cx43, independent of phosphorylation state, occurred. A similar transient increase in cellular Cx43 was observed after exposure to other genotoxic agents, including ultraviolet-C radiation, etoposide, and hydrogen peroxide (data not shown).
Figure 1.

Rapid accumulation of Cx43 protein in response to genotoxic stress with mitomycin C. Cells were treated with prewarmed media containing 5 μM MMC for indicated times in minutes. (A) Western blot for total Cx43 using C-terminal antibody (top) and N-terminal antibody (bottom) from independent experiments. Blots were stripped and reprobed for α-tubulin as a loading control. Quantification below is ratio of Cx43/α-tubulin normalized to values at the “0” time point. Asterisks indicate statistically significant differences from time “0” (ANOVA, P < 0.05; Dunnett's multiple comparison test, P < 0.05). Blots are representative of eight independent experiments and the mean fold change ± SEM is shown in the graph. (B) Alkaline phosphatase (AP) treatments of cell lysates were followed by immunoblotting. Quantification in the graph is the ratio of Cx43/α-tubulin normalized to “0” for either AP– or AP+. Differences between “–AP” and “+AP” at each time point were not statistically significant (2-way ANOVA, P > 0.05). The blot is representative of three independent experiments and the mean fold change ± SEM is shown in the graph.
Immunostaining of CE cells (Fig. 2A) revealed increased Cx43 at cell–cell interfaces, with an increased size of the punctate staining in this locale suggestive of increased accumulation of Cx43 in gap junction plaques. Staining also increased in perinuclear regions of the treated cells, suggesting recently synthesized Cx43 in transit to the cell surface to be increased as well. Cell fractionation of the treated cells found increased Cx43 largely in the Triton X-100 detergent-insoluble fraction (Fig. 2C). Association with gap junction plaques confers detergent insolubility on connexin22; therefore, increases in insoluble Cx43 in response to MMC is consistent with immunostaining results, suggesting that MMC induces transport of the increased Cx43 into cell–cell junctional plaques. Similar changes were not observed in other cell–cell junction proteins, such as ZO-1, cadherin, and β-catenin, based on immunoblotting (data not shown).
Figure 2.

Changes in Cx43 gap junction plaque size and intensity after genotoxic stress. (A) Cells were treated with prewarmed media containing 5 μM MMC for the indicated time in minutes then stained for Cx43 (green). DAPI (nuclei). Orange arrows indicate apparent gap junction plaques. (B) Gap junction plaques were imaged by confocal microscopy and analyzed for area (μm2) and Cx43 fluorescence intensity (arbitrary units) after 60 minutes of 5 μM MMC (n = 25 plaques) or medium alone (n = 25 plaques). Individual data points are shown along with the median. Area and intensity distributions were compared for statistically significant differences using the Mann–Whitney U test (area, P < 0.01; intensity, P < 0.0001). Similar results were obtained in three independent experiments. (C) Western blot for 1% Triton X-100 soluble and insoluble Cx43. Quantification below is insoluble Cx43 normalized to “0.” Asterisks indicate statistically significant differences from time “0” (ANOVA, P < 0.05; Dunnett's multiple comparison test, P < 0.05). Results are representative of three independent experiments and the mean fold change ± SEM is shown in the graph. Scale bar: (A) 20 μm.
Quantitative image analysis of cells treated with MMC confirmed a significant increase in both the gap junction plaque area and Cx43 signal intensity compared to medium alone (Fig. 2B) after 60 minutes of treatment with MMC. Previous studies have shown that interactions between Cx43 and the tight junction protein ZO-1 can regulate the size of Cx43 gap junctions.23–26 To explore the potential involvement of ZO-1, we used coimmunoprecipitation (Co-IP) and colocalization to examine changes in Cx43:ZO-1 association after MMC treatment. Co-IP revealed a relative decrease in the abundance of ZO-1 immunoprecipitating with Cx43 after 60 minutes of MMC treatment (Fig. 3A). This alteration was also evident in images staining for ZO-1 and Cx43 (Fig. 3B). Large Cx43 gap junction plaques displaying reduced ZO-1 colocalization occasionally were evident in untreated cells (Fig. 3B); however, these were present with significantly higher frequency in MMC-treated cells.
Figure 3.

