

Abstract
Background
Following cardiac injury, activated cardiac myofibroblasts can influence tissue electrophysiology. Because mechanical coupling through adherens junctions provides a route for intercellular communication, we tested the hypothesis that myofibroblasts exert tonic contractile forces on the cardiomyocytes and affect electrical propagation via a process of mechanoelectric feedback.
Methods and Results
The role of mechanoelectric feedback was examined in transforming growth factor-beta (TGF-β) treated monolayers of co-cultured myofibroblasts and neonatal rat ventricular cells by inhibiting myofibroblast contraction and blocking mechanosensitive channels (MSCs). Untreated (control) and TGF-β treated (fibrotic) anisotropic monolayers were optically mapped for electrophysiological comparison. Longitudinal conduction velocity (LCV), transverse conduction velocity (TCV), and normalized action potential upstroke velocity (dV/dtmax) significantly decreased in fibrotic monolayers (14.4±0.7 (SEM) cm/s, 4.1±0.3 cm/s, n=53, and 3.1±0.2, n=14, respectively) compared with control monolayers (27.2±0.8 cm/s, 8.5±0.4 cm/s, n=40, and 4.9±0.1, n=12, respectively). Application of the excitation-contraction uncoupler, blebbistatin, or the MSC blocker, gadolinium or streptomycin, dramatically increased LCV, TCV and dV/dtmax in fibrotic monolayers (36±1 cm/s, 10±0.3 cm/s, n=17, and 4.5±0.1, n=14, respectively). Similar results were observed with Cx43-silenced cardiac myofibroblasts. Spiral wave induction in fibrotic monolayers also decreased following the aforementioned treatments. Finally, traction force measurements of individual myofibroblasts showed a significant increase with TGF-β, decrease with blebbistatin, and no change with MSC blockers.
Conclusions
These observations suggest that myofibroblast-myocyte mechanical interactions develop during cardiac injury, and that cardiac conduction may be impaired as a result of increased MSC activation owing to tension that is applied to the myocyte by the myofibroblast.
Keywords: arrhythmia, conduction, electrophysiology, myocardial infarction
Though cardiomyocytes are the undisputed functionally contractile cells in the myocardium, fibroblasts outnumber cardiomyoctes by nearly 2:1.1 While it is well known that cardiac fibroblasts produce and remodel the extracellular matrix (ECM) in the heart,2 their involvement in cell signaling, heterocellular coupling, and mechanoelectric feedback, as well as their contribution to many pathological conditions, has been increasingly recognized.3 After injury, cardiac fibroblasts contribute to wound healing by proliferating and differentiating into myofibroblasts, an α-smooth muscle actin (SMA)-expressing cell adept at contraction. Myofibroblasts utilize contraction and ECM remodeling to replace the necrotic myocardium with mature scar tissue following infarction;4–6 the formed scar tissue, consisting of ECM and persistent myofibroblasts, is electrically unexcitable and ultimately creates a substrate that is vulnerable to arrhythmias.
A potent inducer of cardiac myofibroblast differentiation both in vivo and in vitro is transforming growth factor beta (TGF-β).6 Expression of TGF-β remains low in the normal heart, but is markedly increased after cardiac injury.7 Sustained expression of TGF-β augments the conversion of fibroblasts to myofibroblasts, as well as myofibroblast contraction,8 and ultimately contributes to myocardial fibrosis.9
Until recently, cardiac fibroblasts were believed to act as passive electrical insulators between myocytes, but new data suggest that fibroblasts play a more dynamic role in the electrical activity of the heart. Fibroblast-myocyte electrical coupling has been shown in vitro and in situ in regions of infarcted and healthy myocardium, such as the sinoatrial node; this coupling enables fibroblasts to act as current sinks, short-range conductors, or even long-range conductors.10–12 Though electrical coupling between myocytes and fibroblasts is suspected to be the culprit in slowing conduction velocity in fibroblast-supplemented models, a quantitative study on myocyte-fibroblast cell pairs showed that less than 10% of the 450 studied cell pairs expressed junctional connexin43 (Cx43).13 Furthermore, the limited amount of coupling found in situ suggests that fibroblasts affect cardiomyocyte electrophysiology through a mechanism other than electrical coupling.
