

Summary
The innate and adaptive immune responses that confer resistance to the intracellular pathogen Toxoplasma gondii critically depend on IL-12 production, which drives interferon-γ (IFN-γ) expression. Certain cytokines can activate STAT3 and limit IL-12 production to prevent infection-associated immune pathology but T.gondii also directly activates STAT3 to evade host immunity. We show that Suppressor of Cytokine Signaling molecule 3 (SOCS3), a target of STAT3 which limits signaling by the pleiotropic cytokine IL-6, is upregulated in response to infection but is dispensable for the immune-inhibitory effects of T. gondii. Unexpectedly, mice with targeted deletion of SOCS3 in macrophages and neutrophils have reduced IL-12 responses and succumb to toxoplasmosis. Anti–IL-6 administration or IL-12 treatment blocked disease susceptibility suggesting that in the absence of SOCS3, macrophages are hypersensitive to the anti-inflammatory properties of IL-6. Thus, SOCS3 has a critical role in suppressing IL-6 signals and promoting immune responses to control T. gondii infection.
Introduction
Toxoplasma gondii is an opportunistic pathogen of patients with a variety of primary and secondary deficiencies in T cell function (Denkers and Gazzinelli, 1998). Control of T. gondii is achieved through a combination of innate and adaptive responses that are dependent upon accessory cell production of IL-12 that drives natural killer (NK) and T cells to produce IFN-γ (Sher et al., 2003).These cytokines are critical for long-term resistance to T. gondii as mice deficient in either IL-12 or IFN-γ succumb to the acute phase of this infection (Sher et al., 2003). In current models, the ability of IFN-γ to activate the transcription factor STAT1 synergizes with other factors such as TNF-α, IL-1, and CD40L that utilize NFκB signaling to promote robust anti-microbial responses (Andrade et al., 2005; Lieberman et al., 2004). Together, these stimuli induce the expression of numerous genes, such as those that code for inducible nitric oxide synthase (iNOS) and the Immunity Related GTPases (IRGs), that are critical for resistance to T. gondii (Butcher et al., 2005a; Martens et al., 2005; Scharton-Kersten et al., 1997).
T.gondii has evolved a number of strategies to evade host anti-microbial activity and can interfere with NFκB activation (Butcher et al., 2001; Robben et al., 2004; Shapira et al., 2002), antagonize IFN-γ signaling by inhibiting the phosphorylation and nuclear translocation of STAT1, and also by upregulating Suppressor of Cytokine Signaling Molecule 1, SOCS1, a suppressor of IFN-γ-signaling (Dalpke et al., 2008; Kim et al., 2007; Luder et al., 2001; Zimmermann et al., 2006). This pathogen can also interfere with recruitment of IRGs to the parasitophorous vacuole, thus limiting the ability of IFN-γ-activated cells to control parasite replication (Butcher et al., 2005a; Martens et al., 2005; Zhao et al., 2009). More recent work has revealed that T.gondii injects a rhoptry kinase, ROP16, that directly phosphorylates the host transcription factor STAT3, and this event is associated with impaired IL-12p40 and TNF-α production (Butcher et al., 2005b; Saeij et al., 2007; Yamamoto et al., 2009). Interestingly, STAT3 is also activated by the cytokines IL-6 and IL-10, which antagonize anti-microbial effector function and thereby enhance the growth of the intracellular pathogens T. gondii, Mycobacterium, and Leishmania (Beaman et al., 1994; Bermudez et al., 1992; Murray, 2008). Together, these observations suggest that the ability of T. gondii to directly activate STAT3 limits host responses and promotes parasite survival.
Although STAT3 appears to be a key target of T. gondii, the downstream effects of STAT3 that promote parasite replication are unclear. One candidate gene, upregulated by IL-6 and IL-10-mediated activation of STAT3, is SOCS3 which under normal circumstances can bind to the gp130 subunit of the IL-6 receptor complex and limit IL-6 signaling (Kubo et al., 2003). Because SOCS3 can also limit NFκB signals, act as a ubiquitin ligase and was upregulated in response to T. gondii studies were performed, using mice in which the Socs3 gene is flanked by loxP sites and Cre recombinase is expressed under the control of the Lysozyme M promoter to delete Socs3 from macrophages and neutrophils (LysM-cre Socs3fl/fl), to determine its role in the suppressive effects of T. gondii. Although SOCS3 was not required for the ability of T. gondii to suppress cytokine production in vitro, infection of these mice resulted in overwhelming parasite burdens characterized by impaired neutrophil and monocyte responses. Administration of α-IL-6 or treatment with IL-12 blocked disease susceptibility suggesting that in the absence of SOCS3, macrophages are hypersensitive to the anti-inflammatory properties of IL-6.
Results
Toxoplasma gondii upregulates SOCS3 in macrophages
Infection of macrophages with T. gondii in vitro results in the rapid phosphorylation of STAT3 (Butcher et al., 2005b; Saeij et al., 2007). Since STAT3 activation is normally associated with the induction of SOCS3, we used bone marrow-derived macrophages (BMMϕ) infected with the Type I strain (RH) or a Type II strain (Prugniaud) of T. gondii to assess if infection led to the upregulation of SOCS3. Unstimulated cells had low basal levels of SOCS3 mRNA with no detectable SOCS3 protein, while infection with T. gondii induced a rapid increase in mRNA and protein (Figure 1A,B), an effect comparable to that observed following stimulation with IL-6 (Figure 1A, and data not shown). Similar results were observed using resident peritoneal macrophages infected with either parasite strain (Figure S1). Additionally, macrophages stimulated with STAg or incubated with supernatants from infected cells did not upregulate SOCS3 protein indicating that live parasites are required for this biological activity (Figure 1B).
Figure 1. T. gondii upregulates SOCS3 in macrophages.

Cultures of macrophages infected with T. gondii demonstrate increased SOCS3 mRNA and protein that is correlated with STAT3 phosphorylation and decreased IL-12p40 secretion. SOCS3-deficient macrophages show stronger STAT3 phosphorylation and a defect in IL-12 production. (A) 1×106 BMMϕ from C57/Bl6 mice were infected with RH or Pru at an MOI of 5:1 for the indicated time points. RNA was extracted, reverse transcribed into cDNA and Quantitative Real Time PCR was performed using SOCS3-specific primers. Results are relative to β-Actin and representative of two experiments. (B,E,F) Western blot analysis was performed on lysates of 1×106 BMMϕ infected at 1:1 with RH, Pru, or stimulated with STAg (50 µg/mL) and the lysates were probed with a SOCS3-specific antibody (B), or pSTAT3 and total STAT3 (E,F) and compared to β-Actin. (C,D) BMMϕ were infected at 1:1 for 1 hr with RH-Tomato or RH-Cre-Cherry and then fixed, stained with antibodies to SOCS3 conjugated to Alexafluor 488 and DAPI, and transferred to slides for confocal microscopy. Results are indicative of two independent experiments (G) 2×105 BMMϕ were infected with 5:1 RH or Pru parasites for 1 hr then stimulated with LPS (100ng/mL) for 24 hrs. ELISAS were performed on supernatants for IL-12p40. Error bars +/− SD. Results are representative of at least two experiment.
