

Abstract
The presence of hundreds of copies of mitochondrial (mt) DNA in each human cell poses a challenge for complete characterization of mtDNA genomes by conventional sequencing technologies1. Here, we describe digital sequencing of mtDNA genomes using massively parallel sequencing-by-synthesis. Though the mtDNA of human cells is considered to be homogeneous, we found widespread heterogeneity (heteroplasmy) in the mtDNA of normal human cells. Moreover, the frequency of heteroplasmic variants among different tissues of the same individual varied considerably. In addition to the variants identified in normal tissues, cancer cells harbored additional homoplasmic and heteroplasmic mutations that could also be detected in patient plasma. These studies provide new insights into the nature and variability of mtDNA sequences and have intriguing implications for mitochondrial processes during embryogenesis, cancer biomarker development, and forensic analysis. In particular, they demonstrate that individual humans are characterized by a complex mixture of related mitochondrial genotypes rather than a single genotype.
Mitochondria are pivotal to a large number of basic cellular processes, and as a result of its unique maternal inheritance pattern and relatively high mutation rate, mtDNA is often used in evolutionary biology and population genetics studies. These same attributes, combined with the high copy number of mtDNA in cells, makes mtDNA a favored substrate for forensic analysis2. In typical human cells, there are ~ 50 to hundreds of mitochondria per cell and five to ten copies of mtDNA per mitochondria1. The presence of multiple copies of mtDNA per cell leaves open the possibility that all the copies are not identical. Many studies have shown that mtDNA is homoplasmic in normal cells, i.e., that all of the mtDNA copies are identical not only in an individual cell but also among cells. However, there is apparently a low level of heteroplasmy in the mtDNA of various species, including humans3–14. To further evaluate this issue, we have used massively parallel sequencing-by-synthesis approaches to thoroughly characterize the mtDNA of normal and neoplastic human cells.
Two sets of PCR primers, each resulting in amplicons of ~650 bp in length, were designed to cover the mtDNA genome (Fig. 1a). Sequencing libraries for Illumina GAII made from the PCR products of normal colonic mucosa DNA (Patient #1) yielded 8.5 million tags that matched the mitochondrial genome. Each mtDNA base was sequenced, on average, 16,700 times and less than 11 bases (0.07% of the 16,569 bp in the mtDNA genome) were represented <1000 times (Supplementary Fig. 1a).
Figure 1. Sequencing strategy.
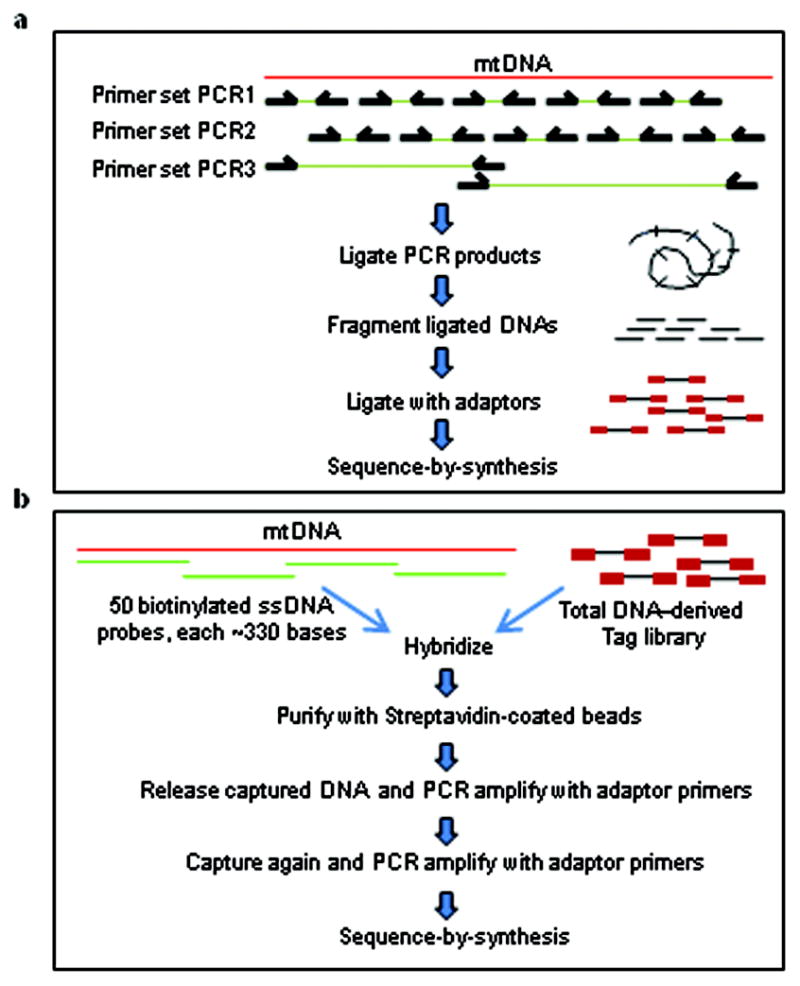
a, PCR amplification for mtDNA enrichment. b, Capture-based method for mtDNA enrichment.
