

Summary
As genetically encoded small molecules, antibiotics are phenotypes that have resulted from mutation and natural selection. Advances in genetics, biochemistry, and bioinformatics have connected hundreds of antibiotics to the gene clusters that encode them, allowing these molecules to be analyzed using the tools of evolutionary biology. This review surveys examples of convergent evolution from microbially produced antibiotics, including the convergence of distinct gene clusters on similar phenotypes and the merger of distinct gene clusters into a single functional unit. Examining antibiotics through an evolutionary lens highlights the versatility of biosynthetic pathways, reveals lessons for combating antibiotic resistance, and provides an entry point for studying the natural roles of these natural products.
Introduction
More than two-thirds of the antibiotics used to treat humans are microbial natural products or semisynthetic derivatives of these molecules [1,2]. Advances in genetics, biochemistry, and bioinformatics have transformed the study of antibiotics and other natural products, not just by revealing how they are synthesized but also by casting them as phenotypes encoded by gene collectives that can be studied through an evolutionary lens [3]. Like other sets of genes that encode adaptive traits, antibiotic-encoding gene collectives can converge evolutionarily on similar phenotypes [4-6]. Due to horizontal gene transfer, they can also arrive in the same genome, allowing distinct gene clusters to merge into a single functional unit. The examples of convergent evolution that form the basis of this review not only hold lessons for how Nature invents and diversifies small molecules, but they also yield information about antibiotic action: which targets have remained effective over evolutionary time, and how combinations of antibiotics can overcome resistance. I begin by surveying examples of convergent evolution from microbially produced antibiotics, continue by examining how distinct gene clusters cohabiting the same genome can merge physically and functionally, and conclude with a discussion of how antibiotic synthesis is distributed among microbial taxa.
Phenotypic convergence
Common selective pressures can lead unrelated enzymes to evolve toward catalyzing the same reaction. For example, while all β-lactamases cleave the same bond in the β-lactam antibiotics, some of these enzymes (the serine hydrolases of classes A, C, and D) likely evolved from the bacterial penicillin-binding proteins, while others (the zinc-dependent hydrolases of class B) are evolutionarily unrelated [7-9]. The functions of unrelated enzyme collectives that produce antibiotics – generally encoded by sets of contiguous genes known as gene clusters – can also converge. Two or more of these self-contained metabolic pathways can produce distinct molecules that target the same biological process (e.g., cell wall synthesis), the same enzyme (e.g., transpeptidase), or even the same binding pocket in an enzyme. Although they don’t share any genes, they can even produce structurally similar molecules to do the job.
Convergence on a target
Many natural antibiotics bind to the ribosome, a 2.3 megadalton nucleoprotein essential for cell growth [10]. Since the ribosome is composed of 55 proteins and 3 RNAs, it is not surprising that antibiotics are known to bind at multiple sites [11,12]. Notably, at least five structurally distinct antibiotics from unrelated gene clusters bind to one of these sites: the peptidyl transferase center of the 50S ribosomal subunit [13,14]. While their bound configurations differ, they occupy overlapping regions of the binding pocket [13], highlighting the ability of distinct biosynthetic pathways to access the molecular diversity required to target a specific site in an enzyme.
Like protein synthesis, peptidoglycan synthesis is an essential multi-step pathway, and many of its steps are targeted by natural antibiotics [15]. Interestingly, one of the converged-upon targets in the peptidoglycan pathway is not an enzyme, but a small molecule: lipid II [16] (Figure 1). While this membrane-anchored peptidoglycan precursor is exposed extracellularly, its use as a target poses a unique challenge. Unlike macromolecular targets, which generally have an active site and other clefts in which small molecules can bind, lipid II is a small molecule, so it must be sequestered instead. Accordingly, the small molecules that target lipid II act as receptor-like cages: vancomycin and other glycopeptides sequester its terminal D-Ala-D-Ala dipeptide [17], while the lantibiotic nisin binds to its pyrophosphate [18]. Still other antibiotics such as the mannopeptimycins [19], ramoplanin [20], plusbacin [21], and katanosin [21] are thought to target lipid II directly, but their binding modes are not yet known.
