

Abstract
An increasingly popular and promising field in functional proteomics is the isolation of proteome subsets based on small molecule-protein interactions. One platform approach in this field are Capture Compounds that contain a small molecule of interest to bind target proteins, a photo-activatable reactivity function to covalently trap bound proteins, and a sorting function to isolate captured protein conjugates from complex biological samples for direct protein identification by liquid chromatography/mass spectrometry (nLC-MS/MS). In this study we used staurosporine as a selectivity group for analysis in HepG2 cells derived from human liver. In the present study, we combined the functional isolation of kinases with different separation workflows of automated split-free nanoflow liquid chromatography prior to mass spectrometric analysis. Two different CCMS setups, CCMS technology combined with 1D LC-MS and 2D LC-MS, were compared regarding the total number of kinase identifications. By extending the chromatographic separation of the tryptic digested captured proteins from 1D LC linear gradients to 2D LC we were able to identify 97 kinases. This result is similar to the 1D LC setup we previously reported but this time 4 times less input material was needed. This makes CCMS of kinases an even more powerful tool for the proteomic profiling of this important protein family.
1. Introduction
Kinases play a pivotal role in cell signaling and human development and diseases, and they are, therefore, a major focus of proteomics research. The low abundance, particularly of protein kinases, makes direct detection of kinases from biological samples a challenging feat. Recently, techniques based on small molecule-protein interactions for the functional enrichment of protein families have been developed [1, 2]. We recently reported the functional isolation of kinases from complex biological mixtures using a kinase-specific trifunctional Capture Compound (Figure 1) with the broadband kinase inhibitor staurosporine as the selectivity function [1].
Figure 1.
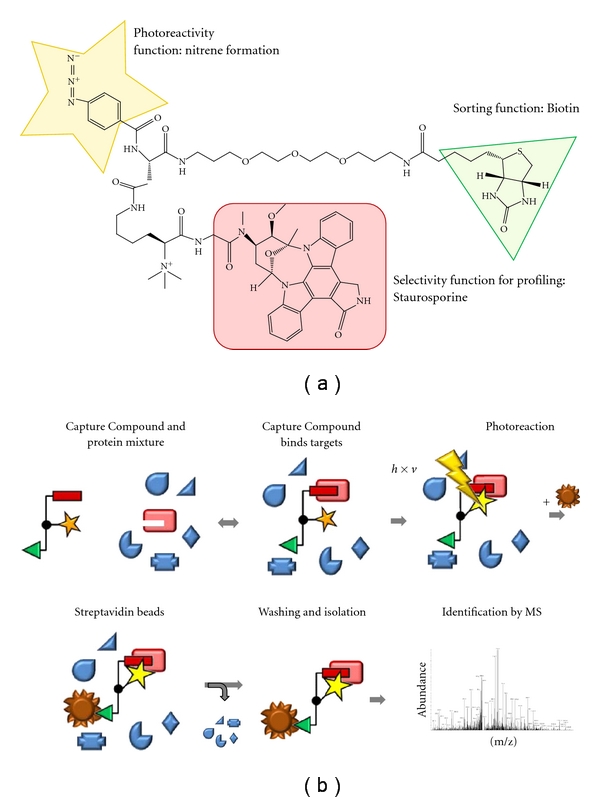
The molecular structure of staurosporine Capture Compound (a) and schematic representation of the capture process (b).
The general design of Capture Compounds consists of three main functionalities (Figure 1(a)): (a) a selectivity function, in this case the ATP mimetic kinase inhibitor staurosporine, (b) an adjacent photoreactive functionality for photocrosslinking, and (c) a sorting function, for example, biotin, for the isolation of captured proteins [3–5]. In order to determine the kinases interacting with staurosporine in a given biological sample, first, incubation leads to an equilibrium binding based on affinity between the Capture Compound and the proteins (Figure 1(b)). This equilibrium is then frozen by a photochemical reaction leading to the formation of a covalent bond between the Capture Compound and the interacting proteins. Addition of streptavidin-coated magnetic beads allows isolation of the CC-protein conjugates via the biotin sorting function of the CC. Isolated proteins are tryptically digested on bead, and the supernatant containing tryptic peptides is then identified by nanoflow liquid chromatography/tandem mass spectrometry (nLC-MS/MS). To discriminate between background proteins and specific interaction partners of the selectivity group, a parallel experiment is carried out in which the lysate is first preincubated with staurosporine (competition experiment). Proteins only present in the capture assay, but not (or significantly weaker) in the competition control, are specific interactors of staurosporine.