To assess if infected cells expressed SOCS3, immunofluorescence was performed on BMMϕ infected for 3 hrs with a transgenic RH parasite that expresses the fluorescent protein Tomato (RH-Tomato). To confirm specificity, macrophages deficient in SOCS3 were used and SOCS3 protein was not detected by immunofluorescence in resting macrophages or those isolated from LysM-cre Socs3fl/fl mice (Figure 1C). Infection resulted in increased SOCS3 expression in wild type cells while fluorescence was absent in knockout cells. However, these images revealed that in wild type macrophages, SOCS3 was upregulated in infected and uninfected cells. Because T. gondii can inject rhoptry proteins into host cells without directly infecting them (Koshy et al., 2010), studies were performed to assess whether this phenomena contributed to the expression of SOCS3 in uninfected cells. Therefore, a transgenic RH parasite that expressed Cre recombinase (RH-Cre-Cherry) was used to infect Socs3fl/fl BMMϕ for 3 hrs and compared to RH-Tomato. If rhoptry proteins are involved in the upregulation of SOCS3, then the injection of the Cre recombinase should excise the Socs3 gene and the infection-induced upregulation of SOCS3 typically observed in these cultures should be reduced. Indeed, while RH-Tomato parasites induced SOCS3 expression, this was markedly reduced (by approximately 10 fold, 55% versus 5%) when the RH-Cre-Cherry parasites were used (Figure 1D). These data are consistent with a model in which the ability of the parasite to inject rhoptry proteins into infected and uninfected cells explains the observed widespread expression of SOCS3.
Since the ability of T. gondii to antagonize IL-12p40 and TNF-α production from LPS-stimulated macrophages is STAT3-dependent, experiments were performed to determine if this effect was also SOCS3-dependent. As previously described (Butcher et al., 2005b), infection of wild type BMMϕ resulted in a rapid phosphorylation of STAT3 that starts to decline by 4 hrs and is nearly absent by 24 hrs in SOCS3-sufficient macrophages infected with T. gondii (Figure 1E,F). In SOCS3-deficient macrophages, STAT3 phosphorylation is enhanced at 1 and 4 hrs post infection and remains high for up to 24 hrs. This observation is similar to reports that IL-6 mediated activation of STAT3 is enhanced in the absence of SOCS3 (Yasukawa et al., 2003), and suggests that the mechanism utilized by SOCS3 to limit IL-6-activated STAT3 also affects STAT3 phosphorylation by T. gondii. To assess whether the presence of SOCS3 was required for the ability of T. gondii to limit IL-12p40 production, control or SOCS3-deficient BMMϕ were infected for 1 hr prior to LPS stimulation, and then incubated for 24 hrs before cytokine analysis. Stimulation of wild type macrophages with LPS resulted in IL-12p40 secretion and this was reduced when SOCS3-deficient macrophages were used (p<0.01) (Figure 1G). Infection with T. gondii resulted in a reduced ability of LPS to induce IL-12p40 and TNF-α in wild type and SOCS3-deficient macrophages (Figure 1G and data not shown). These findings show that infection leads to an upregulation of SOCS3, but this protein is not required for T. gondii to impair cytokine secretion by macrophages.
LysM-cre Socs3fl/fl mice succumb acutely to T.gondii infection
Although SOCS3 was not required for T. gondii to suppress cytokine responses in vitro, studies were performed to assess the role of this regulatory protein in resistance to T. gondii in vivo. Since deletion of Socs3 is embryonic lethal, the cre-loxP system was utilized to generate mice that lack SOCS3 in neutrophil and macrophage lineages (LysM-cre Socs3fl/fl). As shown in Figure 1C, LysM-cre Socs3fl/fl BMMϕ lacked SOCS3 expression upon infection. Similarly, while basal levels of SOCS3 transcripts were present in wild type BMMϕ, infection or IL-6 simulation led to a marked increase, but this was not observed using peritoneal macrophages and BMMϕ from LysM-cre Socs3fl/fl mice whereas levels of SOCS1 were not affected (Figure S1 and data not shown).
To assess the role of SOCS3 in the response to T. gondii, LysM-cre Socs3fl/fl and Socs3fl/fl littermate control mice were challenged with the avirulent Pru strain. While Socs3fl/fl mice survived, LysM-cre Socs3fl/fl mice succumbed by day 10 (Figure 2A). In Socs3fl/fl control mice, there were few infected cells in the peritoneal cavity at day 4 with little increase by day 8 (Figure 2B,C). A similar number of infected cells were observed in the peritoneal cavity of LysMcre Socs3fl/fl at day 4 as compared to Socs3fl/fl mice; however, there was a significant increase in parasite burden by day 8 including the presence of free tachyzoites (bottom right arrow) (Figure 2B,C). Additionally, following infection with Pru-Tomato, there was an elevated percentage of Tomato+ infected cells in the peritoneal cavity in LysM-cre Socs3fl/fl mice that did not vary by cell type (Figure 2D,E). The use of real-time PCR revealed that that the absence of SOCS3 also resulted in higher parasite burdens in the liver at day 4 which had increased dramatically at day 8 (Figure 2F). Histological analysis of tissues from infected mice showed little inflammation in control mice but increased areas of necrosis (arrows) and inflammation in the liver and lungs of LysM-cre Socs3fl/fl mice (Figure 2G) consistent with the increased parasite burdens in these mice.
Figure 2. LysM-cre Socs3fl/fl mice succumb acutely to T. gondii infection.