Myofibroblast contraction is a crucial aspect of wound healing in injured tissues throughout the body,14 and contractile force permits cellular communication to be relayed through intercellular coupling.15 Therefore, we tested the hypothesis that mechanical coupling transmits the contractile force of myofibroblasts to myocytes, and that this interaction activates mechanosensitive channels (MSCs) which alter electrophysiological function and slow conduction. To conduct this study, we used an in vitro co-culture model of neonatal rat cardiomyocytes and myofibroblasts stimulated by TGF-β, together with blockers of excitation-contraction coupling and MSCs.
Methods
An expanded Materials and Methods section is available in the online data supplement. In brief, 20mm diameter anisotropic monolayers of neonatal rat ventricular cells (NRVCs) were obtained by growing cells on parallel, 20μm-wide fibronectin lines formed by microcontact printing. Monolayers were treated with 5ng/ml TGF-β for 48–72 hours to promote the cardiac myofibroblast phenotype. Untreated and TGF-β treated monolayers were compared by immunostaining for cardiac (troponin I, α-actinin) and fibroblast (prolyl-4-hydroxylase, SMA) markers, and optically mapped with 10μM voltage-sensitive dye, di-4-ANEPPS. Activation maps were obtained at 2Hz pacing during constant superfusion (with bath volume exchange every 2 minutes); then, the excitation-contraction uncoupler, blebbistatin, or MSC blocker, gadolinium or streptomycin, was superfused over the monolayer to determine its impact on conduction velocity (CV) in the longitudinal (LCV) and transverse (TCV) directions, minimum cycle length before loss of 1:1 capture (MCL), incidence of spiral waves, pacing cycle length to initiate spiral waves, action potential duration (APD), normalized upstroke velocity (dV/dtmax) and conduction heterogeneity index (HI) in control and fibrotic monolayers. In subsequent experiments, a supplemented model was created in which cardiac fibroblasts were separately pre-treated with 5ng/ml TGF-β for at least 48 hours and then added onto patterned NRVC monolayers at a concentration of 300,000–400,000 cells per monolayer, 24 hours prior to electrophysiological characterization. Fibroblasts were also transduced with Cx43 shRNA lentiviral particles containing a puromycin promoter. Two days later, cells stably expressing shRNA were isolated with puromycin, and Cx43 knockdown was confirmed using Western blots. As a negative control, fibroblasts were transduced with shRNA lentiviral particles encoding a scrambled shRNA sequence. The transduced myofibroblasts were treated with 5ng/ml TGF-β for 48 hours, and 400,000 were added onto control NRVC monolayers for subsequent electrophysiological analysis. Additionally, pure monolayers of untreated fibroblasts and TGF-β treated myofibroblasts were characterized by immunostaining for actin, SMA, pan-cadherin, and Cx43; Cx43 levels were also quantified by western blot. Traction forces of untreated fibroblasts and TGF-β treated myofibroblasts were quantified by elastic micropost arrays of known stiffness.16 All data are expressed as mean±(standard error of mean). Two-tail Student's t-tests were performed for independent data, such as fibrotic and control monolayers, and Wilcoxon signed rank tests were performed for paired data, such as fibrotic monolayers before and after treatment, to determine statistically significant differences (p < 0.05).
Results
Treatment with TGF-β induces a fibrotic NRVC model
The first goal of this project was to create an in vitro model that mimicked tissue-level aspects of cardiac fibrosis, including an excess of contractile myofibroblasts and slowed, heterogeneous conduction in comparison with controls. TGF-β was added to co-cultured monolayers of cardiomyocytes and cardiac fibroblasts. Several concentrations (2.5, 5, and 10ng/ml) of TGF-β were tested to determine the dose-response effect on CV; 5ng/ml was chosen for all subsequent experiments because it was the lowest dose that resulted in a significant and reproducible effect on CV. To mimic physiologic conditions for all subsequent experiments, NRVCs were grown as anisotropic monolayers to evaluate their LCV and TCV. Anisotropic monolayers incubated with TGF-β will be referred to as fibrotic monolayers, and untreated anisotropic monolayers will be referred to as control monolayers.