Figure 1.
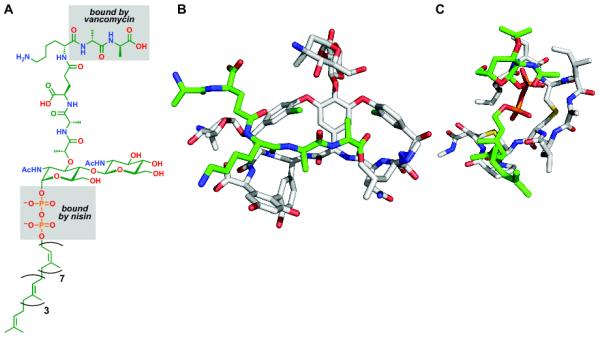
Antibiotics that target lipid II. (A) Chemical structure of lipid II. Gray squares indicate the portions of lipid II bound by vancomycin and nisin. (B) NMR structure of a vancomycin-related glycopeptide bound to the D-Ala-D-Ala terminus of lipid II [17]. (C) NMR structure of the lantibiotic nisin bound to lipid II, forming a cage around its pyrophosphate [18]. The structures were rendered using PyMOL (http://www.pymol.org) from PDB files 1GAC and 1WCO, respectively.
Convergence on a molecular scaffold
In some remarkable cases, unrelated biosynthetic pathways have converged not just on a target, but on the same molecular scaffold for binding the target. The most prominent example of this phenomenon is the β-lactam antibiotic family [22,23]. These molecules inhibit the enzyme transpeptidase, which forms spot welds between the polysaccharide girders of peptidoglycan by fusing their pendant pentapeptide chains [24]. As antibiotic targets go, transpeptidase is a likely candidate for evolutionary convergence: it is extracellular, essential for cell growth, and druggable by a small molecular scaffold that resembles a dipeptide. However, the convergence of at least four unrelated, multi-step pathways on nearly identical molecular scaffolds is a striking demonstration of the ability of evolution to trace different paths to a common answer.
The core of the β-lactam scaffold is a four-membered ring containing an amide bond (hereafter, ring A), which is usually fused to a five- or six-membered ring (hereafter, ring B) (Figure 2). All of the known β-lactam pathways use amino acid building blocks as the β-lactam precursor, but they differ in most other details. In the widely distributed biosynthetic pathway for the penicillins and cephalosporins [22,23], the β-lactam precursor is the tripeptide α-aminoadipoyl-l-cysteinyl-d-valine, which is produced by a nonribosomal peptide synthetase (NRPS) [25]. A single enzyme, isopenicillin N synthase, catalyzes two successive two-electron oxidations to form both the A and B rings [26,27]. The precursor to the monobactam nocardicin is also produced by a NRPS, but it is unrelated to the penicillin NRPS [28]; the mechanism by which the A ring of nocardicin forms is not yet known. Carbapenems have a very similar structure to the penicillins: a 4,5-ring system with a carboxylate at the same position of the B ring and a carbon atom in place of a sulfur. Despite these similarities, they derive from a different amino acid building block, and their formation differs in its connectivity and enzymology. The second connection forms a different bond altogether – the β-lactam amide – and rather than oxidation, this bond is made by the adenylate-mediated activation of the newly installed carboxylate [29,30]. The early steps of the clavulanate/clavam pathway resemble two of those found in the carbapenem pathway: a two-carbon building block is added to an amino acid sidechain, and the A ring forms by adenylate-mediated activation. The B ring of the clavulanate/clavams, however, is formed by two successive oxidative transformations: a hydroxylation to install what becomes the B ring oxygen, and then a second two-electron oxidation to fuse this oxygen to the A ring [31,32]. In sum, the β-lactam precursors can be monomers or polymers of proteinogenic and nonproteinogenic amino acids, one or two rings can form (where two, in either order and with varying connectivity), and ring formation can be catalyzed by oxidative or non-oxidative enzymes. While these β-lactams have functional differences, the ability of such diverse pathways to converge on such similar scaffolds highlights both the importance of the β-lactam scaffold and the evolutionary versatility of biosynthetic pathways. As more microbial genomes are sequenced, a central challenge will be to explore the origins of these pathways: from which other pathways were they derived, and what were the functions of the evolutionary intermediates?