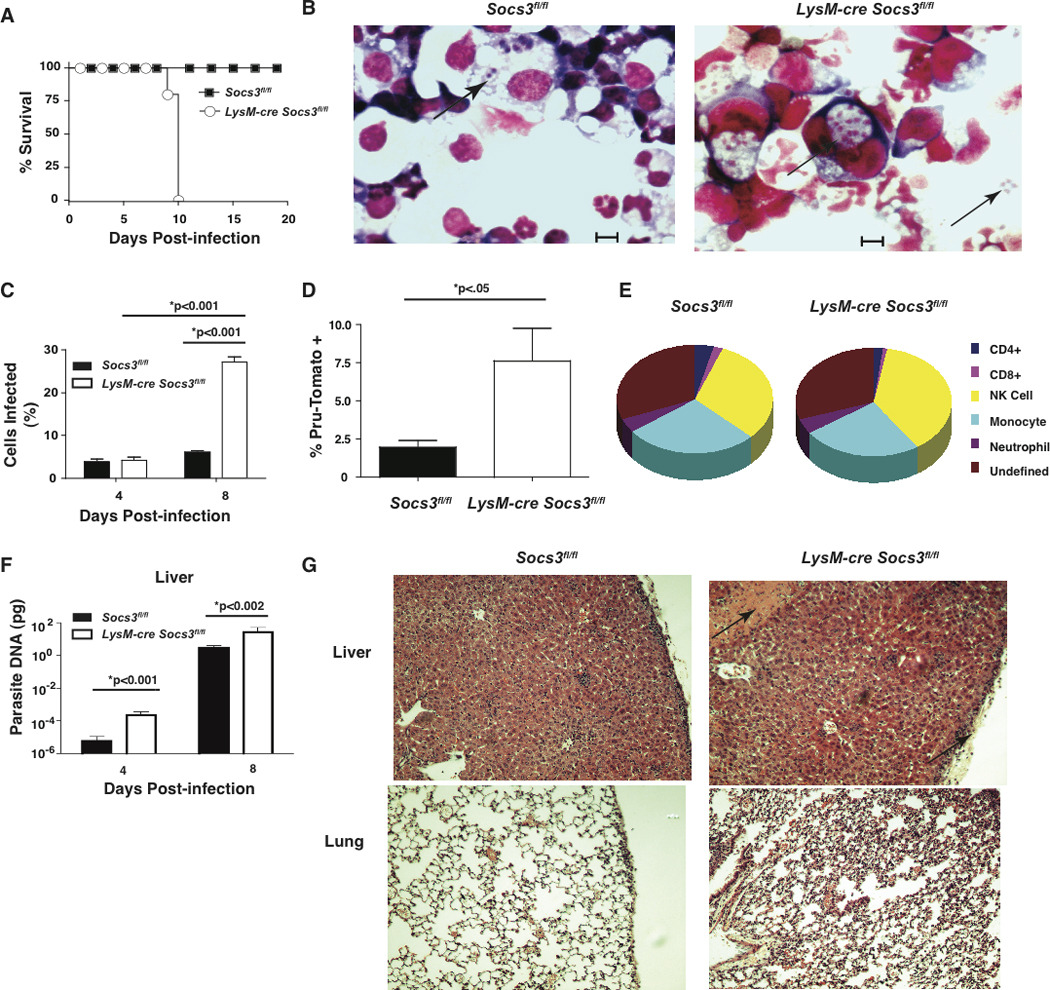
Infection of LysM-cre Socs3fl/fl mice leads to acute mortality associated with increased parasite burden in the peritoneal cavity and liver along with increase pathology in the liver and lungs. (A) Socs3fl/fl (n=5) and LysM-cre Socs3fl/fl (n=5) were infected i.p. with 104 Pru parasites and survival was determined. Infected mice were also sacrificed on day 8 to determine parasite burden. (B,C) PECs were collected, centrifuged onto slides, and visualized using light microscopy for the percentage of infected cells. Arrows denote both intracellular and extracellular tachyzoites although only cells with intracellular parasites, as determined by microscopy, were considered infected. (D,E) Socs3fl/fl (n=5) and LysM-cre Socs3fl/fl (n=5) were infected i.p. with 1×106 Pru-Tomato parasites and PECs were isolated at day 4 post-infection. The total number of live, Pru-Tomato+ cells was counted using flow cytometry and gated on Tomato+ events from the PE channel. (E) From the Pru-Tomato+ cells, the percentage of positive cells that were monocytes (CD11b+, F4/80+, Ly6C+), neutrophils (CD11b+, F4/80−, Ly6Clow, GR1+), T cells (CD3+ and CD4+ or CD8+), and NK cells (CD3−, NK1.1+) were evaluated and depicted. (F) DNA was extracted from spleen and liver tissue from day 8 infected mice (n=5) and analyzed by Real Time PCR for parasite DNA. (G) Lungs of infected (day 8) mice were fixed, sectioned, and imaged using confocal microscopy. Arrows denote regions of increased pathology. Error bars +/− SD. Images and results are representative of two independent experiments. Statistical values were determined by Student’s t tests.
Activation and recruitment of monocytes and neutrophils
Several studies have established that inflammatory monocytes and neutrophils are required for acute resistance to T. gondii (Egan et al., 2008). Phenotypically, inflammatory monocytes are CD11b+, F480+, Ly6Chi, CD62L+, MHCIIhi, and CCR2+, and migrate to sites of infection and contribute to the control of T. gondii (Dunay et al., 2010; Mordue and Sibley, 2003). To test if the enhanced susceptibility of LysMcre Socs3fl/fl mice was due to altered recruitment of these populations, flow cytometry was performed on peritoneal cells from infected mice. While the overall number of peritoneal cells was similar between Socs3fl/fl control and LysMcre Socs3fl/fl mice at day 4, there was a significant increase in recruited cells in LysMcre Socs3fl/fl mice at day 8 (p<0.05) (Figure 3A). Evaluation of CD11b+ cells in the peritoneal cavity of uninfected control and LysM-cre Socs3fl/fl mice revealed that the majority of these cells were resident macrophages (F4/80hi, Ly6Cneg) and that the loss of SOCS3 did not alter the size of this population (Figure 3B, left panel and data not shown). Upon infection, there was recruitment of neutrophil (CD11b+, Ly6Clow, F4/80low, GR1hi) and inflammatory monocytes (CD11b+, F4/80hi, Ly6Chi, Figure 3B top right quadrant) to the peritoneal cavity by day 4, but no significant difference in number was observed between control and LysM-cre Socs3fl/fl mice (Figure 3B,C). Moreover, no significant difference in expression of CD62L, MHCII, CD80, and CD86 was observed between neutrophils or inflammatory monocytes from these mice (Figure 3D–G). To assess functionality, the ability of the monocytes and neutrophils to produce IL-12p40 during infection was measured. Critically, there was a significant reduction in the percentage of IL-12p40+ inflammatory monocytes and neutrophils from LysM-cre Socs3fl/fl mice at day 4 post-infection (Figure 3H,I). Together, these findings indicate that the inability of LysM-cre Socs3fl/fl mice to control parasite replication is not a consequence of a failure to recruit activated neutrophil or inflammatory monocyte populations, but correlates with the reduced production of IL-12 of these cells.
Figure 3. Inflammatory monocytes and neutrophils from LysM-cre Socs3fl/fl mice are deficient in IL-12 production following infection.

LysM-cre Socs3fl/fl mice show impaired IL-12 production from neutrophil and inflammatory monocyte populations recruited to the peritoneal cavity following infection. (A) Socs3fl/fl (n=5) and LysM-cre Socs3fl/fl (n=5) were infected i.p. with 104 Pru parasites and sacrificed at days 4 and 8 post infection and the total number of cells in the peritoneal cavity was quantified along with 2 naïve mice per group. (B,C) Live, singlet, CD19−, CD3−, CD11b+ cells were analyzed for the surface markers Ly6C and F4/80 to identify inflammatory monocytes (F4/80hi,Ly6Chi) and neutrophils (F4/80low,GR1+, Ly6Clow) and quantified. (D–G) Monocyte and neutrophil populations were stained for the surface markers of activation MHC Class II, CD62L, CD80, and CD86 and compared for the overall expression by mean fluorescence intensity (MFI). (H,I) To determine IL-12 production from these cell types, intracellular cytokine staining was performed by incubating 1×106 PECs with BFA and Golgi Stop for 5 hrs, staining with surface markers for monocytes and neutrophils, fixing with paraformaldehyde, staining for intracellular IL-12p40, and analyzing by flow cytometry. MFI was calculated and is depicted in red. (I) The percentage of IL-12p40+ inflammatory monocytes and neutrophils are shown. One-Way ANOVA followed by student’s t test was used to determine statistical significance. Error bars +/− SD. Results are representative of two independent experiments.