TGF-β-induced fibroblast proliferation was signified by an increased number of cells positive for prolyl-4-hydroxylase (Figure 1A,D). Fibroblast density was much higher in fibrotic monolayers compared with control monolayers (70±6% vs. 16.4±2%, n=6, p=10−4), and fibroblast size was much larger compared with that in control monolayers (620±46 μm2 per cell vs. 240±19 μm2, respectively, n=17, p=10−8). Myofibroblast conversion was evaluated by SMA stress-fiber expression; untreated fibroblasts in control monolayers expressed limited amounts of SMA, while TGF-β induced a dramatic up-regulation of SMA-expressing myofibroblasts (Figure 1). Z-stack images revealed that myofibroblasts were directly in contact with the cardiomyocytes, most often beneath the myocytes which allowed the cells to interact in the z-direction, and also in the lateral spaces between the cardiomyocytes (Fig. 1D–1F). Further, purified myofibroblasts treated with TGF-β had more organized SMA stress fibers (Supplemental Figure 1B) than control fibroblasts (Supplemental Figure 1A). Overall, TGF-β treatment increased the fraction of fibroblasts expressing SMA stress fibers from 49% in untreated fibroblasts to 81% (n=795 for control, n=350 for TGF-β treated, p=10−4). Fibrotic monolayers (n=53) had significantly slower LCV and TCV (14.4±0.7 and 4.1±0.3 cm/s, respectively) compared with control monolayers (n=40) (27.2±0.8 and 8.5±0.4 cm/s, respectively) (Figure 1G–H). Furthermore, fibrotic monolayers (n=17) had a significantly larger HI of propagation (1.8±0.11) than control monolayers (n=14) (1.2±0.07) (Figure 1I).
Figure 1.

Characterization of the fibrotic NRVC model. Immunostain images of control NRVC monolayers (A–C) and fibrotic monolayers (D–F) illustrate the relative distributions of cardiomyocytes and fibroblasts. Panels A and D display the dramatic increase in fibroblast density in fibrotic monolayers. The images are stained for prolyl-4-hydroxylase (green) and troponin I (red), and taken in the plane where fibroblasts and myocytes interact (the inset represents the myocyte plane, scale bar is 20μm). Panels B,C,E, and F were stained with antibodies against α-actinin (red), α-smooth muscle actin (green), and DAPI (blue). Undifferentiated fibroblasts in the control monolayer are indicated by the nuclei in cells negative for actinin and SMA (designated by arrows in (C)). Fibrotic monolayers had significantly decreased LCV (G) and TCV (H), as well as significantly higher HI (I), compared with control monolayers. *P<0.001 compared with control monolayers.
Mechanical adherens junctions dominate homo- and heterocellular junctions involving myofibroblasts
Because coupling occurs between cardiomyocytes and fibroblasts in vitro,13 evidence for electrical junctions (Cx43) and mechanical junctions (pan-cadherin) was examined between the two cell types. Heterocellular contacts between adjoining fibroblasts and myocytes in a control monolayer had some Cx43 expression, with a considerable amount of non-junctional Cx43, and little cadherin expression (Figure 2A). However, contacts between adjoining cardiomyocytes and myofibroblasts in a fibrotic monolayer showed that cadherin was more prominently expressed than Cx43 (Figure 2B). In pure monolayers of cardiac fibroblasts (CFs) or cardiac myofibroblasts (CMFs), CFs expressed more junctional Cx43 (Figure 2C,E) while CMFs expressed more junctional cadherin (Figure 2D,E). Both control and fibrotic monolayers were also stained for connexin45, but little expression was observed (Supplemental Figure 2). Cx43 Western blots of the insoluble (Figure 2F) and soluble protein fractions (Figure 2G) revealed that TGF-β treated myofibroblasts had significantly less insoluble Cx43 than untreated fibroblasts, but significantly more soluble Cx43 (n=10 and n=4, respectively, p<0.05).
Figure 2.