MMC induces alteration in Cx43 association with ZO-1. Cells were treated with prewarmed media containing 5 μM MMC for 60 minutes or media alone (CTRL). (A) Coimmunoprecipitation with a rabbit polyclonal anti-Cx43 was performed followed by immunoblotting with mouse monoclonal anti-ZO-1. The blot was stripped and reprobed for Cx43. Quantification below is the ratio of ZO-1:Cx43, which is normalized to “CTRL.” Denatured heavy chain immunoglobulin G (IgG) is indicated as HC IgG. Asterisks indicate a statistically significant difference (Student's t-test, P < 0.001). Results are representative of three independent experiments and mean fold change to “CTRL” ± SEM is shown below. (B) Representative confocal images of a large gap junction plaque after MMC treatment. In the higher magnification inset (below), the areas of Cx43:ZO-1 colocalization are highlighted in white. Cx43 (green), ZO-1 (red), DAPI (blue). Scale bar, 10 μm.
MMC Treatment Increases Cx43 Stability
Connexin protein half-life ranges from 1.5 to 5 hours in many cell types and organ systems.27,28 Treatment of cultured CE with 10 μg/mL cycloheximide (CHX) to inhibit protein synthesis was used to examine decay rates of Cx43. In the untreated control, the half-life of decay was in the expected range of 1 to 2 hours (Fig. 4, left). Treatment with MMC, however, stabilized the Cx43, delaying degradation for up to 120 minutes (Fig. 4, right). After this period of stability, however, the decay rate returned to one similar to that of untreated cells.
Figure 4.

The stability of preexisting Cx43 is altered after MMC treatment. Cells were treated with prewarmed media containing 10 μg/mL CHX with or without 5 μM MMC for indicated times in minutes. Blot shows total Cx43 using a C-terminal–specific antibody. Blots were stripped and reprobed for α-tubulin as a loading control. Quantification below is ratio of Cx43/α-tubulin normalized to the zero time (0) for “CHX” or “CHX + MMC.” Asterisks indicate statistically significant differences between “CHX” and “CHX + MMC” (2-way ANOVA, P < 0.01; Bonferroni multiple comparisons, P < 0.05). Results are representative of three independent experiments and mean fold change ± SEM is shown in the graph.
Gap Junction Internalization is Decreased after MMC Treatment
Degradation of Cx43 present in cell surface plaques occurs via processes involving the internalization of small and/or large endocytic double-membrane vesicles.29–31 These processes can be followed directly by micrography using fluorescently tagged Cx43.21,30,32 We examined this internalization and degradation of Cx43 after MMC treatment by live-cell imaging of CE that had been transiently transfected with a Cx43–GFP fusion protein.24,26 Figure 5 shows a series of images from live-cell video micrography after Cx43–GFP in MMC-treated cells or medium-only controls. Sixty-three individual plaques were identified and followed in the videos starting 24 hours after transfection at the time of MMC treatment. The presence or absence of each of these was scored 3 hours later to assess the relative stability of the plaque-associated Cx43 under the two conditions (Fig. 5A). In medium-treated control transfected cells, 40 of 46 (89.6%) disappeared after 3 hours, whereas in the MMC-treated samples, only 8 of 17 (52.9%) of the plaques had been internalized and vanished (Fig. 5B). This significant (P < 0.01) difference in plaque stability suggests that the reduced degradation of Cx43 identified in Figure 4 relates to alterations in the mechanism by which cells endocytose and degrade the gap junction plaques.
Figure 5.

Genotoxic stress results in reduced internalization of Cx43 plaques. Seventy percent to 80% confluent cells were transfected with a Cx43–GFP plasmid. (A) Twenty-four hours after transfection, cells were treated with either medium (“CTRL,” upper figures) or medium with 5 μM MMC (“MMC,” lower figures) and live-cell imaging was performed. Images were captured every 2 minutes over a 4- to 6-hour period after treatment. Images are stills of Cx43–GFP plaques over time in minutes. “CTRL” images illustrate an example of internalized and degraded plaques. MMC images illustrate a long-lived plaque. (B) To determine internalization and degradation frequency, Cx43–GFP assembled into cell surface plaques were identified and followed frame-by-frame exactly 24 hours after transfection. Plaques were counted if they were internalized and/or degraded by reduced fluorescent signal over a 3-hour timeframe. Statistical comparison of the frequency of internalization/degradation events between medium “CTRL” and MMC-treated cells was performed using Fisher's exact test, P < 0.01. Results are from two independent live-cell experiments tracking a combined 63 plaques (46 CTRLs, 17 MMCs). Scale bar: (A) 20 μm.