LysM-cre Socs3fl/fl mice have impaired NK and T cell responses
Since IL-12 is critical for NK and T cell–mediated resistance to T. gondii, these populations were examined to determine if the loss of SOCS3 in macrophages and neutrophils affected lymphocyte populations and their ability to produce IFN-γ. At day 4, similar numbers of CD4+, CD8+, and NK1.1+ cells were observed in Socs3fl/fl control and LysM-cre Socs3fl/fl mice suggesting that the loss of SOCS3 in macrophages and neutrophils did not impair recruitment of these populations to the site of infection (Figure 4A). Although there was no observable difference in the population of naïve CD4+ T cells in LysM-cre Socs3fl/fl mice, there was a statistically significant decrease in activated CD62Llow, CD44hi CD4+ T cells (Figure 4B,C). Furthermore, CD4+ T and NK cells from LysM-cre Socs3fl/fl mice were significantly impaired in their ability to produce IFN-γ at day 4 as compared to control mice with CD8+ T cells demonstrating the same trend though not statistically significant (Figure 4D,E). Additionally, no defect in IL-17 production of the number of IL-17+ CD4+ cells in LysM-cre Socs3fl/fl mice was observed (Figure S3 and data not shown). To further evaluate the ability of LysM-cre Socs3fl/fl T cells to produce IFN-γ, splenic recall assays were performed. Surprisingly, after 24 hrs of incubation with media alone, αCD3, or STAg, Socs3fl/fl control and LysM-cre Socs3fl/fl splenocytes from day 4 infected mice produced comparable IL-12p40 and IFN-γ (Figure 4F,G). However, impaired production of secreted IL-12p40 was observed in the peritoneal lavage fluid at day 4 and a similar trend was observed in the sera (Figure 5A,B). It is noteworthy that by day 8 post-infection the levels of IL-12p40 detected in the peritoneum were restored but that this correlated with the large increase in parasite burdens (Figure 5B). As the protective effects of IL-12 are most critical during the first 2–3 days of infection (Khan et al., 1994), the elevated concentrations observed at day 8 are not sufficient to confer protection. Similar production of IFN-γ protein was observed at days 4 and 8 post-infection in the serum and peritoneal lavage fluid, suggesting that any defect in the production of IL-12 by monocytes and neutrophils may only affect the earliest production of IFN-γ. Together, these findings demonstrate that the reduced ability of the LysM-cre Socs3fl/fl mice to control growth of T. gondii is associated with an early defect in the ability to produce IL-12.
Figure 4. LysM-cre Socs3fl/fl mice recruit T and NK cells with impaired IFN-γ production.

T and NK cells traffic to the peritoneal cavity in appropriate numbers by day 4 following infection of LysM-cre Socs3fl/fl mice yet exhibit a less activated phenotype with decreased IFN-γ production. Socs3fl/fl (n=5) and LysM-cre Socs3fl/fl (n=5) were infected i.p. with 104 Pru parasites and sacrificed at days 4 and 8 post-infection. (A) The total number of NK cells (B220−, CD3−, NK1.1+), CD4+ (B220−, CD3+, CD4+), and CD8+ (B220−, CD3+, CD8+) T cells were determined by flow cytometry. (A,B) CD4+ T cells were measured for the surface markers CD62L and CD44 and (C) quantified for the number of activated (CD44hi, CD62Llow) and naïve (CD44low, CD62Lhi) cells per mouse. (1×106 (D,E) For intracellular cytokine staining, PECs were incubated with BFA, Golgi Stop, PMA, and ionomycin for 3 hrs and analyzed for IFN-γ production following surface staining for NK cells, and CD4+ and CD8+ T cells. (F,G) 1×106 splenocytes were plated from extracted spleens of infected mice and stimulated with media alone or 50 µg/mL STAg. Concentrations of IL-12p40 and IFN-γ were measured by ELISA following 24 hrs of stimulation. One-Way ANOVA followed by student’s t test was used to determine statistical significance. Error bars +/− SD. Results are representative of two independent experiments.
Figure 5. Infected LysM-cre Socs3fl/fl mice show impaired IL-12 production in the peritoneal cavity at day 4.

Peritoneal lavage fluid from infected LysM-cre Socs3fl/fl mice contains a lower concentration of IL-12p40 at day 4 that corresponds to a similar trend in sera obtained from blood. Although IFN-γ production appears normal at day 4, a diminished IFN-γ response was observed at day 8 in the peritoneal lavage fluid. (A,B) Fluid from intraperitoneal lavage of PECs and serum of infected mice at days 4 and 8 were collected and analyzed by ELISA for IL-12p40 and IFN-γ. PEC fluid results are presented as ng/peritoneum which is calculated by multiplying the ELISA concentration result (ng/mL) by the mL quantity of peritoneal lavage fluid (PBS) recovered following centrifugation. One-Way ANOVA followed by student’s t test was used to determine statistical significance. Error bars +/− SD. Results are representative of two independent experiments.
Effect of SOCS3 deletion on macrophage function
One possible explanation for the increased susceptibility of LysM-cre Socs3fl/fl mice was that SOCS3-deficient macrophages were unable to respond to IFN-γ to control T. gondii. To test whether SOCS3-deficient macrophages could respond appropriately to IFN-γ in vitro, BMMϕ from Socs3fl/fl and LysM-cre Socs3fl/fl mice were incubated with TNF-α and increasing concentrations of IFN-γ overnight and then infected with Pru for 24 hrs. Stimulated Socs3fl/fl control and LysM-cre Socs3fl/fl macrophages limited parasite replication in a concentration-dependent manner (Figure 6A). To determine if IFN-γ signaling was impaired in SOCS3-deficient macrophages, cells were incubated with IFN-γ and the phosphorylation of STAT1 and STAT3 was measured. Unlike control macrophages, SOCS3-deficient macrophages had abbreviated responses to IFN-γ showing less phosphorylated STAT1 at later time points (Figure 6B). Additionally, control macrophages showed an increase in total STAT1 at 20 hrs that was less pronounced in SOCS3-deficient cells. This observation did not correlate with a reduced ability of the LysM-cre Socs3fl/fl macrophages to produce nitric oxide (NO), suggesting that this altered STAT1 signaling pattern did not affect microbicidal activity (Figure 6C). Furthermore, IRGs are critical to resistance to T. gondii and when their expression was measured in wild type and SOCS3 knockout BMMϕ stimulated with IFN-γ, similar levels of IRGM3, IRM1, and IRGA6 were detected (Figure 6D). Thus, deletion of SOCS3 appears to have little impact on the ability of IFN-γ to activate anti-microbial responses.
Figure 6. IL-6 blocks IL-12p40 production from stimulated LysM-cre Socs3fl/fl macrophages.

SOCS3-defiicent macrophages do not display impaired microbicidal activite, nitrite production, or IRG expression despite altered IFN-γ-mediated phosphorylation of STAT1. However, IL-12p40 and TNF-α production by SOCS3-deficient macrophages is inhibited by endogenously-produced IL-6 in these cultures and this effect can be reversed through the addition of neutralizing antibodies to IL-6. (A) 5×105 BMMϕ were plated and infected with Pru (MOI 1:1) for 24 hrs following overnight stimulation with TNF-α (10 ng/mL) with increasing concentrations of IFN-γ (10 fold dilutions, beginning at 100 Units/mL). 2.5×105 macrophages were centrifuged onto coverslips for counts of infected cells. (B,D) 1×106 BMMϕ were treated with IFN-γ (100 Units/mL) for the indicated time points and analyzed by western blot for pSTAT1, IRGM1, IRGM2, IRGM3, IRGD, IRGA6, and IRGB6. (C) Supernatants from (A) were measured for nitrite (nitric oxide) production following the Griess reaction protocol. (E) Similar to (B), macrophages were stimulated with IL-6 (100 ng/mL) for the indicated time points and analyzed for pSTAT3 and pSTAT1. (F) The ability of IL-6 to inhibit IL-12p40 production was evaluated by stimulating 2×105 BMMϕ with IFN-γ and LPS (10 Units/mL and 10 ng/mL respectively) with or without increasing concentrations of IL-6 (10 fold dilutions, beginning at 100 ng/mL) for 24 hrs and analyzed by ELISA. (G,H) 2×105 BMMϕ were given IFN-γ and LPS with or without α-IL-6 and analyzed for IL-12p40 and TNF-α production. Error bars +/− SD. Statistics were performed using Student’s t tests and results are representative of two independent experiments.