Characterization of electrical and mechanical coupling between cells. Magnified view of the heterocellular contact between either a cardiomyocyte and fibroblast (A) or a cardiomyocyte and myofibroblast (B) in a low cell density monolayer, stained for SMA (red), troponin I (blue), connexin43 (white), and pan-cadherin (green). Even though the fibroblast in (A) has substantial C×43 staining, there appear to be only three sites of junctional connexin between the two cells (denoted by red arrows), and little junctional cadherin. Gap junctions between myofibroblasts and cardiomyocytes are scarce as indicated by the limited junctional C×43 staining (denoted by white arrow) while adherens junctions are more prominent as visualized by the intense cadherin staining at the border between the two cells (B). Homocellular mechanical and electrical couplings between fibroblasts (C) and myofibroblasts (TGF-β treated, D) were visualized with antibodies against SMA (red), actin (blue), connexin43 (white), and pancadherin (green). Monolayers of fibroblasts have an abundance of connexin43-containing gap junctions compared with cadherin-containing adherens junctions (C). Conversely, myofibroblasts treated with TGF-β have intense junctional cadherin expression and no apparent junctional C×43 expression (D). A mixed population of fibroblasts and myofibroblasts showed preferential C×43 and cadherin staining in localized regions of fibroblasts and myofibroblasts, respectively (E). Representative Western blots of the insoluble (F) and soluble protein fractions (G) show that control fibroblast monolayers have significantly higher insoluble C×43 than TGF-β treated myofibroblast monolayers, but significantly lower soluble C×43, normalized to the loading control vimentin (n=10 and n=4, respectively, P<0.05) .
Inhibition of contractile force restores conduction
In fibrotic monolayers, the increased presence of myofibroblasts capable of exerting strong contractile forces had functional consequences. To test the hypothesis that myofibroblast contraction influences the conduction properties of the cardiomyocytes, blebbistatin was added to inhibit myosin II and suppress contraction.17 Blebbistatin (5–10μM for 20–30 min) significantly increased CV in fibrotic monolayers, but had no significant effect on control monolayers (Figure 3).
Figure 3.
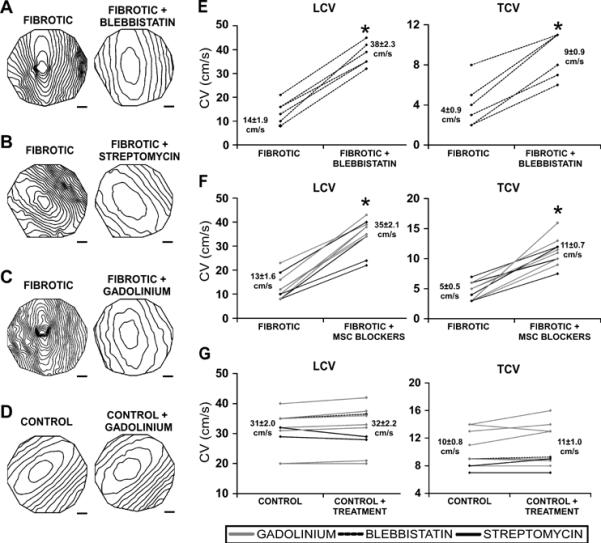
Effects of contraction and mechanosensitive channel inhibition on electrical conduction in fibrotic monolayers. Isochrone maps (10ms spacing) comparing monolayers before and after treatment (A)–(D) and summary line graphs of the same experimental conditions (E)–(G) show that blebbistatin (A,E), streptomycin (B,F), and gadolinium (C,F) significantly increased LCV and TCV of fibrotic monolayers, whereas the treatments had no effect on CV of control monolayers (D,G). Scale bar is 2mm. *denotes significant difference after treatment, P<0.05.
Mechanosensitive channel blockers improve conduction in fibrotic monolayers
Because contractile force has a significant effect on electrical conduction, mechanoelectic coupling was investigated. After 20–30 minutes of treatment with an MSC blocker, gadolinium (20–50μM) or streptomycin (50μM), LCV and TCV increased significantly in fibrotic monolayers, but not in control monolayers (Figure 3). Conduction non-uniformity was common among fibrotic monolayers, as illustrated by the skewed elliptical pattern in Figures 3A,B, and was most likely the result of a heterogeneous density of myofibroblasts in culture, which ranged from 45% to 85%. Blebbistatin, gadolinium, and streptomycin produced more elliptical patterns of propagation and increased the uniformity of conduction in fibrotic monolayers (n=10), decreasing HI from 1.8±0.1 to 1.3±0.1 (p=10−3).