Genotoxic Stress Increases Forward Trafficking of Cx43 into Gap Junctions
To distinguish MMC effects in the formation of new membrane-associated Cx43 from its effects on Cx43 internalization/degradation, we examined transfer of Cx43 from the endoplasmic reticulum (ER) compartment to the cell surface after an 8-hour block with brefeldin A (BFA). BFA has been used in previous studies to investigate gap junction assembly because of its reversible inhibition of vesicle transport from the ER to the Golgi and subsequently to the cell surface.32,33 When BFA is removed, the transport of proteins is restored and the accumulation of proteins at the cell surface becomes briefly representative of the rate at which this forward trafficking process is occurring. Therefore, gap junction formation can be assessed after BFA removal. As shown in Figure 6, BFA treatment significantly reduced Cx43 at the cell surface (Fig. 6A, BFA). After BFA washout and a 2-hour recovery, Cx43 appeared at the cell surface in gap junction plaques and in the perinuclear compartment in both control and MMC-treated cells (Fig 6A, BFA + 2hr, BFA + MMC + 2hr). However, the presence of MMC led to a clear increase of Cx43 in plaques at the cell–cell interfaces (Fig 6A, BFA + MMC + 2hr). These plaques were significantly larger than those formed without MMC (Fig. 6B).
Figure 6.

MMC induces increased assembly of gap junction plaques. Confluent monolayers were treated with prewarmed media containing 5 μg/mL BFA for 8 hours followed by washout with warm medium and a 2-hour recovery. Five μM MMC was added as indicated during the 2-hour recovery period. (A) Representative confocal images of Cx43 (green) before treatment (NOTX), after 8 hour BFA (BFA), after recovery (BFA + 2hr), and after recovery in the presence of MMC (BFA + MMC + 2hr). DAPI (nuclei). (B) Gap junction plaques were imaged by confocal microscopy and analyzed for area (μm2) and Cx43 fluorescence intensity (arbitrary units) similar to Figure 2B. BFA + 2hr (n = 25 plaques) or BFA + MMC + 2hr (n = 25 plaques). Individual data points are shown along with median. Area and intensity distributions were compared using the Mann–Whitney U test (area, P < 0.0001; intensity, P < 0.0001). Similar results were obtained in three independent experiments.
Gap Junction Communication is Stabilized by MMC Treatment
LY dye, which transfers from cell to cell via gap junction channels, was used to assess the temporal rate of GJIC during a 2-minute incubation after scrape loading. As shown in Figure 7A, no significant differences were observed in the amount of dye transferred or the distance the dye moved comparing control and MMC-treated cells (Fig. 7A). FRAP using Calcein AM, a similarly gap junction permeable dye, also revealed identical transfer kinetics between control and MMC-treated CE cells (Fig. 7C). Forskolin is an agent known to maximize GJIC and markedly increases scrape-loaded dye transfer in many cell types.34–36 In CE cells, however, forskolin produced no significant increase on the dye transfer rate (Fig. 7A, far right panel). Both forskolin and MMC, however, increased dye transfer rates in primary bovine corneal fibroblasts (Fig. 7B), suggesting that CE cells may be communicating via gap junctions at near maximal levels, making further increases in GJIC undetectable.
Figure 7.