Figure 2.
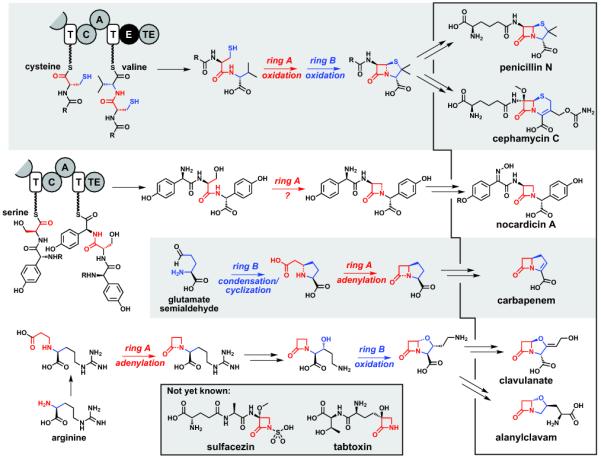
Biosynthetic pathways for β-lactam natural products. Key steps from the known or proposed pathways to penicillin N, cephamycin C, nocardicin A, carbapenem, clavulanate, and alanylclavam are shown. Ring A is colored red and ring B is colored blue, and the steps in which they form are highlighted in the same color. Newly formed ring A and ring B bonds are shown as dashed lines.
Merging gene clusters into super-clusters
Due to horizontal gene transfer, distinct gene clusters can come to cohabit the same microbial genome, an arrangement that can lead to a merger of the two gene clusters. Two types of mergers will be considered in this section: First, distinct gene clusters that produce synergistic antibiotics can be fused into a contiguous cassette; this arrangement favors co-regulation of the two pathways and horizontal transfer of the entire functional unit. Second, distinct gene clusters can merge functionally to form a single pathway that produces a new, hybrid, molecule.
An example of the first type of merger involves two of the β-lactam antibiotics discussed in the previous section. Resistance to the β-lactams is mediated by β-lactamases, bacterial enzymes that cleave these antibiotics [7]. One of the β-lactams described in the previous section, clavulanate, is not an antibiotic itself but rather an inhibitor of β-lactamases [33]. By inhibiting their resistance enzyme, clavulanate restores the effectiveness of the β-lactam antibiotics; indeed, combination therapies comprising a β-lactam and a β-lactamase inhibitor are commonly used in the clinic [34]. Since antibiotic resistance is widespread among soil bacteria [35-38], one might imagine that a β-lactam-producing bacterium would benefit from producing clavulanate at the same time. Indeed, not only does the industrial clavulanate producer Streptomyces clavuligerus also produce the cephalosporin cephamycin [39], but the gene clusters for these two molecules are fused into a contiguous 90 kb super-cluster in the S. clavuligerus genome [40]. This super-cluster also harbors the genes for a third class of clavam metabolites related to clavulanate in structure but not in function [41]. Presumably, contiguous gene clusters enjoy the potential advantage of co-regulation, and are more likely to be transferred horizontally as a single functional unit.
Other bacterial species are known to produce families of functionally related molecules. For example, a single strain of the actinomycete Micromonospora carbonacea produces three unrelated antibiotics that bind to three different sites on the ribosome: everninomicin, a polysaccharide that binds in a cleft between the 23S rRNA and ribosomal protein L16 [12,42]; chloramphenicol, which binds to the peptidyltransferase center of the 50S subunit [13,43]; and Sch 40832, a thiostrepton-related thiopeptide that likely binds in a cleft between the 23S rRNA and ribosomal protein L11 [43,44]. While it is not yet known whether the gene clusters for these antibiotics are physically linked in the M. carbonacea genome, their co-occurrence in the same organism may have functional significance; one possibility is that the combination of three antibiotics prevents the emergence of resistance to any one of them in a target organism.