This high coverage permitted us to identify heteroplasmic variants even when they were relatively rare – theoretically, when present in as few as one per 10,000 mt genomes. However, errors that had accumulated during the PCR and sequencing steps limited the actual sensitivity achieved. Control libraries made from PCR products of nuclear DNA demonstrated that the average fraction of mutations per base was 0.058%, with a standard deviation of 0.057%, and no base was mutated at greater than 0.82% frequency (Supplementary Information). We thus made the very conservative assumption that all variants present in excess of twice this value (1.6%) represented true heteroplasmies rather than sequencing artifacts. Using these criteria, we detected 28 homoplasmic alleles and eight heteroplasmic alleles in this sample of normal colonic mucosa (Patient #1). Homoplasmic alleles were defined as any allele not present in the standard mtDNA reference sequence of humans but present in >98.4% (=100% − 1.6%) of the mtDNA sequences analyzed. All homoplasmic alleles identified in Patient #1 were previously identified in normal individuals. The less frequent (minor) allele at the heteroplasmic sites represented 1.6% to 29.7% of the total alleles at that site (Table 1). Interestingly, all (100%) of these eight heteroplasmic alleles were listed as normal variants in mtDNA databases, while only 3,601 bases (21.7%) of the 16,569 bases in the mt genome are reported to have variants in the same databases (P<0.01, χ2).
Table 1.
Heteroplasmic variants in the normal mucosa of Patient #1
| Position | Allele 1 | Allele 2 | Frequency of allele 1 in PCR1# library | Frequency of allele 1 in PCR2# library | Frequency of allele 1 in PCR3# library | Frequency of allele 1 as assessed by capture$ library |
|---|---|---|---|---|---|---|
| 60 | C | T* | 1.90% | 2.00% | 1.70% | 1.40% |
| 72 | C | T* | 4.50% | 4.50% | 4.10% | 4.10% |
| 94 | A | G* | 2.70% | 2.50% | 2.70% | 3.10% |
| 189 | G | A* | 1.60% | 1.50% | 1.40% | 1.40% |
| 228 | G* | A | 2.0% | 2.0% | 1.9% | 2.5% |
| 1888 | A | G* | 2.70% | 2.20% | 2.30% | 2.40% |
| 14566 | G | A* | 29.6% | 29.7% | 26.7% | 26.8% |
| 16126 | T* | C | 4.5% | 4.7% | 4.1% | 4.2% |
Two control experiments were performed to verify that these heteroplasmic allelic variants were not artifactually generated. First, we performed an independent PCR analysis of the mtDNA genome using nine sets of primers (PCR primer set 3 in Fig. 1a) that produced longer PCR products and have been shown not to amplify homologous nuclear DNA sequences15. Sequencing of these PCR products on an Illumina GAII confirmed the identical 28 homoplasmic and heteroplasmic alleles at similar frequencies to those identified with the two other PCR primer sets (Table 1). Second, we employed a capture-based approach for enrichment rather than a PCR-based approach (Fig. 1b). This approach yielded 3.4 million tags and each mtDNA base was sequenced, on average, 7,300 times (Supplementary Fig. 1b). The homoplasmic and heteroplasmic alleles discovered with the PCR-based enrichment strategy were again observed (Table 1).
To determine whether the relatively high number of heteroplasmic variants in this sample was unusual, we analyzed nine additional samples of normal colorectal mucosae. Heteroplasmic allelic variants were observed in each of these additional samples, with an average of 28 homoplasmic and four heteroplasmic alleles per sample. The minor allele frequencies of the heteroplasmic variants ranged from 1.6% to 43.6% (Supplementary Table 1).
To test whether this relatively high level of heteroplasmy was specific to colorectal mucosa tissue, we studied ten tissues obtained from autopsy of a single individual. All homoplasmic alleles in one tissue were found to be homoplasmic in all other tissues. But, remarkably, the fraction and number of heteroplasmic variants varied from tissue to tissue. Each tissue harbored at least one heteroplasmic variant and four tissues harbored ≥ four heteroplasmic variants, each with minor allele frequencies >1.6% (Table 2). As a result of this variation, there were twice as many heteroplasmic alleles than homoplasmic alleles observed in the tissues on an individual (14 and 7, respectively), the reverse of what was found in any single tissue such as normal colorectal mucosa.
Table 2.
Heteroplasmic variants in different organs of the same individual (patient #11, 59 year old)
| Allele 1 frequency | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | allele 1 | allele 2 | Cerebrum | Heart | Skeletal Muscle | Lung | Kidney | Spleen | Liver | Pancreas | Colon | Cerebellum | Number of tissues with variant | Min% | Max% |
| 60 | C | T* | <0.35% | <0.35% | <0.35% | <0.35% | 2.16% | <0.35% | 1.96% | <0.35% | <0.35% | <0.35% | 2 | <0.35% | 2.16% |
| 64 | A | C* | <0.35% | <0.35% | 1.73% | <0.35% | 0.36% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | 1 | <0.35% | 1.73% |
| 72 | C | T* | <0.35% | <0.35% | <0.35% | <0.35% | 18.4% | <0.35% | 11.7% | <0.35% | <0.35% | <0.35% | 2 | <0.35% | 18.4% |
| 73 | G | A* | <0.35% | <0.35% | 2.27% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | 1 | <0.35% | 2.27% |
| 74 | G | T* | <0.35% | <0.35% | <0.35% | <0.35% | 2.64% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | 1 | <0.35% | 2.64% |
| 189 | G | A* | 1.06% | 0.92% | 9.77% | 0.37% | <0.35% | <0.35% | 0.91% | 0.39% | <0.35% | <0.35% | 1 | <0.35% | 9.77% |
| 408 | A | T* | <0.35% | <0.35% | 3.64% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | 1 | <0.35% | 3.64% |
| 1983 | C | T* | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | 1.76% | <0.35% | <0.35% | <0.35% | <0.35% | 1 | <0.35% | 1.76% |
| 6078 | C | A* | 0.52% | 0.49% | 0.82% | 1.23% | 0.44% | 1.10% | 2.99% | 0.92% | 0.54% | 1.60% | 2 | 0.44% | 2.99% |
| 8021 | G | A* | 0.37% | <0.35% | <0.35% | 1.42% | <0.35% | 1.87% | <0.35% | 0.78% | 0.39% | 0.83% | 1 | <0.35% | 1.87% |
| 11090 | C | A* | 0.88% | 0.67% | 1.63% | 1.12% | 1.04% | 1.65% | 1.46% | 1.62% | 1.02% | 1.24% | 3 | 0.67% | 1.65% |
| 14274 | C | A* | 0.54% | 1.6% | 0.41% | <0.35% | <0.35% | <0.35% | 0.92% | 0.61% | 1.18% | 0.84% | 1 | <0.35% | 1.60% |
| 16092 | C | T* | 0.60% | 0.53% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | <0.35% | 0.54% | 2.83% | 1 | <0.35% | 2.83% |
| 16093 | C | T* | 67.4% | 43.6% | 7.44% | 73.0% | 90.9% | 81.6% | 70.9% | 63.8% | 62.2% | 60.1% | 10 | 7.44% | 90.9% |
| Total number of heterolasmic variants | 1 | 2 | 6 | 1 | 4 | 4 | 4 | 2 | 1 | 3 | |||||
Indicates the reference allele. Allele 1 frequencies equal or greater than 1.6% are shaded.