Previously, we enriched in one CCMS from 400 μg of cell lysate as starting material approximately 20 kinases in a single run of the experiment. The number of proteins annotated as kinases was increased by repetitive performance of the capture experiments followed by 1D nLC-MS/MS runs. However, this was only possible by increasing the amount of required biological material at the same time [1]. Due to the identification of additional unspecific proteins during the capture experiment, we asked ourselves whether the number of specifically enriched kinases can be raised by changing chromatographic conditions to either a longer linear gradient or even to a combination of two different chromatographic separation methods. Multidimensional chromatography was applied to our CCMS technology to get more information about the detection limit of our CCMS technology. In this study, we demonstrate that a much higher number of kinases is specifically captured and identified by combining CCMS with 2D LC in comparison to 1D LC and keeping the same low amount of input material. Moreover, the confidence with which each kinase could be identified was increased by getting a higher number of spectral counts per protein. Performing relative quantification based on spectral counts of identified proteins from assay versus competition experiments reflects directly the detection limit of our CCMS technology. The potential of the CCMS technology is significantly enhanced by combining capture experiments with 2D LC-MS/MS analysis.
2. Materials and Methods
The HepG2 cell line was purchased from the German collection of microorganisms and cell lines (DSMZ) and growth and lysate preparation was contracted to InVivo BioTech (Berlin, Germany). Cells were lysed in cell opening buffer (6.7 mM MES, 6.7 mM NaOAc, 6.7 mM HEPES, 1 mM EDTA, 10 mM β-mercaptoethanol, 200 mM NaCl, pH 7.5, Roche Complete Protease Inhibitor EDTA free) and homogenized three times in a French press (>50,000 and <150,000 kPa). Debris was removed by centrifugation. To remove free ATP as well as other small molecules from the supernatant, the HepG2 lysate was dialysed with a molecular weight cut off of 6–8 kDa overnight. Protein concentrations were determined by the Bradford assay [2]. Naturally biotinylated proteins were removed from the HepG2 cell lysates by preclearing with streptavidin-coated magnetic beads.
Capture experiments were carried out as previously described [1]. Briefly, in one capture assay or competition control, respectively, 400 μg protein of precleared HepG2 cell lysate was supplemented with 5x capture buffer. To the competition reaction, 20 μL of 1 mM staurosporine competitor was added, while to the capture reaction an equal volume of water was added. After incubation for 2 h at 4°C, the CC was added (10 μL of a 50 μM solution in water) giving a final reaction volume of capture assay and competition control of 100 μL, respectively. Samples were then irradiated for 10 min at 4°C at using the caproBox, a device for the controlled irradiation of biological samples. The maximal irradiation is at λ = 312 nm, and a UV filter with a cutoff below λ = 330 nm was applied. The covalently cross-linked protein-CC conjugates were then incubated with streptavidin coated magnetic beads. The beads were washed successively with wash buffer, 80% acetonitrile/water, and water. Washing steps were carried out using the caproMag, a device for handling magnetic beads. Isolated proteins were tryptically digested as described (Luo et al. [6]). We carried out two sets of biological replicates, each set consisting of 3 capture assay, and 3 competition controls. The three replicates were then analyzed either by 1D LC-MS/MS (90 min gradient), 1D LC-MS/MS (180 min gradient), or 2D LC-MS/MS.
Tryptic digests were analyzed on a LTQ-Orbitrap XL instrument online coupled to an EASY-nLC system (ThermoScientific, Bremen, Germany). 1D chromatographic peptide separation was performed on a reversed phase (RP) precolumn (5 cm, 100 μm, 100 Å) connected to an RP analytical column (15 cm, 75 μm, 100 Å) using either a 90 min or 180 min gradient (0–35% acetonitrile, 0.1% formic acid). In the 2D LC approach, samples were first loaded onto a biphasic strong cation-exchange SCX/RP precolumn (3.5 cm, 150 μm) and eluted with 5 successive salt steps (50 mM–100 mM–150 mM–250 mM–500 mM ammonium acetate (8 μL each)) onto a RP for subsequent peptide separation using a 120 min linear gradient. Mass spectrometric detection was performed in the data-dependent mode allowing to automatically switch between Orbitrap-MS and LTQ-MS/MS (MS2) acquisition. The most intense ions (up to five, depending on signal intensity) were sequentially isolated for fragmentation in the linear ion trap using collisionally induced dissociation (CID) at a target value of 10,000 charges. Target ions, already mass selected for CID, were dynamically excluded for the duration of 60 s. The minimal signal required for MS2 was 500 counts. An activation q of 0.25 and an activation time of 30 min were applied for MS2 acquisitions.