It was previously reported that deletion of SOCS3 from macrophages converted IL-6 signals into anti-inflammatory IL-10-like signals due to enhanced STAT3 activation (El Kasmi et al., 2006; Yasukawa et al., 2003). Indeed, IL-6 induced sustained phoshorylation of STAT1 and STAT3 in LysM-cre Socs3fl/fl macrophages as compared to Socs3fl/fl macrophages, yet demonstrated similar kinetics of activation of the MAP kinase ERK suggesting that only STAT signaling is altered in these macrophages (Croker et al., 2003) (Figure 6E, Figure S2). LysM-cre Socs3fl/fl macrophages also demonstrated impaired IL-12p40 production in response to IFN-γ and LPS and both Socs3fl/fl and LysM-cre Socs3fl/fl macrophages were sensitive to increasing concentrations of IL-6 (Figure 6F). Addition of neutralizing antibodies to IL-6 restored IL-12p40 production from LysM-cre Socs3fl/fl macrophages and increased its production over stimulation alone (Figure 6G), indicating that endogenous IL-6 is an antagonist of IL-12 production in these cultures. A similar effect of IL-6 on TNF-α production was observed after 48 hrs of culture while IL-6 had no effect on nitric oxide production (Figure 6H and data not shown).
Anti-IL-6 or IL-12 treatment restores resistance to T. gondii
The data described above indicate that in the absence of SOCS3 IL-6 becomes a potent antagonist of IL-12 production. During acute infection, IL-6 is upregulated in both Socs3fl/fl control and LysM-cre Socs3fl/fl mice and is detected in the peritoneal cavity and sera by day 4 (data not shown). To test whether endogenous IL-6 contributed to the observed susceptibility of LysM-cre Socs3fl/fl mice, Socs3fl/fl and LysM-cre Socs3fl/fl mice were treated with neutralizing antibodies to IL-6. Socs3fl/fl and LysM-cre Socs3fl/fl mice showed an increase in IL-6 production at day 8 post-infection; however the LysM-cre Socs3fl/fl mice produced higher levels of IL-6 (Figure 7A). Furthermore, neutralization of IL-6 markedly reduced the available IL-6 in Socs3fl/fl and LysM-cre Socs3fl/fl mice (Figure 7A). When these mice were evaluated for survival, control mice given the α-IL-6 treatment survived up to 40 days after challenge (Figure 7B) whereas LysM-cre Socs3fl/fl mice treated with rat IgG succumbed acutely. However, when these mice were treated with α-IL-6, 80% of the mice survived acute challenge and progressed into the chronic phase of this infection (Figure 7B). Indeed, treatment with α-IL-6 resulted in a marked reduction in the number of infected cells in the peritoneal cavity at day 8 in Socs3fl/fl and LysM-cre Socs3fl/fl mice (Figure 7C). Similarly, IL-17, IL-10, IL-12p40, and IFN-γ levels were reduced in the peritoneal cavity (Figure 7D) and serum (data not shown) of Socs3fl/fl and LysM-cre Socs3fl/fl mice. The reduced cytokine production observed following α-IL-6 treatment is likely the result of an overall decrease in T cell and monocyte recruitment to the peritoneal cavity associated with lower parasite burdens. Given the reduced inflammation following α-IL-6 treatment, it is difficult to determine if this treatment restored IL-12 production in this model. Therefore, to determine if neutralizing IL-6 in vivo alters IL-12 responses, control and LysM-cre Socs3fl/fl mice were injected with thioglycollate and treated with IgG or α-IL-6. As predicted, thioglycollate-elicited macrophages from LysM-cre Socs3fl/fl mice demonstrated impaired IL-12p40 production but this effect was reversed when these mice were treated with neutralizing antibodies to IL-6 (Figure 7E). Overall, these data suggest that IL-6 is an inhibitor of IL-12 production during toxoplasmosis and that LysM-cre Socs3fl/fl mice are hypersensitive to these effects.
Figure 7. Administration of IL-12 or neutralizing antibodies to IL-6 reduces parasite burdens and overall inflammation in LysM-cre Socs3fl/fl mice.

In vivo neutralization of IL-6 reduces acute mortality of infected LysM-cre Socs3fl/fl mice that correlates with reduced parasite burdens and indicators of Th1 inflammation. (A–D) 200 µg/mouse of rat IgG or α-IL-6 was administered i.p. to 5 mice per group on the day preceding infection as well as 2 days post infection with 104 Pru parasites. (A) Mice were sacrificed at day 8 p.i. and IL-6 was measured in the peritoneum to confirm neutralization. (B) Survival of infected mice was performed on 5 mice per group and carried out to 40 days. (C) The percentage of infected cells in the peritoneal cavity was measured similarly to Figure 2C and described in the methods. (D) IL-12p40 and IFN-γ production was also measured in the peritoneum as described in Figure 5 and the methods. (E) Thioglycollate-elicited macrophages were used to determine if a-IL-6 treatments could restore IL-12 production from LysM-cre Socs3fl/fl macrophages. IL-12p40 was measured by ELISA from 24 hr cultures of 2.5×105 thioglycollate-elicited macrophages isolated from the peritoneal cavity on day 4 following the antibody regimen described above. Experiments were repeated twice. To test whether addition of IL-12 could rescue lethality in LysM-cre Socs3fl/fl mice, 200 ng of IL-12 was injected on days 0,1,2 and 3 post-infection with 104 Pru for mice sacrificed on day 4 and days 0–5 for mice sacrificed on day 7. (F) Mice were infected and monitored for signs of mortality and 7F depicts a modified survival curve for control and LysM-cre Socs3fl/fl mice given PBS or IL-12. Day 7 was chosen as the day for sacrifice as only untreated LysM-cre Socs3fl/fl mice exhibited behavior indicating imminent mortality (i.e. hind-limb paralysis, malaise, ocular opacity, etc.). (G) The peritoneum of mice was washed and cells were centrifuged onto slides for counts of infected cells as previously described. Results are indicative of two independent experiments with at least 3 mice per group. (H) Liver sections were taken and DNA was isolated as previously described. Real Time analysis was performed on parasite DNA in liver sections from control and LysM-cre Socs3fl/fl mice at the given time points and measured against a standard curve. (I,J) Peritoneal cells from day 4 LysM-cre Socs3fl/fl mice, either treated with IL-12 or PBS were measured by flow cytometry for the presence of T cells, NK cells, monocytes, and neutrophils and depicted. Error bars +/− SD. Statistics were performed using Student’s t tests and results are representative of two independent experiments.
To determine if LysM-cre Socs3fl/fl mice could survive challenge when provided with exogenous IL-12, mice were infected and given 200 ng of IL-12 i.p. for 5 days. By day 7, control LysM-cre Socs3fl/fl mice were moribund associated with an 8% loss in body weight while IL-12 treated LysM-cre Socs3fl/fl mice appeared healthy and lost significantly less weight (4%, that was comparable to infected wild type controls). Further analysis on days 4 and 7 showed that IL-12 treatment resulted in a dramatic reduction in parasite loads in the peritoneum and liver (Figure 7F,G) to levels comparable to wild type controls (data not shown). This was accompanied by a reduction in the overall numbers of neutrophils, monocytes, T cells, and NK cells at day 4 (Figure 7H,I and data not shown) and reduced levels of pro-inflammatory cytokines (Figure S4). Thus, treatment of infected LysM-cre Socs3fl/fl mice with IL-12 is sufficient to overcome the inhibitory effects mediated by IL-6.