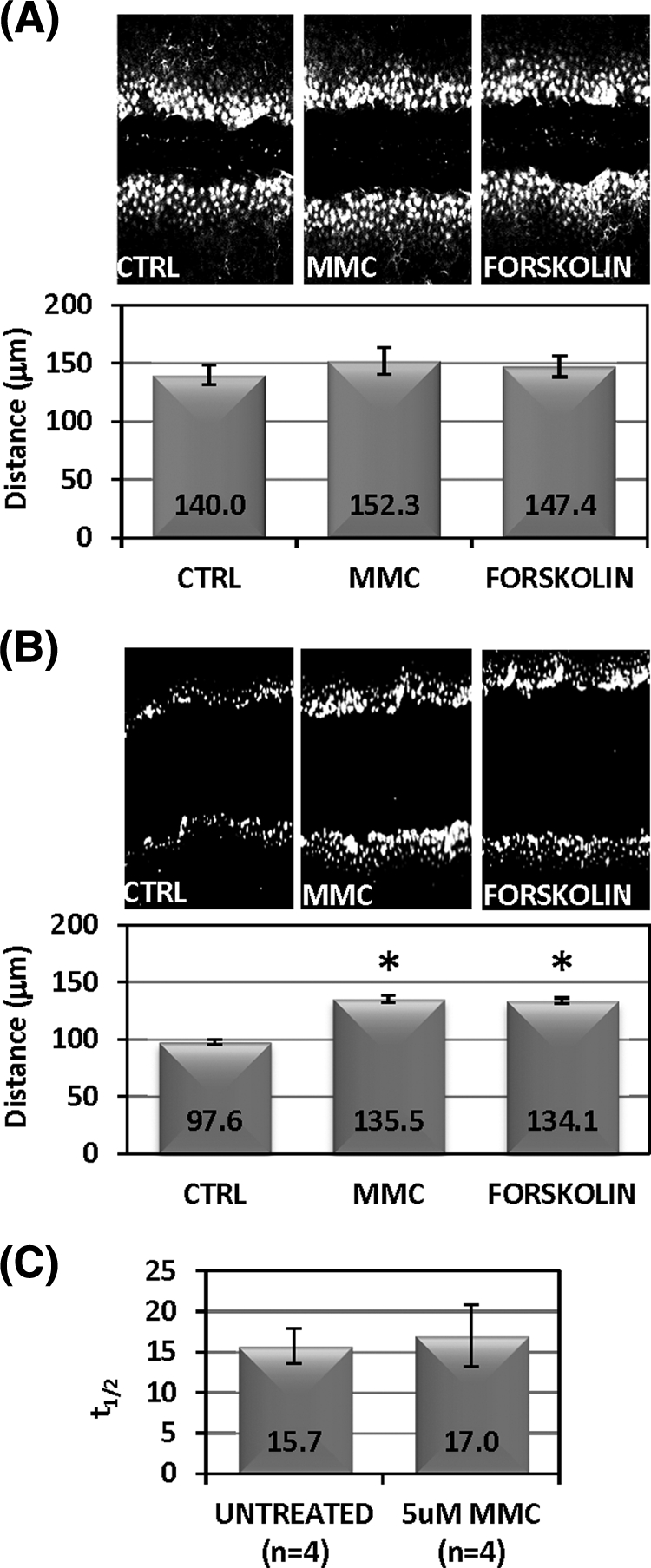
MMC and forskolin do not significantly increase GJIC. (A) Confluent monolayers were treated with prewarmed media containing 5 μM MMC, 10 μM forskolin, or medium alone (“CTRL”) for 30 minutes Medium was replaced with 1 mg/mL Lucifer yellow (LY) dye in PBS and scrape-load dye transfer assay was performed as described in the Methods section. Ten individual sites were measured along different regions of the scrape wound for distance of LY dye transfer to determine cell–cell communication as shown in the graph below. Data points are mean distance ± SD. There was no statistically significant difference between treatments (ANOVA [P = 0.3]). (B) Confluent monolayers of bovine corneal fibroblasts were treated similar to (A). Asterisks indicate statistically significant differences (ANOVA [P < 0.001]; Dunnett's test: medium versus Forksolin [P < 0.01], medium versus MMC [P < 0.01]). (C) FRAP was performed after loading confluent CE monolayers with 1 μM calcein AM and observing dye recovery. Half-life of recovery in seconds (t1/2) shown on the y-axis was compared between untreated (n = 4 cells) and MMC-treated (n = 4) cells. Mean ± SD t1/2 shown. No statistically significant difference was found between treatments (Student's t-test [P = 0.8]).
To determine whether the increased Cx43 at the cell surface induced any alteration in gap junction function of CE cells, we inhibited GJIC using epidermal growth factor (EGF). EGF treatment has been shown to decrease GJIC in cultured cells through activation of the mitogen-activated kinase pathway.37 This response was clearly detected in CE cells using the LY dye transfer assay (Fig. 8). MMC-treated cultures partially rescued the EGF-induced GJIC inhibition, suggesting that increases in cell surface Cx43 induced by MMC are indeed functional in promoting intercellular communication.