One of the most tantalizing aspects of these data was that five of the fourteen heteroplasmic variants were found in more than one tissue. For example, the most common heteroplasmic variant, at base 16,093, was identified in all tissues but its allelic frequency varied from 7.4% to 90.9%. The variant at codon 72 was present in kidney and liver at 18.4% and 11.7% allelic frequencies, but was not detectable in eight other tissues (Table 2 and Supplementary Figs 2 and 3).
To verify that this tissue-specific heteroplasmy was a general phenomenon, we analyzed the mtDNA of five tissues from another individual (Supplementary Table 2). The same patterns were evident, with a high ratio of heteroplasmic to homoplasmic alleles (11 to 7) and variant frequencies differing by more than a 100-fold among different tissues. As an independent test of the observed heteroplasmy, we determined the sequence of the most abundant variants using conventional dye-terminator chemistry. Though not nearly as quantitative as the digital approach afforded by sequencing-by-synthesis, the chromatograms confirmed the existence and relative extent of heteroplasmy in each case (Supplementary Fig. 3).
There are three potential ways to explain the heteroplasmic variants observed here and in other studies14, 16:(i) Paternal - heteroplasmic variants might represent mtDNA inherited from the father; (ii) Maternal - heteroplasmic variants might be inherited from the mother, with bottlenecks during embryonic development resulting in tissue-specific variations; and (iii) De novo - Heteroplasmic variants might represent new mutations that occurred during embryonic development.
To distinguish among these possibilities, we analyzed the mtDNA from lymphocytes of the parents and two children in each of two CEPH families These analyses confirmed the conclusions made above in that heteroplasmic variants were observed in all but one sample (Supplementary Tables 3 and 4). Additionally, they informed the interpretation of these heteroplasmies in the following way.
Paternal - There were 30 homoplasmic alleles in the fathers of these two families that were not found in their spouses (examples in Table 3; complete data in Supplementary Tables 3 and 4). None of these variants were passed on to any of the four children. This excludes the possibility that the heteroplasmies observed in the progeny’s human cells were transmitted from sperm.
Maternal - The results in Table 3 provide conclusive evidence that a portion of the heteroplasmy observed in the progeny was derived from the mother. Thus, the mother of CEPH family 1377 had two heteroplasmic variants, each present at ~56%, that were both passed on to both her children. Importantly, both these heteroplasmic variants were present in both children at the same allelic frequency (~56%). These heteroplasmic variants were thus unequivocally present in the maternal germ-line as well as in her lymphoid cells. In the other kindred (CEPH family 45), there were three heteroplasmic variants in the mother, and none were passed on to the children. The fact that these were not inherited is consistent with a bottleneck during maternal germ-cell development thought to be responsible for the maintenance of homoplasmy in general. An alternative explanation is that the three maternal heteroplasmic variants were not present in the germ-line of the mother of CEPH family 45 and instead were tissue-specific mutations confined to lymphocytes and other somatic cells. We favor this latter possibility for two reasons. First, it is consistent with the large amount of tissue-specific heteroplasmy documented in Table 2 and described in the following paragraph. Second, even with a severe bottleneck, it is unlikely that a heteroplasmic variant present in a majority of the alleles would be excluded by random segregation from two separate oocyte precursors. Note that one of the heteroplasmic variants was present in 78% of the CEPH family 45 maternal mtDNA copies (Supplementary Table 3). Even if the postulated bottleneck stage contained only one mt genome, the probability that this allele would be excluded from two randomly chosen oocytes is <5% (calculated from binomial distribution).
De novo - The results in Table 3 provide evidence that most of the heteroplasmic variants identified in lymphocytes arose during development. There were no variants present in both children that were not present in one of the parents. On the other hand, there were two or three variants in each child that were not present in the sibling or the parents. The allelic frequencies of these variants were as high as 36%. Though some of these variants may have arisen during expansion of the lymphoid cells in culture, it is notable that similar degrees of allelic variation in heteroplasmic variants were present in several tissues taken directly from patients without further culturing (Table 2 and Supplementary Table 2).
Table 3.