Sometimes, gene clusters that merge physically take their association one step further: they fuse functionally to form a single pathway that produces a hybrid antibiotic. For example, simocyclinone is a hybrid between aminocoumarins and anthracycline glycosides, and accordingly its gene cluster harbors sub-clusters that resemble aminocoumarin and anthracycline gene clusters [3,45,46]. These two moieties are linked by a polyene dicarboxylate, which may represent a third gene cluster that merged with the first two (the evolutionary order of events is not known). The function of simocyclinone is likely distinct from that of aminocoumarins and anthracyclines: while it has been reported to inhibit DNA gyrase like the aminocoumarin antibiotics, there are differences in its activity that may arise from the ability of its anthracycline moiety to intercalate between DNA base pairs [47]. The functional relevance of merging gene clusters is more apparent for siderophore-antibiotic conjugates like albomycin [48] and microcin E492 [49]; these Trojan Horse molecules are composed of an antibacterial moiety that inhibits cell growth and a siderophore moiety that facilitates transport into target cells through siderophore receptors [50,51].
Conclusion: How is antibiotic synthesis distributed among microbial taxa?
Analyses of bacterial genome sequences suggest that only ~10% of the natural products produced by screened strains have been discovered, and perhaps ~1% of the molecules from the global consortium of microbial producers are known [52,53]. Why, then, has rediscovery been such a common problem in industrial antibiotic discovery programs [54]?
One contributing factor is that most industrial screening programs have focused on soil actinomycetes [52]; these bacteria represent a limited portion of the extant microbial diversity, both phylogenetically and ecologically. Given that a single actinomycete or myxobacterial genome encodes twenty to thirty different gene clusters for natural products [55-59], a second contributing factor is that a single known antibiotic can mask the activity of new antibiotics in a crude or partially purified extract. But a third factor likely plays an equally important role: unrelated bacteria often produce the same antibiotics. One estimate is that 1 out of 100 actinomycetes produce streptomycin, 4 out of 1000 produce tetracycline, 1.5 out of 100,000 produce vancomycin, and 5 out of 1,000,000 produce erythromycin [52].
The cosmopolitan distribution of antibiotic production is not limited to actinomycetes. Andrimid and its relative moiramide, antibiotics that block fatty acid synthesis by inhibiting acetyl-CoA carboxylase [60], have been isolated from a tunicate-associated Pseudomonas strain from Alaska [61], an Enterobacter endosymbiont of a brown planthopper from Thailand [62], a marine Vibrio from southern California [63], and a strain of Pantoea from upstate New York [64]. The andrimid gene clusters from the latter two strains have been sequenced; they are nearly identical and both gene clusters harbor a transposase pseudogene, suggesting that they entered their hosts’ lineages by horizontal gene transfer [3].
More strikingly, certain classes of natural product antibiotics are produced by two or more microbial taxa that diverged hundreds of millions of years ago (Figure 4). Antibiotics as complex as aminoglycosides and thiopeptides are produced by both actinomycetes and Bacillus spp., and their biosynthetic genes are homologous and even partly syntenic [65-67]. In an extreme example of the cross-taxa production of a single antibiotic class, cephalosporins are produced by actinomycetes, Gammaproteobacteria, and fungi, and their biosynthetic gene clusters are remarkably similar [22,23]. An important challenge in the coming years will be to investigate the role horizontal gene transfer has played in the cosmopolitan distribution of antibiotic production. These observations could be useful for synthetic biology and metabolic engineering; since Gammaproteobacteria and Firmicutes are usually easier to manipulate genetically than actinomycetes, strains in the former taxa may be particularly well suited to serving as hosts for the heterologous production of widely distributed natural product classes [68].