All MS/MS data were analyzed using SEQUEST implemented in Bioworks Browser 3.3.1 SP1 (Thermo Fisher Scientific) and X!Tandem (http://www.thegpm.org/; version 2007.01.01.1, Proteome Software, Portland, Ore, USA). Automated database searching against the human UniProtKB/Swiss-Prot database (release 2010_07 contains 517 802 sequence entries and human entries) was performed with 5 ppm precursor tolerance, 1 amu fragment ions tolerance, and full trypsin specificity allowing for up to 2 missed cleavages. Methionine oxidation was used as fixed and phosphorylation as variable modification in the database search.
Spectra were annotated via human UniProtKB/Swiss-Prot database and analyzed using the software Scaffold (version Q+ 3_00_02, Proteome Software, Portland, Ore, USA). First Proteins were selected if the peptide probability was ≥95% assigned by the Peptide Prophet algorithm [3], and the protein was identified by at least 2 unique peptides. The estimated false discovery rate of peptide identifications was determined using the reversed protein database approach and was ≤1%. Subsequently, the spectra of kinases identified by only one unique peptide with ≥95% were manually validated (Supplementary Figures available at doi: 10.1155/2011/850589). Gene ontology annotation was carried out for all identified proteins via BioMart on the Ensembl web site (http://www.ensembl.org) using the build Ensembl 58 Homosapiens GRCh 37. All gene ontologies were then manually validated via SwissProt, and only proteins annotated as kinases in both databases were accepted.
3. Results
We carried out CCMS experiments in HepG2 whole cell lysates and compared the results combined with different LC-MS techniques. We identified the isolated kinases by performing gene ontology mapping of all identified proteins and then searching in the term “molecular function” for kinases. The number of kinases identified in the various setups and the number of the corresponding unique peptides and unique spectra are illustrated as Venn diagrams in Figure 2. Across all experiments, 101 kinases were identified; however, three of these were only detected in the competition experiments and therefore rejected. Out of the remaining 98 kinases, only 7 kinases were exclusively identified in 1D LC experiments. The number of identified kinases in the capture assays increased from 23 to 39 to 91 out of the total number of proteins from 386 to 617 to 1274 when comparing 90 min gradient 1D LC to 180 min gradient 1D LC to 2D LC experiment (Table 1 and Supplementary Table). This represents at least 38% of the 253 kinases that have been reported to be targeted by staurosporine [4] and not all of which may be expressed in HepG2 cells. The identified kinases cover a broad functional range and are representative for the different classes of kinases.
Figure 2.
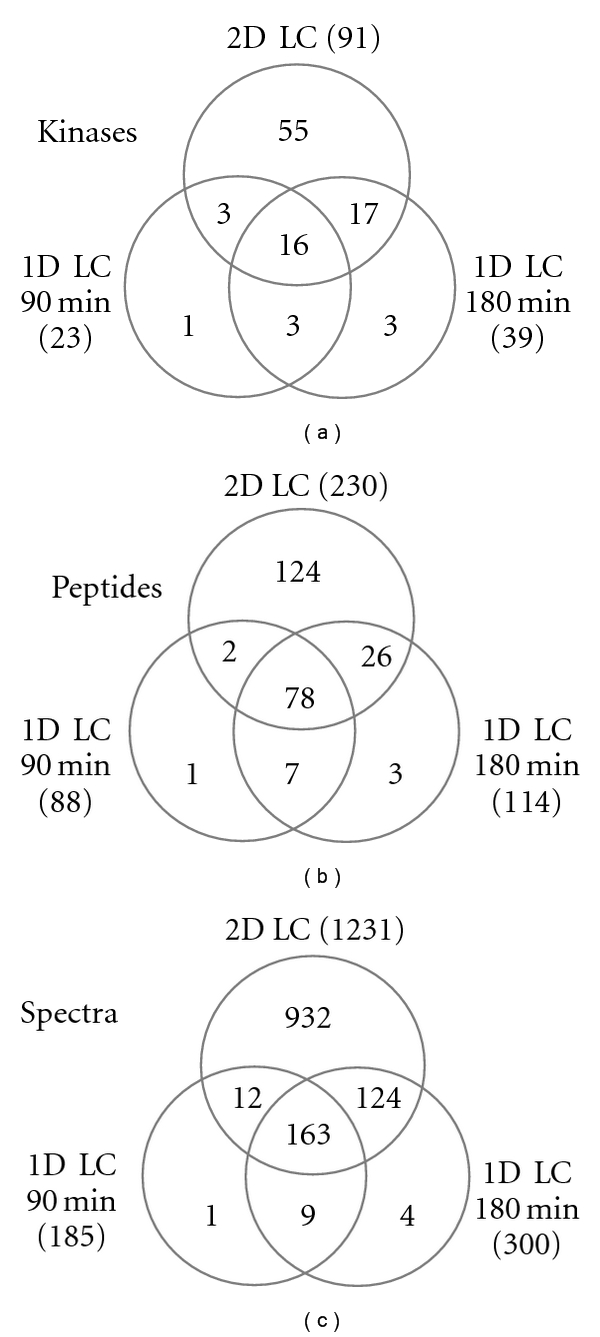
Venn-diagrams of kinases (with ≥2 peptides) (a), peptides (b), spectra (c) derived from each two technical replicates: 1D LC-MS (90 and 180 min gradient and 2D LC-MS).