Examples of CEPH family data relating to mechanisms underlying heteroplasmy*
| Position | Allele 1 | Allele 2 | Allele 2 frequency in mother | Allele 2 frequency in father | Allele 2 frequency in child #1 | Allele 2 frequency in child #2 | CEPH family # |
|---|---|---|---|---|---|---|---|
| 9055 | G* | A | <0.35% | >98.4% | <0.35% | <0.35% | 1377 |
| 13967 | C* | T | <0.35% | >98.4% | <0.35% | <0.35% | 1377 |
| 12308 | A* | T | <0.35% | >98.4% | <0.35% | <0.35% | 1377 |
| 3010 | G* | A | <0.35% | >98.4% | <0.35% | <0.35% | 45 |
| 8245 | A* | G | <0.35% | >98.4% | <0.35% | <0.35% | 45 |
| 12618 | G* | A | <0.35% | >98.4% | <0.35% | <0.35% | 45 |
| 16187 | C* | T | 56.5% | <0.35% | 56.7% | 56.4% | 1377 |
| 16186 | C* | T | 55.9% | <0.35% | 56.7% | 56.9% | 1377 |
| 13619 | T* | C | <0.35% | <0.35% | 32.9% | <0.35% | 1377 |
| 12889 | G* | A | <0.35% | <0.35% | 14.7% | <0.35% | 1377 |
| 12775 | G* | A | <0.35% | <0.35% | <0.35% | 25.8% | 45 |
| 3899 | T* | C | <0.35% | <0.35% | <0.35% | 5.3% | 45 |
| 5043 | G* | A | <0.35% | <0.35% | 7.6% | <0.35% | 45 |
| 14615 | G* | A | <0.35% | <0.35% | 36.0% | <0.35% | 45 |
| 567 | A* | C | <0.35% | <0.35% | <0.35% | 5.7% | 45 |
Indicates the reference allele. Values of particular interest in each quartet are in bold typeface.
In summary, heteroplasmy was observed in nearly all of the normal tissues from the individuals examined in the current study (Tables 1–3 and Supplementary Tables 1–4). Furthermore, the degree of heteroplasmy varied in a tissue-specific manner. These findings are consistent with previous observations of tissue-specific mtDNA deletions17 and mtDNA mutations in human stem cells18,19. A small fraction of the heteroplasmic variants were clearly inherited from the mother, but the remainder were apparently somatic mutations that likely occurred during very early embryonic development, as evidenced by the fact that the same heteroplasmic variant was present in two different tissues (e.g., liver and kidney) but not in other tissues.
We next turned our attention to colorectal cancers arising from the colonic mucosae of the ten individuals described above (Supplementary Table 1). Each of the 279 homoplasmic alleles identified in the colonic mucosae were also found to be homoplasmic in the colorectal cancers from the respective patient. Additionally, 90% of the cancers harbored at least one point mutation not present in the matched normal mucosa (Supplementary Tables 5 and 6). This result is consistent with prior studies documenting that somatic mutations of mtDNA are frequently found in human cancers20–23. However, with the precise quantification achieved with deep sequencing, it was clear that most of these cancer-specific somatic mutations were heteroplasmic rather than homoplasmic. Indeed, of the 20 cancer-specific point mutations identified, only ~10% were homoplasmic (Supplementary Table 5).
A comparison between the heteroplasmic variants present in normal colonic mucosae with those in the cancers permitted us to test a model of mtDNA somatic mutation development proposed by Thilly and colleagues24. These authors suggested that any heteroplasmic mutation present in a stem cell should become either homoplasmic or be lost within 70 generations, given reasonable estimates of mtDNA copy number and segregation patterns. As cancer cells evolve through hundreds of generations over decades25, we would thereby expect that any heteroplasmic mutation in normal colorectal mucosa would not exist in the same heteroplasmic state in the cancer developing from it. Our results strongly support this model in that, of 41 heteroplasmic mutations in normal colorectal mucosae, 28 (68%) were absent from the cancer and 9 (22%) were present in the homoplasmic state (Supplementary Table 6).
Using similar reasoning, it was possible to determine the genealogy of individual mtDNA molecules during tumor progression in some cases. For example, in patient #7, the cancer cell mtDNA underwent a bottleneck wherein a single mtDNA molecule harboring heteroplasmic variants at nt 59 and 225 was eventually able to become the dominant mtDNA species in the cancer cell (Supplementary Table 6). Another mtDNA molecule present in 91.8% of the alleles of the normal mucosae was lost during this bottleneck or in subsequent cell divisions (Supplementary Table 6). Note that we cannot determine whether the bottleneck occurred during neoplastic development rather than in a normal colorectal epithelial stem cell from which the cancer developed. However, at some point later during tumor development, two other mutations, at nt 1576 and 12009 arose. Given the relative frequencies of these two mutations (7.8% and 68.8% of the cancer alleles) it is reasonable to suggest that the nt 12009 mutation arose earlier than the 1576 mutation. In both cases, an insufficient number of generations has passed to take these two variants to the homoplasmic state (or to oblivion).
There were two differences between the somatic mutations identified in cancers and the heteroplasmic variants identified in normal tissues. First, 85% of the cancer-specific somatic mutations were in protein- or RNA-coding regions, while only 33% of the heteroplasmic variants were in protein- or RNA-coding regions (p=0.0005, Fisher’s exact test). Second, only 15% of the cancer-specific somatic mutations were present in the mtDNA databases while 75% of the heteroplasmic variants in normal tissues were present in these databases (p<0.0001, Fisher’s exact test). One interpretation of these data is that the process responsible for generating somatic mutations in tumors, whether due to exogenous or endogenous carcinogens, is different than that responsible for mutations during embryonic development (the period in which most heteroplasmic variants in normal tissues appear to generated, as described above). Another interpretation is that a major fraction of the somatic mutations in cancers are intrinsically selected for their tumor-promoting effect rather than representing random segregation events such as those that presumably occur during embryogenesis. Yet a third possibility is that variants in protein coding regions would have been selected against during organismal development but not during tumor development.