Figure 4.
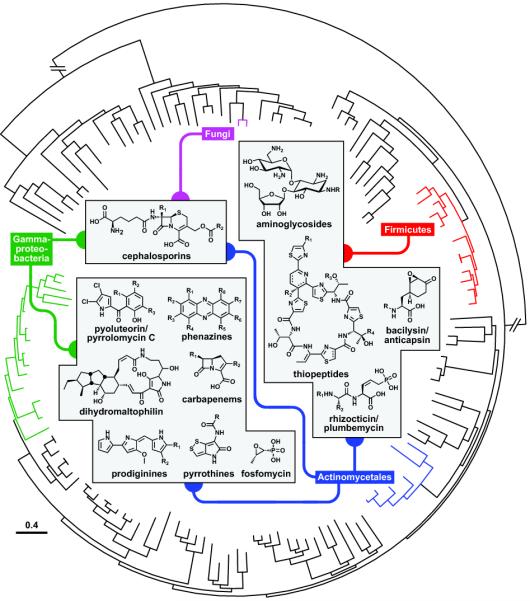
Widely distributed antibiotics. The antibiotics shown are produced by at least two of the following microbial taxa: Actinomycetales, Firmicutes, Gammaproteobacteria, and Fungi. The phylogenetic tree was obtained from iTOL (http://itol.embl.de/) [69].
Figure 3.

Merging gene clusters into super-clusters. Four molecules or pairs of molecules that have resulted from known or presumed gene cluster mergers are shown.
Acknowledgments
I am indebted to Laura Brown, Jon Clardy, and Christopher Walsh for helpful discussions and comments on the manuscript, and to the Department of Molecular Biology and the Center for Computational and Integrative Biology at Massachusetts General Hospital for research funding and support.
References
- 1.Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington, DC: 2003. [Google Scholar]
- 2.Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nat Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 3.Fischbach MA, Walsh CT, Clardy J. The evolution of gene collectives: How natural selection drives chemical innovation. Proc Natl Acad Sci U S A. 2008;105:4601–4608. doi: 10.1073/pnas.0709132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Challis GL, Hopwood DA. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci U S A. 2003;100(Suppl 2):14555–14561. doi: 10.1073/pnas.1934677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conant GC, Wagner A. Convergent evolution of gene circuits. Nat Genet. 2003;34:264–266. doi: 10.1038/ng1181. [DOI] [PubMed] [Google Scholar]
- 6.Fernald RD. Casting a genetic light on the evolution of eyes. Science. 2006;313:1914–1918. doi: 10.1126/science.1127889. [DOI] [PubMed] [Google Scholar]
- 7.Jacoby GA, Munoz-Price LS. The new beta-lactamases. N Engl J Med. 2005;352:380–391. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 8.Garau G, Garcia-Saez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frere JM, Dideberg O. Update of the standard numbering scheme for class B beta-lactamases. Antimicrob Agents Chemother. 2004;48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joris B, Ghuysen JM, Dive G, Renard A, Dideberg O, Charlier P, Frere JM, Kelly JA, Boyington JC, Moews PC, et al. The active-site-serine penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. Biochem J. 1988;250:313–324. doi: 10.1042/bj2500313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 2008;9:242–253. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 11.Zimmerman E, Yonath A. Biological implications of the ribosome’s stunning stereochemistry. Chembiochem. 2009;10:63–72. doi: 10.1002/cbic.200800554. [DOI] [PubMed] [Google Scholar]
- 12.Hermann T. Drugs targeting the ribosome. Curr Opin Struct Biol. 2005;15:355–366. doi: 10.1016/j.sbi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol. 2003;330:1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 14.Moore PB, Steitz TA. The ribosome revealed. Trends Biochem Sci. 2005;30:281–283. doi: 10.1016/j.tibs.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Silver LL. Novel inhibitors of bacterial cell wall synthesis. Curr Opin Microbiol. 2003;6:431–438. doi: 10.1016/j.mib.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5:321–332. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 17.Prowse WG, Kline AD, Skelton MA, Loncharich RJ. Conformation of A82846B, a glycopeptide antibiotic, complexed with its cell wall fragment: an asymmetric homodimer determined using NMR spectroscopy. Biochemistry. 1995;34:9632–9644. doi: 10.1021/bi00029a041. [DOI] [PubMed] [Google Scholar]