Figure 8.

MMC treatment generates GJIC resistant to EGF-mediated downregulation. (A) Confluent monolayers were pretreated for 60 minutes with either prewarmed media containing 5 μM MMC or medium (“CTRL”) alone. Medium was replaced with 100 ng/mL EGF for an additional 30 minutes and then GJIC was assessed with a scrape-load dye transfer assay as described in the Methods section. Ten individual sites were measured along different regions of the scrape wound for distance of LY dye transfer to determine cell–cell communication as shown in the graph. Data points are mean distance ± SD of the dye transfer. ANOVA was used for statistical comparison among treatment groups (P < 0.0001). Tukey's multiple comparison test reported untreated versus MED + EGF (P < 0.001), untreated versus MMC + EGF (P > 0.05), MED + EGF versus MMC + EGF (P < 0.001). Results are representative of two independent experiments.
Discussion
The CE is a nonregenerating tissue in which the total number of cells declines throughout life. As a result of damage or various acute stressors, this process can occur more rapidly. With cell loss, CE cells spread and flatten, covering a greater area. Apart from this process, little of the physiologic response to stressors in these cells is known, particularly regarding how cell–cell junctions are maintained. In the present study, we document several novel observations regarding how CE cells respond to an acute stress: Cx43 rapidly accumulates in detergent-insoluble plaques at cell–cell junctions; the plaques are stabilized, showing reduced endocytosis and degradation; transport of Cx43 from the ER to the cell surface is increased; and gap junction communication is stabilized against downregulation by EGF. These observations describe a previously unidentified response of CE cells to stress, rapidly increasing the abundance of functional gap junctions in the tissue.
The mechanism mediating this response is not straightforward. In initial experiments, we found no statistically significant increase in Cx43 mRNA (data not shown); therefore, the rapid change in Cx43 abundance in response to MMC seemed likely to involve decreased Cx43 degradation. A temporary halt to Cx43 degradation after MMC treatment was indeed observed (Fig. 4), as was a significant reduction in endocytosis and dispersion of plaque-associated Cx43 (Fig, 5), providing a mechanism that could be responsible for the observed increase in total cellular Cx43. Cx43 stabilization has been described in other cell types in response to stress. Long-lived gap junctions after arsenite and heat-shock exposure were observed in in CHO and S180 cell lines.38 In the case of CHO cells, cell surface Cx43 was stabilized by a mechanism dependent on functional proteasomal action.38 The CE response to MMC observed in this study could, therefore, be typical of a generalized cellular mechanism by which cells stabilize GJIC in response to stress.
Another potential effector of GJIC stabilization is ZO-1. ZO-1 is present in gap junctions, binding the C-terminal intracellular region of Cx43.25 We observed Cx43 but not ZO-1 to increase in response to MMC, and subsequently found a decreased association of ZO-1 with Cx43 by coimmunoprecipitation (Fig. 3). There is reason to believe that this reduced ZO-1:Cx43 ratio could contribute to the stress-induced Cx43 plaque stabilization. Association of Cx43 with ZO-1 has been linked to an increased endocytosis of Cx4339–41; therefore, reduced association of Cx43 with Z0–1 in gap junction plaques (such as we observed) could delay or prevent Cx43 endocytosis and internalization. ZO-1 may also have an effect on the gap junction plaque size (Fig. 2). ZO-1 has been reported to regulate gap junction size by limiting access of ZO-1-associated Cx43 into preexisting gap junctions.23–26 Reduced ZO-1 association with cell surface Cx43 is therefore consistent with the increased gap junction plaques we observed.
The in vitro CE layers used in this study did not appear to restrict the intercellular transmission of dyes via gap junctions (Fig. 7). However, in the presence of EGF—an agent that disrupts GJIC—it was clear that GJIC in MMC-treated cells was more resistant to this inhibition than control cells (Fig. 8). Therefore, the rapid alterations in Cx43 protein in response to genotoxic stress produce a cell–cell communication array that is more robust than that in nonstressed cells. This response gains relevance in light of the documentation that exposure to genotoxic stressors releases cytokines and growth factors42–45 that could compromise GJIC in the CE.