Discussion
Several reports have highlighted the ability of T. gondii to induce the phosphorylation of STAT3 associated with the ability to suppress cytokine production (Butcher et al., 2005b; Saeij et al., 2007). This phenomena has been linked to the activity of a parasite kinase, ROP16, that is injected into host cells where it can directly phosphorylate STAT3 (Saeij et al., 2007; Yamamoto et al., 2009). How these events limit cytokine production is unclear but the observation that T. gondii also upregulates SOCS3, a well characterized target of STAT3 linked to inhibition of cytokine signaling, suggested a possible mechanism. However, in vitro studies revealed that SOCS3 was not required for T. gondii to limit macrophage cytokine production. Rather, the use of the LysM-cre Socs3fl/fl mice revealed that SOCS3 was essential for appropriate innate responses mediated by neutrophils and monocyte/macrophages, required for the control of T. gondii.
While little is known about the role of SOCS3 in resistance to infection, the ability of this protein to limit cytokine signaling through the receptor gp130 has been described in detail (Murray, 2007). Thus, while cytokines such as IL-6 and IL-10 can activate STAT3 and upregulate SOCS3, this inhibitory protein binds directly to the gp130 subunit of the IL-6 receptor complex but not the IL-10R and so SOCS3 acts to inhibit IL-6 but not IL-10 signaling (El Kasmi et al., 2006; Yasukawa et al., 2003). However, our data show that the absence of SOCS3 resulted in enhanced T. gondii-induced STAT3 signaling. This suggests that either the ROP16 secreted by T. gondii following infection associates with the gp130 subunit and is inhibited by SOCS3 (similarly to an IL-6 stimulus) or that SOCS3 has an additional mechanism to limit phosphorylation of STAT3. Thus, the ROP16-mediated mechanism by which T. gondii activates STAT3 may be more complex than just direct phosphorylation of this substrate.
The significance of the inhibitory action of IL-6 has been documented in reports that in the absence of SOCS3, IL-6 induces a sustained STAT3 signal similar to that of IL-10 and becomes a potent inhibitor of macrophage production of TNF-α and IL-12 (Yasukawa et al., 2003). Indeed, addition of α-IL-6 to cultures of SOCS3-deficient macrophages stimulated with IFN-γ/LPS resulted in enhanced IL-12p40 production. Consistent with this observation, the neutralization of IL-6 in the LysM-cre Socs3fl/fl mice allowed these mice to control the acute challenge with T. gondii and develop the protective responses required for long-term resistance to this infection. Thus, it appears that in the absence of SOCS3, the IL-6 produced during infection has a profound inhibitory effect on the innate response similar to the observation that LysM-cre Socs3fl/fl mice have increased resistance to sepsis (Yasukawa et al., 2003).
One of the themes that emerged from these studies was that IL-6, even in wild type cells, has significant but specific inhibitory effects. For example, when wild type macrophages were treated with IL-6, their ability to produce IL-12p40 was selectively inhibited while effector molecules such as nitric oxide were unaffected. Additionally, when control mice were treated with neutralizing antibodies to IL-6, their ability to control early parasite replication was enhanced suggesting that IL-6 is a natural inhibitor of the innate mechanisms that contribute to early control of T. gondii. IL-6, similarly to IL-10, can antagonize the ability of IFN-γ-activated macrophages to limit the replication of several intracellular organisms (Beaman et al., 1994; Bermudez et al., 1992; Hatzigeorgiou et al., 1993). Based on these in vitro studies, the ability of IL-6 to antagonize anti-microbial activities might have explained why LysM-cre Socs3fl/fl mice were unable to control parasite burden. However, the observation that the relative percentage of infected macrophages and neutrophils is identical between control and LysM-cre Socs3fl/fl mice indicates that the absence of SOCS3 does not result in a cell intrinsic defect in the ability to control T. gondii. Thus, SOCS3-deficient macrophages can control parasite replication adequately when provided appropriate signals and so the enhanced susceptibility observed in the absence of SOCS3 is most consistent with an early defect in the innate response.
While this study has focused on the contribution of IL-6 to the regulation of inflammatory responses during toxoplasmosis, this cytokine is also important in many other settings where pathogens appear to benefit from the inhibitory effects of IL-6. For example, IL-6 promotes growth of Leishmania, T. gondii, and Mycobacterium spp. (Beaman et al., 1994; Bermudez et al., 1992; Hatzigeorgiou et al., 1993; Murray, 2008) and has been associated with enhanced lytic replication of Human Herpes Virus-8 (HHV-8) and exacerbated disease in patients coinfected with HIV (Uldrick et al., 2010). Moreover, HHV-8 encodes a viral IL-6 gene that has been shown to impair neutrophil recruitment and promote viral growth in vitro (Fielding et al., 2005; Sun et al., 1998). From a host perspective, resistance to many pathogens is characterized by the need to balance the development of a strong cell mediated immune response with the ability to limit collateral damage caused by the immune system. Indeed, there are many examples of inhibitory pathways that prevent infection-induced immune hyperactivity and the SOCS proteins also limit signals that promote excessive inflammation. SOCS1 is best described as a regulator of Type I and II IFN-mediated phosphorylation of STAT1 (Fenner et al., 2006; Gingras et al., 2004) and has been shown to diminish IFN-γ responsiveness to T. gondii, L. major, and Hepatitis C Virus (Alexander et al., 1999; Vlotides et al., 2004; Zimmermann et al., 2006). However, while SOCS3 can regulate IFN responses, its main role during toxoplasmosis, and possibly other intracellular pathogens, is to limit the anti-inflammatory actions of IL-6. Thus, SOCS3 differentiates itself from other SOCS family members and its signaling pathway can be considered among those that promote appropriate and effective Th1 immunity.
Experimental procedures
Mice and infections
LysM-cre Socs3fl/fl mice were generated as previously described (Croker et al., 2003). C57/Bl6 mice were obtained from Taconic Farms Inc (Germantown, NY). C57/Bl6, Socs3fl/fl, and LysM-cre Socs3fl/fl mice were bred in the University Laboratory Animal Resources facilities at the University of Pennsylvania. All procedures were performed in accordance to the guidelines of the University of Pennsylvania Institutional Animal Care and Use Committee. RH and Prugniaud (Pru) parasites were grown in Human Foreskin Fibroblast (HFF) monolayers cultured with D10 media (DMEM, 25% M199 Supplement, 10% Fetal Bovine Serum, 0.5% penicillin/streptomycin (10,000 µg/mL), 0.5% gentamycin (50 µg/mL)) as previously described (Wille et al., 2004). RH-Cre-Cherry parasites were obtained from John Boothroyd and passaged as described (Koshy et al., 2010). Injections of 200 µg of α-IL-6 (BioXCell, West Lebanon, NH) or rat IgG (Sigma, St. Louis, MO) were administered i.p. at days −1 and 2 post-infection. Injections of 200 ng of IL-12 (Peprotech, Rocky Hill, NJ) were administered on the day of the infection as well as for the first 3 or 5 days prior to sacrificing the mice on day 4 or 7 respectively. Thioglycollate injections were performed i.p. with 0.5 mL of 4% Brewer’s thioglycollate medium (Sigma) and peritoneal exudates cells (PECs) were harvested on day 4 as described (Wille et al., 2004). 2.5×105 thioglycollate-elicited macrophages were then plated for 24 hrs in 200 µl total volume and supernatants were used for detection of IL-12p40 production by ELISA.