- 18.Hsu ST, Breukink E, Tischenko E, Lutters MA, de Kruijff B, Kaptein R, Bonvin AM, van Nuland NA. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11:963–967. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- 19.Ruzin A, Singh G, Severin A, Yang Y, Dushin RG, Sutherland AG, Minnick A, Greenstein M, May MK, Shlaes DM, et al. Mechanism of action of the mannopeptimycins, a novel class of glycopeptide antibiotics active against vancomycin-resistant gram-positive bacteria. Antimicrob Agents Chemother. 2004;48:728–738. doi: 10.1128/AAC.48.3.728-738.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Helm JS, Chen L, Ye XY, Walker S. Ramoplanin inhibits bacterial transglycosylases by binding as a dimer to lipid II. J Am Chem Soc. 2003;125:8736–8737. doi: 10.1021/ja035217i. [DOI] [PubMed] [Google Scholar]
- 21.Maki H, Miura K, Yamano Y. Katanosin B and plusbacin A(3), inhibitors of peptidoglycan synthesis in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2001;45:1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liras P, Martin JF. Gene clusters for beta-lactam antibiotics and control of their expression: why have clusters evolved, and from where did they originate? Int Microbiol. 2006;9:9–19. [PubMed] [Google Scholar]
- 23.Liras P, Rodriguez-Garcia A, Martin JF. Evolution of the clusters of genes for beta-lactam antibiotics: a model for evolutive combinatorial assembly of new beta-lactams. Int Microbiol. 1998;1:271–278. [PubMed] [Google Scholar]
- 24.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 25.Byford MF, Baldwin JE, Shiau C, Schofield CJ. The mechanism of ACV synthetase. Chem Rev. 1997;97:2631–2649. doi: 10.1021/cr960018l. [DOI] [PubMed] [Google Scholar]
- 26.Roach PL, Clifton IJ, Hensgens CM, Shibata N, Schofield CJ, Hajdu J, Baldwin JE. Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation. Nature. 1997;387:827–830. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- 27.Baldwin JE, Bradley M. Isopenicillin N synthase: mechanistic studies. Chem Rev. 1990;90:1079–1088. [Google Scholar]
- 28.Gunsior M, Breazeale SD, Lind AJ, Ravel J, Janc JW, Townsend CA. The biosynthetic gene cluster for a monocyclic beta-lactam antibiotic, nocardicin A. Chem Biol. 2004;11:927–938. doi: 10.1016/j.chembiol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 29.Nunez LE, Mendez C, Brana AF, Blanco G, Salas JA. The biosynthetic gene cluster for the beta-lactam carbapenem thienamycin in Streptomyces cattleya. Chem Biol. 2003;10:301–311. doi: 10.1016/s1074-5521(03)00069-3. [DOI] [PubMed] [Google Scholar]
- 30.Coulthurst SJ, Barnard AM, Salmond GP. Regulation and biosynthesis of carbapenem antibiotics in bacteria. Nat Rev Microbiol. 2005;3:295–306. doi: 10.1038/nrmicro1128. [DOI] [PubMed] [Google Scholar]
- 31.Kershaw NJ, Caines ME, Sleeman MC, Schofield CJ. The enzymology of clavam and carbapenem biosynthesis. Chem Commun (Camb) 2005:4251–4263. doi: 10.1039/b505964j. [DOI] [PubMed] [Google Scholar]
- 32.Townsend CA. New reactions in clavulanic acid biosynthesis. Curr Opin Chem Biol. 2002;6:583–589. doi: 10.1016/s1367-5931(02)00392-7. [DOI] [PubMed] [Google Scholar]
- 33.Charnas RL, Fisher J, Knowles JR. Chemical studies on the inactivation of Escherichia coli RTEM beta-lactamase by clavulanic acid. Biochemistry. 1978;17:2185–2189. doi: 10.1021/bi00604a025. [DOI] [PubMed] [Google Scholar]
- 34.Lee N, Yuen KY, Kumana CR. Clinical role of beta-lactam/beta-lactamase inhibitor combinations. Drugs. 2003;63:1511–1524. doi: 10.2165/00003495-200363140-00006. [DOI] [PubMed] [Google Scholar]