Somatic mutations provide exquisitely specific biomarkers for cancer diagnostics26. Somatic mutations in mitochondrial DNA are particularly well-suited for such diagnostic applications because, when homoplasmic, they are 500 to 1000 times more numerous in the cell than nuclear DNA mutations21,23,27. We wondered whether heteroplasmic mutations could be identified in blood samples from cancer patients despite their relatively low representation compared to homoplasmic mutations. To test this idea, we studied the plasma of two colorectal cancer patients prior to and following surgery to remove their cancers (Supplementary Fig. 4). In both patients, nuclear DNA mutations were evident in the plasma before but not after surgery28. Mutant mtDNA template molecules were also identified in the plasma of these two patients at much higher concentrations before surgery than after surgery, confirming their tumor-specificity. The concentration of mutant mtDNA molecules in the plasma greatly exceeded the concentration of the mutant nuclear genes, even though both mtDNA mutations were heteroplasmic. Accordingly, only 25 ul of plasma was required to reliably detect the tumor-specific mtDNA mutations, while at least 2 ml of plasma is generally required to search for nuclear DNA mutations28.
Because mtDNA template molecules are so numerous compared to nuclear DNA template molecules, they are also useful for forensic applications. Previous studies have shown variations in the length of mononucleotide tracts in mtDNA from hair roots compared to blood29,30. Our new results clearly demonstrate that heteroplasmies affect the entire mitochondrial genome, are common in normal individuals and vary dramatically from tissue to tissue. Thus an individual, and perhaps even a single cell, does not have a single mtDNA genotype. Instead, tissues have a mixture of genotypes, a few of which may be maternally inherited and the remaining ones the result of somatic mutations. This suggests caution in excluding identity on the basis of a single or small number of mismatched alleles when the tissue in evidence (e.g., sperm) is not the same as the reference tissue of the suspect (e.g., blood or hair).
METHODS SUMMARY
Mitochondrial DNA from total cellular DNA was enriched by PCR-based or by capture-based strategies. In PCR-based enrichment, three sets of primers were designed to amplify amplicons that cover the mtDNA genome. Purified, blunted-ended PCR products were ligated to form high molecule weight DNA that was subsequently fragmented by sonication. Fragmented DNA was then end-repaired, A-tailed and ligated with a paired-end adapter oligonucleotide from the Illumina genomic DNA library preparation kit following the manufacturer’s instructions. Adapter-ligated products were then size-selected by gel purification and amplified to obtained genomic DNA libraries that were suitable for the Illumina Genome Analyzer. For the capture-based approach, llumina genomic DNA library was prepared from total cellular DNA. To capture mtDNA fragments, 50 biotinylated, single strand DNA probes (baits) of ~300–360 base in size were prepared. These baits were then used for hybridization with the total cellular DNA-derived library. After two rounds of hybridization, the captured library DNA (enriched for mtDNA fragments) was amplified prior to analysis on the Illumina GAII. The 36 base tags obtained from sequencing were aligned to the human mtDNA sequence (AC_000021) using Eland software from Illumina. Tags that passed a set of filters described in the full methods were used for determining coverage and mutation analyses. Background mutation rates derived from PCR and sequencing was determined by sequencing PCR products amplified from nuclear DNA. The MitoMap (http://www.mitomap.org) and mtDB (http://www.genpat.uu.se/mtDB/) databases were used to identify sequence variants. Confirmation of a subset of variants (from both cancer cells and normal tissues) was performed by PCR amplification and conventional Sanger dye terminator sequencing. Detection of circulating mutant APC gene and mutant mtDNA in plasma by BEAMing as previously described28.
Methods
PCR-based enrichment strategy
Three to six ng of total cellular DNA (equivalent to ~ 500 – 1000 cells) was used as a template for each PCR reaction. Amplicons generated using primer sets PCR1 and 2 (Fig. 1a and Supplementary Table 7) were in general produced using HotStart Phusion polymerase (NEB, Beverly, MA) in reactions of 30 ul containing 1 × Phusion HF buffer, 0.2 mM dNTPs, 0.5uM forward and 0.5 uM reverse primers, 5% DMSO and 0.6 u Phusion polymerase. The following cycling conditions were used: 1 cycle of 98°C for 30 sec; 25 cycles of 98°C for 10 sec, 60°C for 30 sec, 72°C for 30 sec. Amplicons generated with primer set PCR3 (Fig. 1a and previously described15) were produced in the same fashion, with the exception that the 72°C elongation step was for 90 sec.
For each DNA sample, PCR products representing 25 amplicons (PCR1 or PCR2) or nine amplicons (PCR3) were pooled. PCR1 and PCR2 amplicons were purified using a kit from Agencourt (Beverly, MA) while the longer PCR products generated with primer set PCR3 were purified with a kit from Omega Bio-Tek (Norcross, GA). Two to five ug of the pooled, purified PCR product was blunt-ended at 20°C for 30 min in a total volume of 100 ul containing 1 × T4 ligation buffer, 10 mM ATP, 1 mM of each dNTP, 5 ul T4 DNA polymerase, 1 ul Klenow polymerase, and 5 ul T4 PNK (all enzymes were obtained from NEB). Blunt-ended DNA was PCR-purified and ligated using Quick Ligase (NEB), in a total volume of 100 ul, as described by the manufacturer. Ligated DNA was purified with a Qiagen PCR purification kit (Cat# 28104) and eluted with 100 ul of 70 °C elution buffer. Purified, ligated DNA was fragmented with a Bioruptor sonicator (Diagenode, Sparta, NJ) at low power, by cycling for 15 sec on, then 15 sec off, for 2×15 minutes while cooled in an ice bath. This sonication reduced the size of the ligated DNA from >12 kb to 100 to 400 bp. The fragmented DNA was purified and used for preparation of an Illumina DNA library as described in Illumina genomic DNA library preparation manual.