The tight junctions in CE cells serve to control movement of water and nutrients into the cornea,46 but there is not yet a clearly demonstrated role for gap junctions in corneal physiology. Our results suggest that alterations in Cx43 may serve as survival factor for CE cells. In other biologic systems in which cells are subjected to physical stress or injury, gap junction coupling activation results in the mediation of injury or increased survival.47,48 In the CE, upregulation of Cx43 mRNA and protein, via release of ciliary neurotrophic factor and its receptor, promote CE cell survival during oxidative stress and ex vivo corneal storage.49–51 Also, point mechanical stimulation of CE cell monolayers results in a Cx43-dependent increase in ATP release and calcium-wave propagation between cells.52 Therefore, Cx43 and GJIC provide a response in CE to changes in cellular environment that is likely to represent a survival response.
The rapid rate at which CE responds to MMC suggests a mechanism not reliant on changes in gene expression but instead on posttranslation modifications of Cx43. Cx43 C-terminal phosphorylation controls multiple aspects of this protein, including channel gating, subcellular localization, secondary structure, and stability.53 Increased phosphorylation of Cx43 C-terminal, for example is responsible for decreased ZO-1 binding.54 Site-specific Cx43 phosphorylation has also been linked to internalization and degradation of this protein.55 Casein kinase 1 is a likely candidate for an active participant in the response to MMC. Phosphorylation of Cx43 by casein kinase 1 has been proposed to stimulate incorporation of Cx43 into gap junction plaques,56 and it appears to exert a general role in regulating the stability of its numerous substrates.57 In addition, casein kinase 1 acutely responds to genotoxic stress resulting in increased kinase activity, changes in subcellular location, and increased mRNA and protein levels.57–60
In conclusion, we have documented rapid changes in Cx43 in response to genotoxic stress and identified the changes in binding partners and reduced degradation of Cx43. Mitomycin C is widely used in ocular surgery, and we recently showed that such treatment generates DNA cross-linking in CE.19 MMC, however, may not be the only genotoxic agent to which these cells are exposed. The high metabolic rate of the CE generates reactive oxygen species, which are capable of DNA damage,61 and as a possible consequence, CE exhibits an accumulation of DNA modifications with increasing age.3,4 The implications of the stabilization of Cx43 and GJIC on CE cell homeostasis/survival are yet to be fully elucidated but may serve to protect against detrimental effects of natural DNA damage and acute stress generated by agents such as MMC or ultraviolet light. Future areas of interest include determining the functional consequences of stabilized cell surface Cx43 and GJIC on CE cell viability. One potential function influencing cell survival may be to increase the intercellular transfer of cytoprotective molecules that increase survival and function of interconnected cells and/or disperse cytotoxic molecules and prevent their accumulation in any one cell.
Footnotes
Supported by National Institutes of Health Grants EY016415 (JLF), EY009368 (JLF), P30-EY008098, F30-AG035443 (DSR), Research to Prevent Blindness, and the Eye and Ear Foundation of Pittsburgh.
Disclosure: D. Roh, None; J. Funderburgh, None.
References
- 1. Bourne WM. Biology of the corneal endothelium in health and disease. Eye (Lond). 2003;17:912–918 [DOI] [PubMed] [Google Scholar]
- 2. Bourne WM, McLaren JW. Clinical responses of the corneal endothelium. Exp Eye Res. 2004;78:561–572 [DOI] [PubMed] [Google Scholar]
- 3. Joyce NC, Harris DL, Zhu C. Age-related gene response of human corneal endothelium to oxidative stress and DNA damage. Invest Ophthalmol Vis Sci. 2011;52:1641–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joyce NC, Zhu CC, Harris DL. Relationship among oxidative stress, DNA damage, and proliferative capacity in human corneal endothelium. Invest Ophthalmol Vis Sci. 2009;50:2116–2122 [DOI] [PubMed] [Google Scholar]