Table 1.
Proteins and kinases identified in the different LC-MS set-ups.
| Total number of identified proteins in capture assays (with ≥2 peptides) |
Kinases identified in capture assays (with ≥2 peptides) |
Number of competed kinases* (with ≥2 peptides) |
|
|---|---|---|---|
| 1D LC-MS 90 min | 386 (158) | 23 (10) | 20 (8) |
| 1D LC-MS 180 min | 617 (265) | 39 (19) | 31 (16) |
| 2D LC-MS | 1274 (489) | 91 (48) | 66 (32) |
*A kinase is considered to be competed if the number of spectral counts in the capture assay divided by number of spectral counts in the competition control is ≥2 or the number of spectral counts in the competition is = 0.
Among the 98 kinases identified the majority are members of the serine/threonine kinase family (36) followed by small molecule kinases (28). Furthermore, 34 kinases belonging to the tyrosine kinase family such as proto-oncogene kinases and dual-specific kinases were identified.
Comparison of the capture assays with the competition controls distinguished specific interactors of staurosporine from unspecific background. We defined an interaction to be specific if the protein was exclusively present, or with twice as many spectral counts in the capture assay than in the competition control (Table 1). Approximately, two-thirds of the kinases (66) identified in the 2D LC-MS runs were specifically enriched. The remaining kinases mainly have metabolic function such as bifunctional ATP-dependent dihydroxyacetone kinase (DHAK) or 6-phosphofructokinase (K6PL). These kinases are highly expressed and can also be detected in shotgun experiments without prior enrichment as previously shown [1].
Out of a total number of 1274 proteins, 144 non-kinases were also specifically enriched performing the 2D LC-MS; these can be mainly classified into three groups: ATP-binding proteins, detoxification enzymes, and nucleic acid-binding proteins. The majority is ATP-binding proteins, which is plausible, as staurosporine is an ATP mimetic kinase inhibitor. The most highly enriched non-kinase protein with a fold change of 2.8 between the assay and the competition analyzed by 2D LC-MS was the nucleic acid-binding FUBP2; the next following are the two ATP-binding proteins ACACA and PCCA. These non-kinase target proteins of staurosporine can shed light on additional pathways disturbed when using staurosporine as kinase inhibitor, for example, in cell-based assays.
4. Discussion
The results presented in this study demonstrate that combining CCMS technology with 2D LC-MS enables identification of enriched kinases with enhanced confidence and much lower amounts of input material. In our previous study [1], using 1D LC-MS, eight experiments were needed to obtain nearly as much enriched kinases, and consequently four times as many capture experiments (each set of assay and competition requiring approximately one workday) were needed requiring four times as much input material, while the overall MS analyses time remained comparable. There are alternative approaches for the comprehensive profiling of the kinome published: the most successful is the use of multi-inhibitor beads that address a very large range of kinases ([5], “kinobeads”). We were therefore also interested to use the stauro-CC in such a solid support approach. In previous studies [6, 7] using other Capture Compounds we also used a second experimental setup termed “On-bead CCMS configuration” and obtained results complementing the off-bead setup presented here. (on-bead: CCs are first immobilized on beads, then added to protein mixture and irradiated, off-bead: CCs are first added to protein mixture, irradiated and then isolated via magnetic beads). Using the stauro-CC in the on-bead configuration, however, no kinases were enriched and identified. Possibly the hydrophobic staurosporine selectivity function buries itself in the hydrophobic bead surface, or the beads disturb the photoreaction for this particular Capture Compound. In the case of the kinobeads, seven different kinase inhibitors are covalently attached to a solid support to create affinity chromatography material for kinase binding. With these kinobeads, 269 human kinases could be isolated from a total of six different cell lines and two human tissues. Up to 25 mg of total protein was used as input for the affinity pulldown experiments. Another successful study has been published by Oppermann et al. [8]. This study is based on kinase inhibitor affinity chromatography beads as well, in this case with a single inhibitor attached to a solid support. From sample sized 1.5 mg protein of lysate each, a total of 170 kinases were identified across three different-cultured cancer cell lines. Samples of affinity-purified target proteins were analyzed after fractionation via SDS-PAGE.