Capture-based enrichment strategy
Step 1. Preparation of probes
Fifty amplicons were designed to cover the mtDNA genome (Supplementary table 8). For every amplicon, the 5′ end of the forward primer was tagged with M13-Forward sequence (5′-gtaaaacgacggccagt-3′). PCR was performed as described above using normal human colon mucosa DNA as template and primers without the biotin tag. These PCR products were then used as templates for a second PCR using double-biotinylated M13-Forward sequence as forward primers (Supplementary Table 7) to obtain biotinylated PCR products. The resultant PCR products were pooled and purified with a Qiagen kit (cat# 28104). The purified PCR products were then mixed with an equal volume of 2 × Bind-and-Wash buffer (10 mM Tris-Cl, pH7.5, 1 mM EDTA, 2 M NaCl) containing 250 ul Dynal MyOne streptavidin-coated beads (product# 650.02, Invitrogen). After incubation at room temperatures for one hour, the beads were washed twice with 1 × PCR buffer (20 mM Tris-Cl, pH 8.4, 50 mM KCl). Bead-attached DNA was then denatured in 0.2N NaOH for 5 min at room temperature to remove the non-biotinylated strands, and washed in 1xPCR buffer. The biotinylated single-stranded DNA remaining attached to the beads was released by incubation in 200 ul Hi-Di formamide (product# 4311320, ABI, Foster City, CA) at 95°C for three minutes. ssDNA probe mixture was precipitated by adding 3M Sodium Acetate, pH5.2, DNA carrier and 2.5 volumes of ethanol, then centrifuged for five minutes in a microfuge at ~12,000 g. After washing the pellet with 75% ethanol, the single strand DNA was resuspended in H2O.
Step 2. Hybrizidation-based capture
Total cellular DNA was used to prepare a paired-end library by slight modifications of the standard Illumina protocol. 2.5 ug of the library DNA was then mixed with 2.5 ug of the single-stranded biotinylated DNA in 50ul containing 0.2% SDS and 4.5 × SSPE (1xSSPE: 0.15 M NaCl, 0.01 M NaH2PO4, and 0.001 M EDTA; from Amresco, Solon, Ohio), covered with mineral oil and incubated at 95°C for 3 min, followed by incubation at 65°C for 12 hours to allow hybridization. DNA was then purified with NucleoSpin Extract II with NTB buffer (Clontech, Mountain View, CA). The DNA was eluted from the NucleoSpin column in 100 ul elution buffer and was captured on streptavidin-coated MyOne beads as described above. The beads were washed twice with 1 × PCR buffer; resuspended in washing buffer composed of 0.1% SDS in 2 × SSPE and incubated at 68°C with shaking for 30 min. The beads were collected on a magnet and washed with fresh washing buffer for another 30 min at 68°C, then washed twice with 1x PCR buffer. The captured target DNA was released by denaturation in 0.2N NaOH at room temperature for 5 min, ethanol-precipitated as described above, then resuspended in 15 ul elution buffer (5mM Tris-HCl, pH8.5). Two ul was used as template for HotStart Phusion PCR (18-cycle) amplification to obtain a 1°-enriched mtDNA library. Approximately three ug of 1°-enriched mtDNA library DNA was then mixed with 2.5 ug of the biotinylated ss DNA described above in 110 ul of 1xPCR buffer (from 10xPCR buffer, cat#53286, Invitrogen). Hybridization was performed by incubating the DNA at 95°C for 1 min; then cooled to 50°C at 0.1°C/sec, then incubated at 50°C for 1min. The hybrids were captured with MyOne beads, washed once in 1 × PCR buffer; twice with wash solution I (2 × SSC, 0.05% SDS), using a 15 min incubation at room temperature for each wash; beads were then washed twice, for 15 minutes each, with wash solution II (0.1 × SSC, 0.1% SDS) at 50°C, then washed once with 1 × PCR buffer. Captured target DNA was released and ethanol-purified as described above, resuspended in 15 ul elution buffer (5mM Tris-HCl, pH8.5). Three ul was used as a template for HotStart Phusion PCR (18-cycle) amplification to obtain a 2°-enriched mtDNA library, which was subsequently used for sequencing with a Genome Analyzer II instrument.
Genome Analyzer sequence data analysis
The 36 base tags obtained from the Genome Analyzer II reads aligned to the human mtDNA sequence of 16569 bp (AC_000021) using Eland software from Illumina. This alignment allowed up to two mismatches at the first 32 out of the total 36 bases in each tag. Four bases in the human mtDNA genome were excluded from analysis: bases 309–311 (in a long stretch of “Cs”) and base 3107 (“N” base in AC_000021). Three quality filters were used for selecting tags for further analysis: (i) all 36 bases in a tag were required to have a Phred score of at least 23 (meaning there was ~0.5% probability of a base calling error); (ii) no “N” base was allowed anywhere in the 36 bases; and (iii) no more than three mismatches were permitted in the 36 bases (e.g., the last four bases could have no more than 2 mismatches if there was one mismatch in the first 32 bases). Subsequent analysis was performed with database software SQL 2005. Each position of the mtDNA genome was assigned a coverage depth, representing the number of quality-filtered tags containing the base was observed, and a mutation fraction, representing the fraction of tags containing that base in mutant form. To be categorized as a mutation, the identical mutation had to be identified (i) in at least ten “distinct” tags, i.e., tags whose first base was at ten different positions (for clarity, there are 16569 distinct tags possible in the mitochondrial genome) and (ii) in at least three tags sequenced from the forward direction and at least three from the reverse direction. These criteria were applied to normal mucosa samples, cancer samples and samples from CEPH families. For analysis of mtDNA from normal human organs, mutations were defined by at least five “distinct” tags, and at least one tag sequenced from the forward direction and at least one from the reverse direction.