- 5. Jurkunas UV, Bitar MS, Funaki T, Azizi B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am J Pathol. 2010;177:2278–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jurkunas UV, Rawe I, Bitar MS, et al. Decreased expression of peroxiredoxins in Fuchs' endothelial dystrophy. Invest Ophthalmol Vis Sci. 2008;49:2956–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joyce NC, Meklir B, Neufeld AH. In vitro pharmacologic separation of corneal endothelial migration and spreading responses. Invest Ophthalmol Vis Sci. 1990;31:1816–1826 [PubMed] [Google Scholar]
- 8. Alexander DB, Goldberg GS. Transfer of biologically important molecules between cells through gap junction channels. Curr Med Chem. 2003;10:2045–2058 [DOI] [PubMed] [Google Scholar]
- 9. Lin JH, Yang J, Liu S, et al. Connexin mediates gap junction-independent resistance to cellular injury. J Neurosci. 2003;23:430–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramachandran S, Xie LH, John SA, Subramaniam S, Lal R. A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS One. 2007;2:e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hutnik CM, Pocrnich CE, Liu H, Laird DW, Shao Q. The protective effect of functional connexin43 channels on a human epithelial cell line exposed to oxidative stress. Invest Ophthalmol Vis Sci. 2008;49:800–806 [DOI] [PubMed] [Google Scholar]
- 12. Albright CD, Kuo J, Jeong S. cAMP enhances Cx43 gap junction formation and function and reverses choline deficiency apoptosis. Exp Mol Pathol. 2001;71:34–39 [DOI] [PubMed] [Google Scholar]
- 13. Nakase T, Fushiki S, Naus CC. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke. 2003;34:1987–1993 [DOI] [PubMed] [Google Scholar]
- 14. Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc Res. 2004;61:386–401 [DOI] [PubMed] [Google Scholar]
- 15. Tekpli X, Rivedal E, Gorria M, et al. The B[a]P-increased intercellular communication via translocation of connexin-43 into gap junctions reduces apoptosis. Toxicol Appl Pharmacol. 2010;242:231–240 [DOI] [PubMed] [Google Scholar]
- 16. Decrock E, Vinken M, De Vuyst E, et al. Connexin-related signaling in cell death: to live or let die? Cell Death Differ. 2009;16:524–536 [DOI] [PubMed] [Google Scholar]
- 17. Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res. 2004;62:256–267 [DOI] [PubMed] [Google Scholar]
- 19. Roh DS, Cook AL, Rhee SS, et al. DNA cross-linking, double-strand breaks, and apoptosis in corneal endothelial cells after a single exposure to mitomycin C. Invest Ophthalmol Vis Sci. 2008;49:4837–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Long CJ, Roth MR, Tasheva ES, et al. Fibroblast growth factor-2 promotes keratan sulfate proteoglycan expression by keratocytes in vitro. J Biol Chem. 2000;275:13918–13923 [DOI] [PubMed] [Google Scholar]
- 21. Falk MM. Connexin-specific distribution within gap junctions revealed in living cells. J Cell Sci. 2000;113(pt 22):4109–4120 [DOI] [PubMed] [Google Scholar]
- 22. Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hunter AW, Jourdan J, Gourdie RG. Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun Adhes. 2003;10:211–214 [DOI] [PubMed] [Google Scholar]
- 25. Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934 [DOI] [PubMed] [Google Scholar]
- 26. Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Musil LS, Cunningham BA, Edelman GM, Goodenough DA. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol. 1990;111:2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635 [DOI] [PubMed] [Google Scholar]
- 29. Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW., 3rd Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009;20:3342–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Segretain D, Falk MM. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta. 2004;1662:3–21 [DOI] [PubMed] [Google Scholar]
- 32. Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A. 2002;99:10446–10451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laird DW, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in brefeldin A-treated rat mammary tumor cells. J Cell Biol. 1995;131:1193–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paulson AF, Lampe PD, Meyer RA, et al. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci. 2000;113(pt 17):3037–3049 [DOI] [PubMed] [Google Scholar]
- 35. Romanello M, Moro L, Pirulli D, Crovella S, D'Andrea P. Effects of cAMP on intercellular coupling and osteoblast differentiation. Biochem Biophys Res Commun. 2001;282:1138–1144 [DOI] [PubMed] [Google Scholar]
- 36. Dowling-Warriner CV, Trosko JE. Induction of gap junctional intercellular communication, connexin43 expression, and subsequent differentiation in human fetal neuronal cells by stimulation of the cyclic AMP pathway. Neuroscience. 2000;95:859–868 [DOI] [PubMed] [Google Scholar]