The Capture Compound approach has been optimized as a method for biological samples of limited abundance such as tissue biopsies. Therefore, our workflow is designed for relatively small volumes (such as 100 μL) containing moderate amounts of protein mixture (∼400 μg per assay). This is roughly a quarter of the material used by Oppermann et al., and less than 2% of the material used by Bantscheff et al. In this study, we were able to demonstrate using even less input material and still raise the overall number of specifically enriched proteins. The rationale behind that is that we want to establish our assay in an as robust and simple and straightforward way as possible.
5. Conclusions
Using staurosporine as selectivity function, a broad range of kinases belonging to different pathways could be identified. As a unique feature, the photoreactivity group forms a covalent bond between the Capture Compound and the captured kinases, so that even weak interactors of the selectivity function are covalently attached allowing stringent washing conditions. This allows endogenous low-affinity and low-abundant proteins to be isolated and identified by CCMS. Our results demonstrate that CCMS is a powerful new technology for the investigation of kinases, and its investigative power can be even enhanced when combined with two-dimensional liquid chromatography connected to a high scan speed mass spectrometer for a better separation and detection of the digested peptides resulting in a higher number of identified proteins and more confident results. Our findings open a perspective for CCMS as tool for charting kinases in the postgenome area and in biomarker discovery and research.
Supplementary Material
Supplementary Table: Detailed MS data of kinases (Kinases identified in the different LC-MS setu-ups (1D LC, 90 min gradient; 1D LC, 180 min gradient; 2D LC, 120 min gradient) in assay (A) and competition (C): unique peptides, spectral counts, and calculated fold change A/C based on spectral counts. Kinases belong either to the serine/threonine/tyrosine kinase family (S/T/Y) or small molecule kinases (small mol)).
Supplementary Figures: MS spectra of kinases identified by only one unique spectrum.
Acknowledgment
The authors thank Erik Duelsner for graphical design support. O. Graebner and J. J. Fischer contributed equally to the paper.
References
- 1.Fischer JJ, Graebner OY, Dalhoff C, et al. Comprehensive identification of staurosporine-binding kinases in the hepatocyte cell line HepG2 using capture compound mass spectrometry (CCMS) Journal of Proteome Research. 2010;9(2):806–817. doi: 10.1021/pr9007333. [DOI] [PubMed] [Google Scholar]
- 2.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Analytical Biochemistry. 1976;72(1-2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical Chemistry. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 4.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nature Biotechnology. 2008;26(1):127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 5.Bantscheff M, Hopf C, Kruse U, Drewes G. Proteomics-based strategies in kinase drug discovery. Ernst Schering Foundation Symposium Proceedings. 2007;(3):1–28. doi: 10.1007/2789_2007_060. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, Blex C, Baessler O, et al. The cAMP capture compound mass spectrometry as a novel tool for targeting camp-binding proteins: from protein kinase A to potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channels. Molecular and Cellular Proteomics. 2009;8(12):2843–2856. doi: 10.1074/mcp.M900110-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lenz T, Poot P, Gräbner O, et al. Profiling of methyltransferases and other S-adenosyl-L-homocysteine-binding proteins by Capture Compound mass spectrometry (CCMS) Journal of Visualized Experiments. 2010;(46):1–7. doi: 10.3791/2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oppermann FS, Gnad F, Olsen JV, et al. Large-scale proteomics analysis of the human kinome. Molecular and Cellular Proteomics. 2009;8(7):1751–1764. doi: 10.1074/mcp.M800588-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table: Detailed MS data of kinases (Kinases identified in the different LC-MS setu-ups (1D LC, 90 min gradient; 1D LC, 180 min gradient; 2D LC, 120 min gradient) in assay (A) and competition (C): unique peptides, spectral counts, and calculated fold change A/C based on spectral counts. Kinases belong either to the serine/threonine/tyrosine kinase family (S/T/Y) or small molecule kinases (small mol)).
Supplementary Figures: MS spectra of kinases identified by only one unique spectrum.