Determination of background error rates
PCR products used for Genome Analyzer II sequencing were generated through two PCR steps. In the first step, 96 pairs of PCR primers, each with a universal tag (5′-ACACGACGCTCTTCCGATCT-3′ for forward primers, 5′-GCATACGAGCTCTTCCGATCT-3′ for reverse primers), were designed to amplify 96 different regions on chromosome 13, 18 and 21. Genomic DNA from human B lymphoblastoid cells was used as PCR template. After 30 cycles of PCR using Phusion high fidelity polymerase (NEB), the PCR product was mixed and diluted 1000 times in TE buffer. In the second step PCR, one pair of PCR primers were designed to contain the universal sequence used in the first step and the Illumina grafting primers (Forward Primer: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′; Reverse Primer: 5′-CAAGCAGAAGACGGCATACGAGCTCTTCCGATCT-3′). The PCR was conducted in a single PCR tube using this pair of primers and the diluted PCR product mixture from first step as template. After 15 cycles of PCR using Phusion Flash master mix (NEB), the product was purified with a Qiaquick purification column (cat# 28104, Qiagen) and quantified by absortion at 600 nm.
For sequencing data analysis, a Phred score of at least 23 for each base in the 36-base tags was applied to select high quality tags for mutation analysis. For every non-heterozygous position, coverage of the non-reference base and reference base was extracted from the sequencing result. The sequencing error for each base was calculated as the coverage of non-referenced base divided by the coverage of the reference base. Analysis of 2746 bases by two independent experiments showed that the average fraction of mutations per base was 0.063% and 0.052%, with a standard deviation of 0.058% in both experiments, and the highest mutation rate was 0.82% and 0.7%, respectively.
Sanger sequencing and BEAMing confirmation of mutation
Three to six ng of total cellular DNA was used as a template for each PCR reaction using HotStart Phusion polymerase (NEB) in reactions of 30 ul containing 1 × Phusion HF buffer, 0.2 mM dNTPs, 0.5uM forward and 0.5 uM reverse primers, 5% DMSO and 0.6 u Phusion polymerase. The following cycling conditions were used: 1 cycle of 98°C for 30 sec; 25 cycles of 98°C for 10 sec, 60°C for 30 sec, 72°C for 30 sec. PCR products were purified with PCR purification kit (cat# 28104, Qiagen) and conventional dye terminator sequencing with forward and reverse PCR primers were performed by MWG biotech (High Point, NC). Beaming confirmation of mutation was performed as described previously28.
Detection of circulating mutant mtDNA in plasma
Collection of plasma samples from colorectal cancer patients and isolation of circulating DNA was performed as previously described with modifications28. Specifically, we used a virus vacuum kit (Qiagen #57714) for plasma DNA purification. Five hundred ul of plasma was mixed with 40 ul of proteinase K (Invitrogen #25530-049). 5.6 ul of 1ug/ul RNA carrier was mixed with 500 ul of AL buffer and subsequently this mixture of 506 ul was added to the plasma/proteinase mixture, mixed by vortexing and incubated for one hour at 60°C. Following incubation, 600 ul of ice cold ethanol was added and the solution incubated at room temperature for 5 minutes. The mixture was then transferred into the virus vacuum kit column and spun in a microfuge at 7000 rpm for one minute. This loading and centrifugation step was repeated several times until the whole mixture was processed. The column was washed with 600 ul AW1 buffer, 700 ul AW2 buffer and 700 ul ethanol by spinning for one minute at 7000 rpm at each step. Finally, a spin at maximal speed for three minutes was performed to dry the column. DNA was finally eluted in 35 ul AVE buffer.
The nuclear DNA copy number (genome equivalent) per milliliter of plasma was determined by quantitative PCR as previously described28. To measure the copy number of circulating mtDNA in plasma, we used forward primer 5′-ctccagcgtctcgcaatg-3′ and reverse primer 5′-tcacaggtctatcacc-3′ to amplify and obtain mtDNA fragment of 101 bp from normal human total cellular DNA. This PCR product was quantified by Picogreen and subsequently used as a copy number standard for quantitative PCR using the same set of primers to determine mtDNA copy number per milliliter of plasma.
The determination of circulating mutant APC gene frequency was previously described28. Primers used for BEAMing detection of mutant mtDNA frequency: for patient #8 (base 4097) - forward primer 5′-TCCCGCGAAATTAATACGACacgcactctcccctgaact-3′, and reverse primer 5′-GCTGGAGCTCTGCAGCTAgtagcggaatcgggggtat-3′; for patient #9 (base 16291) – forward primer 5′-TCCCGCGAAATTAATACGACcacccctcacccactaggat-3′, and reverse primer 5′-GCTGGAGCTCTGCAGCTAgggacgagaagggatttgac-3′. First step PCR was performed as described above with Phusion polymerase with an input template DNA of about 100 genome equivalent. Second step amplification (Beaming with universal primer – forward 5′-TCCCGCGAAATTAATACGAC-3′, and reverse 5′-GCTGGAGCTCTGCAGCTA-3′) and detection of mutant mtDNA allele frequency by single base extension was performed as previously described28. Primers used for single base extension: for base 4097 5′-cataagaacagggaggttagaagt-3′; and for base 16291 5′-tatgtactatgtactgttaagggtg-3′. The percentages of circulating mutant mtDNA was determined to be 0.65% (before surgery) or 0.004% (after surgery) for patient #8; and 2.87% (before surgery) or 0.06% (after surgery) for patient #9.