Immunoblotting
Bone-marrow derived macrophages (BMMϕ) were grown in DMEM containing 1% sodium pyruvate, 1% penicillin/streptomycin, 10% FBS, 30% L929 cell supernatant, and 1% HEPES (Shapira et al., 2005). 1×106 BMMϕ were cultured in 96 well plates overnight and then infected with 5:1 Pru. Lysates were prepared using TNT buffer (0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20) combined with complete protease inhibitors (Roche, Mannheim, Germany) at 1:100 for 20 minutes on ice and then stored at −20°C. Samples were electrophoresed and transferred to Immobulon-P membrane (Millipore, Bedford, MA) and blocked for 1 hr in 5% nonfat dry milk and probed overnight with mouse monoclonal antibodies to SOCS3 (Invitrogen, Carlsbad, CA), β-actin, pSTAT3, total STAT3, pSTAT1, total STAT1, pERK, and total ERK (Cell Signaling, Danvers, MA) and IRGM1, IRGM2, IRGM3, IRGD, IRGA6, or IRGB6 (Santa Cruz Biotechnology, Santa Cruz, CA). Blots were washed in TBS-Tween and probed with α-mouse or α-rabbit secondary antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) at 1:10,000 for 1 hr at room temperature. Blots were washed and developed with enhanced chemiluminescence and exposed to film.
Confocal immunofluorescence microscopy
5×105 BMMϕ were isolated and cultured in 96 well plates and infected at a multiplicity of infection (MOI) of 1:1 RH-Tomato or RH-Cre-Cherry parasites. Cells were blocked in 5% BSA, fixed with 2% paraformaldehyde, incubated with monoclonal α-SOCS3 (Invitrogen) overnight, washed, and detected by incubation with biotinylated anti-mouse antibodies and streptavidin conjugated to Alexafluor 488 (Invitrogen). Cells were then washed and visualized using the Nikon Eclipse E600 microscope (Melville, NY) equipped with a Photometrics Cool Snap EZ CCD camera (Tucson, AZ). Results are representative of two independent experiments.
RNA isolation and quantitative Real Time PCR
RNA was isolated using the RNeasy kit (Qiagen) according to manufacturer’s instructions and reverse transcribed into cDNA using Superscript II (Invitrogen) using standard protocols. Quantitative PCR was performed with customized primer sets for Socs3 (Forward 5’-ACCAGCGCCACTTCTTCACG-3’, Reverse 5’-GTGGAGCATCATACTGATCC-3’) and Socs1 (Forward 5’-ACTTCTGGCTGGAGACCTCA-3’, Reverse 5’-CCCAGACACAAGCTGCTACA-3’) using Power SYBR green reagents and an AB7500 Fast Real Time PCR thermal cycler (Applied Biosystems, Warrington UK). The values for Socs3 were normalized to β-Actin (Qiagen, Germantown, MD). Parasite DNA levels were measured by real time PCR of the T. gondii B1 gene using DNA isolated from tissue samples using the High Pure PCR Template Purification Kit (Roche) and SYBR Green PCR Master mix (Applied Biosystems) in an AB7500 fast real-time PCR machine (Applied Biosystems) using published conditions (Wilson et al., 2005).
Cytokine and flow cytometric analysis
Splenocytes were washed and resuspended in complete RPMI 1640 (Invitrogen) containing 10% FBS (Serum Source International), 1% penicillin/streptomycin (Invitrogen), 1 mM sodium pyruvate (Mediatech, Manassas, VA), 2 mM L-glutamine (Mediatech), 0.1% 2-mercaptoethanol (Invitrogen) before being plated at 1×106 cells per well in a volume of 200 µl in 96-well plates (Costar, New York). Cells were stimulated with or without soluble Toxoplasma antigen (STAg) at 50 µg/mL, or α-CD3 at 1µg/mL (eBioscience) for 24 hr at 37 C in 5% CO2. Peritoneal cells were isolated as previously described (Wilson et al., 2005) and cell-free peritoneal wash fluid was used for cytokine analysis. Concentrations were normalized to the amount of peritoneal fluid recovered from each mouse. Blood was collected via cardiac puncture. Concentrations of IFN-γ, IL-12p40, IL-17, IL-10, and IL-6 were determined by ELISA using antibodies from eBioscience (San Diego, CA). Freshly isolated PECs from infected mice were incubated with Fc block (0.1 µg/mL 24G2 antibody) for 15 min prior to surface staining with Ly6C–PerCP 5.5 (HK1.4), CD11b–FITC (M1/70), F4/80-eFluor 780 (BM8), GR1-PECy7 (RB6–8C5), CD86-APC (16-10A1), CD80-PE (GL1), NK1.1-PE (PK136), CD4- eFluor. 780 (RM4–5), CD8-PerCP 5.5 (53-6.7), CD19-PECy7 (eBio1D3 (1D3)), CD11c–APC (N418), CD62L–APC (MEL-14), and CD44-PE (IM7) (eBioscience, San Diego, CA), MHC Class II I-A/I-E (Biolegend, M5/114.15.2), CD3ε-Pacific Blue (500A2), and CD19-Pacific Blue (1D3) (BD Biosciences). For intracellular cytokine staining, 1×106 PECs were plated in RPMI 1640 and stimulated with PMA (50 ng/mL) and ionomycin (500 ng/mL) in the presence of brefeldin A (10 µg/mL) (Sigma) for 4 hrs at 37 °C with 5% CO2 unless otherwise noted. The cells were fixed with 2% paraformaldehyde following surface stain. (Electron Microscopy Science, Hatfield, PA) for 10 min. For intracellular staining, the cells were permeabilized with 0.5% saponin (Sigma, St. Louis, MO) and stained with anti-IFN-γ-APC (XMG1.2) or IL-12p40-APC (17.8) (eBioscience). All flow cytometry was performed on a FACsCanto using FACsDIVA 6.0 software (BD Biosciences, San Jose, CA). Analysis was performed using FloJo software (Treestar Inc., Ashland, OR).
In vitro killing assay and inhibition assays
5×105 BMMϕ were harvested and cultured in 96 well plates and pretreated overnight with media alone or IFN-γ (R&D Systems, Minneapolis, MN) and TNF-α (eBioscience) at indicated concentrations. Cells were then infected with Pru at 1:1 and incubated overnight. Cells were then centrifuged onto microscope slides and the number of infected macrophages was counted. Nitrite analysis was performed using the Griess protocol as previously described (Stuehr and Nathan, 1989). For parasite cytokine-inhibition assays, 2×105 BMMϕ were plated and infected with either RH or Pru parasites at an MOI of 1:1 for 1 hr prior to LPS stimulation (100 ng/mL, Sigma) for 24 hours. For IL-6 inhibition assays, BMMϕ were stimulated with IFN-γ (10ng/mL) and LPS (10ng/mL) ± IL-6 at 10 fold increments beginning at 0.001 ng/mL (eBioscience) or α-IL-6 (100 ng/mL, BioXcell) and IL-12p40 and TNF-α were assayed by ELISAs (eBioscience).
Statistical analysis
Statistical analysis was conducted on normally distributed data with Students’ t test or One-Way ANOVA, and correlations were calculated with Prism software (Graphpad Software).
Highlights.
Neutrophils and monocytes require SOCS3 for resistance to T. gondii.
IL-6 inhibits IL-12 production from neutrophils and macrophages.