- 37. Abdelmohsen K, Sauerbier E, Ale-Agha N, et al. Epidermal growth factor- and stress-induced loss of gap junctional communication is mediated by ERK-1/ERK-2 but not ERK-5 in rat liver epithelial cells. Biochem Biophys Res Commun. 2007;364:313–317 [DOI] [PubMed] [Google Scholar]
- 38. VanSlyke JK, Musil LS. Cytosolic stress reduces degradation of connexin43 internalized from the cell surface and enhances gap junction formation and function. Mol Biol Cell. 2005;16:5247–5257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Segretain D, Fiorini C, Decrouy X, Defamie N, Prat JR, Pointis G. A proposed role for ZO-1 in targeting connexin 43 gap junctions to the endocytic pathway. Biochimie. 2004;86:241–244 [DOI] [PubMed] [Google Scholar]
- 40. Barker RJ, Price RL, Gourdie RG. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ Res. 2002;90:317–324 [DOI] [PubMed] [Google Scholar]
- 41. Bruce AF, Rothery S, Dupont E, Severs NJ. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc Res. 2008;77:757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prise KM, O'Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer. 2009;9:351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen TC, Chang SW. Effect of mitomycin C on IL-1R expression, IL-1-related hepatocyte growth factor secretion and corneal epithelial cell migration. Invest Ophthalmol Vis Sci. 2010;51:1389–1396 [DOI] [PubMed] [Google Scholar]
- 44. Chang SW, Chou SF, Yu SY. Dexamethasone reduces mitomycin C-related inflammatory cytokine expression without inducing further cell death in corneal fibroblasts. Wound Repair Regen. 2010;18:59–69 [DOI] [PubMed] [Google Scholar]
- 45. Novakova Z, Hubackova S, Kosar M, et al. Cytokine expression and signaling in drug-induced cellular senescence. Oncogene. 2010;29:273–284 [DOI] [PubMed] [Google Scholar]
- 46. Srinivas SP. Dynamic regulation of barrier integrity of the corneal endothelium. Optom Vis Sci. 2010;87:E239–E254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chew SS, Johnson CS, Green CR, Danesh-Meyer HV. Role of connexin43 in central nervous system injury. Exp Neurol. 2010;225:250–261 [DOI] [PubMed] [Google Scholar]
- 48. Qi J, Chi L, Bynum D, Banes AJ. Gap junctions in IL-1β-mediated cell survival response to strain. J Appl Physiol. 2011;110:1425–1431 [DOI] [PubMed] [Google Scholar]
- 49. Koh SW, Cheng J, Dodson RM, Ku CY, Abbondandolo CJ. VIP down-regulates the inflammatory potential and promotes survival of dying (neural crest-derived) corneal endothelial cells ex vivo: necrosis to apoptosis switch and up-regulation of Bcl-2 and N-cadherin. J Neurochem. 2009;109:792–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koh SW, Celeste J, Ku CY. Functional CNTF receptor alpha subunit restored by its recombinant in corneal endothelial cells in stored human donor corneas: connexin-43 upregulation. Invest Ophthalmol Vis Sci. 2009;50:1801–1807 [DOI] [PubMed] [Google Scholar]
- 51. Koh SW. Ciliary neurotrophic factor released by corneal endothelium surviving oxidative stress ex vivo. Invest Ophthalmol Vis Sci. 2002;43:2887–2896 [PubMed] [Google Scholar]
- 52. Gomes P, Srinivas SP, Vereecke J, Himpens B. Gap junctional intercellular communication in bovine corneal endothelial cells. Exp Eye Res. 2006;83:1225–1237 [DOI] [PubMed] [Google Scholar]
- 53. Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen J, Pan L, Wei Z, Zhao Y, Zhang M. Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. EMBO J. 2008;27:2113–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cooper CD, Lampe PD. Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem. 2002;277:44962–44968 [DOI] [PubMed] [Google Scholar]
- 57. Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675–689 [DOI] [PubMed] [Google Scholar]
- 58. Knippschild U, Milne DM, Campbell LE, et al. p53 is phosphorylated in vitro and in vivo by the delta and epsilon isoforms of casein kinase 1 and enhances the level of casein kinase 1 delta in response to topoisomerase-directed drugs. Oncogene. 1997;15:1727–1736 [DOI] [PubMed] [Google Scholar]
- 59. Inuzuka H, Tseng A, Gao D, et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell. 2010;18:147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene. 2008;27:3653–3661 [DOI] [PubMed] [Google Scholar]
- 61. Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res. 2003;531:5–23 [DOI] [PubMed] [Google Scholar]