Supplementary Material
Acknowledgments
The authors acknowledge Melissa Whalen, Janine Ptak, Lisa Dobbyn, and Natalie Silliman for expert technical assistance. This work was supported by The Virginia and D.K. Ludwig Fund for Cancer Research and NIH grants CA57345, CA 43460, CA 62924 and CA121113.
Footnotes
Supplementary Information is available online.
Author Contributions Y.H., K.W.K., B.V. and N.P. designed and performed experiments, analyzed data and wrote the paper. J.W. and D.C.D. performed experiments and analyzed data. C.I.-D., S.D.M. and L.A.D. provided critical materials and reagents. V.E.V. analyzed data and provided input to the manuscript.
References
- 1.Legros F, Malka F, Frachon P, Lombes A, Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117 (Pt 13):2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 2.Schneider PM. Scientific standards for studies in forensic genetics. Forensic Sci Int. 2007;165 (2–3):238–243. doi: 10.1016/j.forsciint.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 3.Wong LJ, Boles RG. Mitochondrial DNA analysis in clinical laboratory diagnostics. Clin Chim Acta. 2005;354 (1–2):1–20. doi: 10.1016/j.cccn.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 4.White HE, et al. Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genet Test. 2005;9 (3):190–199. doi: 10.1089/gte.2005.9.190. [DOI] [PubMed] [Google Scholar]
- 5.Maitra A, et al. The Human MitoChip: a high-throughput sequencing microarray for mitochondrial mutation detection. Genome Res. 2004;14 (5):812–819. doi: 10.1101/gr.2228504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobrowolski SF, Gray J, Miller T, Sears M. Identifying sequence variants in the human mitochondrial genome using high-resolution melt (HRM) profiling. Hum Mutat. 2009;30 (6):891–898. doi: 10.1002/humu.21003. [DOI] [PubMed] [Google Scholar]
- 7.Kraytsberg Y, Nicholas A, Caro P, Khrapko K. Single molecule PCR in mtDNA mutational analysis: Genuine mutations vs. damage bypass-derived artifacts. Methods. 2008;46 (4):269–273. doi: 10.1016/j.ymeth.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kajander OA, et al. Human mtDNA sublimons resemble rearranged mitochondrial genoms found in pathological states. Hum Mol Genet. 2000;9 (19):2821–2835. doi: 10.1093/hmg/9.19.2821. [DOI] [PubMed] [Google Scholar]
- 9.Santos C, et al. Frequency and pattern of heteroplasmy in the control region of human mitochondrial DNA. J Mol Evol. 2008;67 (2):191–200. doi: 10.1007/s00239-008-9138-9. [DOI] [PubMed] [Google Scholar]
- 10.Greaves LC, et al. Quantification of mitochondrial DNA mutation load. Aging Cell. 2009;8 (5):566–572. doi: 10.1111/j.1474-9726.2009.00505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraytsberg Y, et al. Quantitative analysis of somatic mitochondrial DNA mutations by single-cell single-molecule PCR. Methods Mol Biol. 2009;554:329–369. doi: 10.1007/978-1-59745-521-3_21. [DOI] [PubMed] [Google Scholar]
- 12.Osborne A, Reis AH, Bach L, Wangh LJ. Single-molecule LATE-PCR analysis of human mitochondrial genomic sequence variations. PLoS One. 2009;4 (5):e5636. doi: 10.1371/journal.pone.0005636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286 (5440):774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Natl Acad Sci U S A. 2001;98 (7):4022–4027. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramos A, Santos C, Alvarez L, Nogues R, Aluja MP. Human mitochondrial DNA complete amplification and sequencing: a new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification. Electrophoresis. 2009;30 (9):1587–1593. doi: 10.1002/elps.200800601. [DOI] [PubMed] [Google Scholar]
- 16.Zsurka G, et al. Recombination of mitochondrial DNA in skeletal muscle of individuals with multiple mitochondrial DNA heteroplasmy. Nat Genet. 2005;37 (8):873–877. doi: 10.1038/ng1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marzuki S, et al. Developmental genetics of deleted mtDNA in mitochondrial oculomyopathy. J Neurol Sci. 1997;145 (2):155–162. doi: 10.1016/s0022-510x(96)00241-9. [DOI] [PubMed] [Google Scholar]
- 18.Taylor RW, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112 (9):1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coller HA, et al. Clustering of mutant mitochondrial DNA copies suggests stem cells are common in human bronchial epithelium. Mutat Res. 2005;578 (1–2):256–271. doi: 10.1016/j.mrfmmm.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Polyak K, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20 (3):291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 21.Jones JB, et al. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001;61 (4):1299–1304. [PubMed] [Google Scholar]
- 22.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25 (34):4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 23.Wong LJ, Lueth M, Li XN, Lau CC, Vogel H. Detection of mitochondrial DNA mutations in the tumor and cerebrospinal fluid of medulloblastoma patients. Cancer Res. 2003;63 (14):3866–3871. [PubMed] [Google Scholar]
- 24.Coller HA, et al. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet. 2001;28 (2):147–150. doi: 10.1038/88859. [DOI] [PubMed] [Google Scholar]
- 25.Jones S, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105 (11):4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2 (3):210–219. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- 27.Fliss MS, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287 (5460):2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 28.Diehl F, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14 (9):985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekiguchi K, Kasai K, Levin BC. Inter-and intragenerational transmission of a human mitochondrial DNA heteroplasmy among 13 maternally-related individuals and differences between and within tissues in two family members. Mitochondrion. 2003;2 (6):401–414. doi: 10.1016/S1567-7249(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 30.Sekiguchi K, Sato H, Kasai K. Mitochondrial DNA heteroplasmy among hairs from single individuals. J Forensic Sci. 2004;49 (5):986–991. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.