SOCS3 limits IL-6 signals, indirectly promoting IL-12 production.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants to CAH and CMG. CAH: State of Pennsylvania and NIH AI42334. CMG: University of Pennsylvania NIHTG T32 GM07229
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Andrade RM, Portillo JA, Wessendarp M, Subauste CS. CD40 signaling in macrophages induces activity against an intracellular pathogen independently of gamma interferon and reactive nitrogen intermediates. Infection and immunity. 2005;73:3115–3123. doi: 10.1128/IAI.73.5.3115-3123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaman MH, Hunter CA, Remington JS. Enhancement of intracellular replication of Toxoplasma gondii by IL-6. Interactions with IFN-gamma and TNF-alpha. Journal of immunology. 1994;153:4583–4587. [PubMed] [Google Scholar]
- Bermudez LE, Wu M, Petrofsky M, Young LS. Interleukin-6 antagonizes tumor necrosis factor-mediated mycobacteriostatic and mycobactericidal activities in macrophages. Infection and immunity. 1992;60:4245–4252. doi: 10.1128/iai.60.10.4245-4252.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher BA, Greene RI, Henry SC, Annecharico KL, Weinberg JB, Denkers EY, Sher A, Taylor GA. p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infection and immunity. 2005a;73:3278–3286. doi: 10.1128/IAI.73.6.3278-3286.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher BA, Kim L, Johnson PF, Denkers EY. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-kappa B. Journal of immunology. 2001;167:2193–2201. doi: 10.4049/jimmunol.167.4.2193. [DOI] [PubMed] [Google Scholar]
- Butcher BA, Kim L, Panopoulos AD, Watowich SS, Murray PJ, Denkers EY. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-αlpha in host macrophages. Journal of immunology. 2005b;174:3148–3152. doi: 10.4049/jimmunol.174.6.3148. [DOI] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nature immunology. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Dalpke A, Heeg K, Bartz H, Baetz A. Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology. 2008;213:225–235. doi: 10.1016/j.imbio.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Denkers EY, Gazzinelli RT. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunay IR, Fuchs A, Sibley LD. Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infection and immunity. 2010;78:1564–1570. doi: 10.1128/IAI.00472-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan CE, Sukhumavasi W, Bierly AL, Denkers EY. Understanding the multiple functions of Gr-1(+) cell subpopulations during microbial infection. Immunologic research. 2008;40:35–48. doi: 10.1007/s12026-007-0061-8. [DOI] [PubMed] [Google Scholar]
- El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y, et al. General nature of the STAT3-activated anti-inflammatory response. Journal of immunology. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, Sheehan K, Hilton DJ, Alexander WS, Hertzog PJ. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nature immunology. 2006;7:33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- Fielding CA, McLoughlin RM, Colmont CS, Kovaleva M, Harris DA, Rose-John S, Topley N, Jones SA. Viral IL-6 blocks neutrophil infiltration during acute inflammation. Journal of immunology. 2005;175:4024–4029. doi: 10.4049/jimmunol.175.6.4024. [DOI] [PubMed] [Google Scholar]
- Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. The Journal of biological chemistry. 2004;279:54702–54707. doi: 10.1074/jbc.M411043200. [DOI] [PubMed] [Google Scholar]
- Hatzigeorgiou DE, He S, Sobel J, Grabstein KH, Hafner A, Ho JL. IL-6 down-modulates the cytokine-enhanced antileishmanial activity in human macrophages. Journal of immunology. 1993;151:3682–3692. [PubMed] [Google Scholar]
- Khan IA, Matsuura T, Kasper LH. Interleukin-12 enhances murine survival against acute toxoplasmosis. Infection and immunity. 1994;62:1639–1642. doi: 10.1128/iai.62.5.1639-1642.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Fouts AE, Boothroyd JC. Toxoplasma gondii dysregulates IFN-gamma-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. Journal of immunology. 2007;178:5154–5165. doi: 10.4049/jimmunol.178.8.5154. [DOI] [PubMed] [Google Scholar]
- Koshy AA, Fouts AE, Lodoen MB, Alkan O, Blau HM, Boothroyd JC. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nature methods. 2010;7:307–309. doi: 10.1038/nmeth.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nature immunology. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- Lieberman LA, Banica M, Reiner SL, Hunter CA. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. Journal of immunology. 2004;172:457–463. doi: 10.4049/jimmunol.172.1.457. [DOI] [PubMed] [Google Scholar]
- Luder CG, Walter W, Beuerle B, Maeurer MJ, Gross U. Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1alpha. Eur J Immunol. 2001;31:1475–1484. doi: 10.1002/1521-4141(200105)31:5<1475::AID-IMMU1475>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard JC. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS pathogens. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. Journal of leukocyte biology. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- Murray HW. Accelerated control of visceral Leishmania donovani infection in interleukin-6-deficient mice. Infection and immunity. 2008;76:4088–4091. doi: 10.1128/IAI.00490-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ. The JAK-STAT signaling pathway: input and output integration. Journal of immunology. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Robben PM, Mordue DG, Truscott SM, Takeda K, Akira S, Sibley LD. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. Journal of immunology. 2004;172:3686–3694. doi: 10.4049/jimmunol.172.6.3686. [DOI] [PubMed] [Google Scholar]
- Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharton-Kersten TM, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med. 1997;185:1261–1273. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira S, Harb OS, Margarit J, Matrajt M, Han J, Hoffmann A, Freedman B, May MJ, Roos DS, Hunter CA. Initiation and termination of NF-kappaB signaling by the intracellular protozoan parasite Toxoplasma gondii. Journal of cell science. 2005;118:3501–3508. doi: 10.1242/jcs.02428. [DOI] [PubMed] [Google Scholar]
- Shapira S, Speirs K, Gerstein A, Caamano J, Hunter CA. Suppression of NF-kappaB activation by infection with Toxoplasma gondii. J Infect Dis. 2002;185 Suppl 1:S66–S72. doi: 10.1086/338000. [DOI] [PubMed] [Google Scholar]
- Sher A, Collazzo C, Scanga C, Jankovic D, Yap G, Aliberti J. Induction and regulation of IL-12-dependent host resistance to Toxoplasma gondii. Immunologic research. 2003;27:521–528. doi: 10.1385/IR:27:2-3:521. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Nathan CF. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldrick TS, Wang V, O’Mahony D, Aleman K, Wyvill KM, Marshall V, Steinberg SM, Pittaluga S, Maric I, Whitby D, et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin Infect Dis. 2010;51:350–358. doi: 10.1086/654798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlotides G, Sorensen AS, Kopp F, Zitzmann K, Cengic N, Brand S, Zachoval R, Auernhammer CJ. SOCS-1 and SOCS-3 inhibit IFN-alpha-induced expression of the antiviral proteins 2,5-OAS and MxA. Biochem Biophys Res Commun. 2004;320:1007–1014. doi: 10.1016/j.bbrc.2004.06.051. [DOI] [PubMed] [Google Scholar]
- Wille U, Nishi M, Lieberman L, Wilson EH, Roos DS, Hunter CA. IL-10 is not required to prevent immune hyperactivity during memory responses to Toxoplasma gondii. Parasite immunology. 2004;26:229–236. doi: 10.1111/j.0141-9838.2004.00704.x. [DOI] [PubMed] [Google Scholar]
- Wilson EH, Wille-Reece U, Dzierszinski F, Hunter CA. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. Journal of neuroimmunology. 2005;165:63–74. doi: 10.1016/j.jneuroim.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, Kubo E, Ito H, Takaura M, Matsuda T, et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med. 2009;206:2747–2760. doi: 10.1084/jem.20091703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nature immunology. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. Journal of immunology. 2009;182:3775–3781. doi: 10.4049/jimmunol.0804190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Murray PJ, Heeg K, Dalpke AH. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. Journal of immunology. 2006;176:1840–1847. doi: 10.4049/jimmunol.176.3.1840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.