- 35.D’Costa VM, Griffiths E, Wright GD. Expanding the soil antibiotic resistome: exploring environmental diversity. Curr Opin Microbiol. 2007;10:481–489. doi: 10.1016/j.mib.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 37.D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 38.Dantas G, Sommer MO, Oluwasegun RD, Church GM. Bacteria subsisting on antibiotics. Science. 2008;320:100–103. doi: 10.1126/science.1155157. [DOI] [PubMed] [Google Scholar]
- 39.Alexander DC, Jensen SE. Investigation of the Streptomyces clavuligerus cephamycin C gene cluster and its regulation by the CcaR protein. J Bacteriol. 1998;180:4068–4079. doi: 10.1128/jb.180.16.4068-4079.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward JM, Hodgson JE. The biosynthetic genes for clavulanic acid and cephamycin production occur as a ’super-cluster’ in three Streptomyces. FEMS Microbiol Lett. 1993;110:239–242. doi: 10.1111/j.1574-6968.1993.tb06326.x. [DOI] [PubMed] [Google Scholar]
- 41.Zelyas NJ, Cai H, Kwong T, Jensen SE. Alanylclavam biosynthetic genes are clustered together with one group of clavulanic acid biosynthetic genes in Streptomyces clavuligerus. J Bacteriol. 2008;190:7957–7965. doi: 10.1128/JB.00698-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinstein MJ, Luedemann GM, Oden EM, Wagman GH. Everninomicin, a New Antibiotic Complex from Micromonospora Carbonacea. Antimicrob Agents Chemother (Bethesda) 1964;10:24–32. [PubMed] [Google Scholar]
- 43.Puar MS, Chan TM, Hegde V, Patel M, Bartner P, Ng KJ, Pramanik BN, MacFarlane RD. Sch 40832: a novel thiostrepton from Micromonospora carbonacea. J Antibiot (Tokyo) 1998;51:221–224. doi: 10.7164/antibiotics.51.221. [DOI] [PubMed] [Google Scholar]
- 44.Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, Spahn CM, Fucini P. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol Cell. 2008;30:26–38. doi: 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Galm U, Schimana J, Fiedler HP, Schmidt J, Li SM, Heide L. Cloning and analysis of the simocyclinone biosynthetic gene cluster of Streptomyces antibioticus Tu 6040. Arch Microbiol. 2002;178:102–114. doi: 10.1007/s00203-002-0429-z. [DOI] [PubMed] [Google Scholar]
- 46.Trefzer A, Pelzer S, Schimana J, Stockert S, Bihlmaier C, Fiedler HP, Welzel K, Vente A, Bechthold A. Biosynthetic gene cluster of simocyclinone, a natural multihybrid antibiotic. Antimicrob Agents Chemother. 2002;46:1174–1182. doi: 10.1128/AAC.46.5.1174-1182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flatman RH, Howells AJ, Heide L, Fiedler HP, Maxwell A. Simocyclinone D8, an inhibitor of DNA gyrase with a novel mode of action. Antimicrob Agents Chemother. 2005;49:1093–1100. doi: 10.1128/AAC.49.3.1093-1100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferguson AD, Braun V, Fiedler HP, Coulton JW, Diederichs K, Welte W. Crystal structure of the antibiotic albomycin in complex with the outer membrane transporter FhuA. Protein Sci. 2000;9:956–963. doi: 10.1110/ps.9.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nolan EM, Walsh CT. Investigations of the MceIJ-catalyzed posttranslational modification of the microcin E492 C-terminus: linkage of ribosomal and nonribosomal peptides to form “trojan horse” antibiotics. Biochemistry. 2008;47:9289–9299. doi: 10.1021/bi800826j. [DOI] [PubMed] [Google Scholar]
- 50.Fischbach MA, Lin H, Liu DR, Walsh CT. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol. 2006;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 51.Pramanik A, Braun V. Albomycin uptake via a ferric hydroxamate transport system of Streptococcus pneumoniae R6. J Bacteriol. 2006;188:3878–3886. doi: 10.1128/JB.00205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baltz RH. Antibiotic discovery from actinomycetes: Will a renaissance follow the decline and fall? SIM News. 2005;55:186–196. [Google Scholar]
- 53.Watve MG, Tickoo R, Jog MM, Bhole BD. How many antibiotics are produced by the genus Streptomyces? Arch Microbiol. 2001;176:386–390. doi: 10.1007/s002030100345. [DOI] [PubMed] [Google Scholar]
- 54.Baltz RH. Marcel Faber Roundtable: Is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol. 2006;33:507–513. doi: 10.1007/s10295-005-0077-9. [DOI] [PubMed] [Google Scholar]
- 55.Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M, Horinouchi S. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol. 2008;190:4050–4060. doi: 10.1128/JB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol. 2007;25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- 57.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci U S A. 2001;98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO, Bartels D, Bekel T, Beyer S, Bode E, et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol. 2007;25:1281–1289. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 59.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci U S A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freiberg C, Brunner NA, Schiffer G, Lampe T, Pohlmann J, Brands M, Raabe M, Habich D, Ziegelbauer K. Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity. J Biol Chem. 2004;279:26066–26073. doi: 10.1074/jbc.M402989200. [DOI] [PubMed] [Google Scholar]
- 61.Needham J, Kelly MT, Ishige M, Andersen RJ. Andrimid and Moiramides A-C, metabolites produced in culture by a marine isolate of the bacterium Pseudomonas fluorescens: Structure elucidation and biosynthesis. J Org Chem. 1994;59:2058–2063. [Google Scholar]
- 62.Fredenhagen A, Tamura SY, Kenny PT, Komura H, Naya Y, Nakanishi K, Nishiyama K, Sugiura M, Kita H. Andrimid, a new peptide antibiotic produced by an intracellular bacterial symbiont isolated from a brown planthopper. J Am Chem Soc. 1987;109:4409–4411. [Google Scholar]
- 63.Long RA, Rowley DC, Zamora E, Liu J, Bartlett DH, Azam F. Antagonistic interactions among marine bacteria impede the proliferation of Vibrio cholerae. Appl Environ Microbiol. 2005;71:8531–8536. doi: 10.1128/AEM.71.12.8531-8536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jin M, Fischbach MA, Clardy J. A biosynthetic gene cluster for the acetyl-CoA carboxylase inhibitor andrimid. J Am Chem Soc. 2006;128:10660–10661. doi: 10.1021/ja063194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flatt PM, Mahmud T. Biosynthesis of aminocyclitol-aminoglycoside antibiotics and related compounds. Nat Prod Rep. 2007;24:358–392. doi: 10.1039/b603816f. [DOI] [PubMed] [Google Scholar]
- 66.Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, Chen D, Shen B, Yu Y, Liu W. Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. Chem Biol. 2009;16:141–147. doi: 10.1016/j.chembiol.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown LC Wieland, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc Natl Acad Sci U S A. 2009;106:2549–2553. doi: 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H, Wang Y, Pfeifer BA. Bacterial hosts for natural product production. Mol Pharm. 2008;5:212–225. doi: 10.1021/mp7001329. [DOI] [PubMed] [Google Scholar]
- 69.Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]