Inhibition of contraction and mechanosensitive channels increases upstroke velocity and shortens action potential duration in fibrotic monolayers
In addition to slowed, discontinuous conduction, fibrotic monolayers also have slowed dV/dtmax in comparison with control monolayers (3.1±0.2, n=14 vs. 4.9±0.1, n=12, p=10−10, respectively) that increased following application of blebbistatin, gadolinium or streptomycin (to 4.5±0.1, n=14) (Figure 4A,B). Calculations indicated that some, but not all, of the changes in dV/dtmax could be accounted for by changes in CV. APD was also prolonged in fibrotic monolayers (186±4ms, n=16 vs. 148±7ms, n=8, p=10−5 in control monolayers) and shortened significantly in response to the aforementioned drug treatments (to 161±4ms, n=10) (Figure 4A,C).
Figure 4.
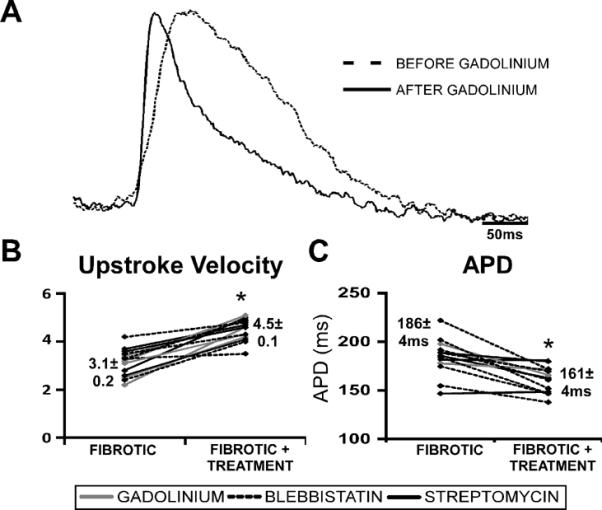
Effects of contraction and mechanosensitive channel inhibition on dV/dtmax and APD in fibrotic monolayers. Representative AP traces comparing fibrotic monolayers before and after treatment with gadolinium at 2Hz pacing show that dV/dtmax increased and APD decreased significantly (A). Summary dV/dtmax (B) and APD (C) line graphs of drug treatments for fibrotic monolayers. Blebbistatin and MSC blockers significantly increased dV/dtmax (n=5 and 9, respectively) and significantly decreased APD (n=6 and 4, respectively) in fibrotic monolayers (B). * denotes significance after treatment, P<0.05.
Supplemented model corroborates results with fibrotic model
In the experiments described so far, TGF-β was added directly to a mixed population of fibroblasts and cardiomyocytes, which may affect the electrophysiology of the cardiomyocytes directly. To eliminate this potentially confounding effect, CMFs were separately treated with TGF-β and then added onto untreated anisotropic NRVC monolayers (supplemented model). Untreated CFs were also added at similar numbers onto untreated monolayers for comparison. The addition of CMFs or CFs decreased CV compared with control, although CMFs had a more dramatic effect than CFs, decreasing LCV from 32.6±1.2 cm/s in control monolayers (n=26) to 15.3±1.3 cm/s (n=26), compared with a decrease to only 24.5±2.0 cm/s with addition of CFs (n=17) (Figure 5A). Similar differences were observed for TCV (10.6±0.5 cm/s for control monolayers, 5.1±0.5 cm/s with addition of CMFs, and 7.5±0.6 cm/s with addition of CFs) (Figure 5B). Finally, the addition of CMFs had a more dramatic effect on dV/dtmax and APD than the addition of CFs. DV/dtmax decreased from 4.9±0.1 (n=12) in control monolayers to 3.6±0.2 (n=8), p=10−7 with CMFs but to only 3.9±0.3 (n=8), p=10−4 with CFs, while APD increased from 145±6ms (n=21) in control monolayers to 185±7ms (n=11), p=10−4 with CMFs but insignificantly to only 150±5ms (n=7) with CFs.
Figure 5.
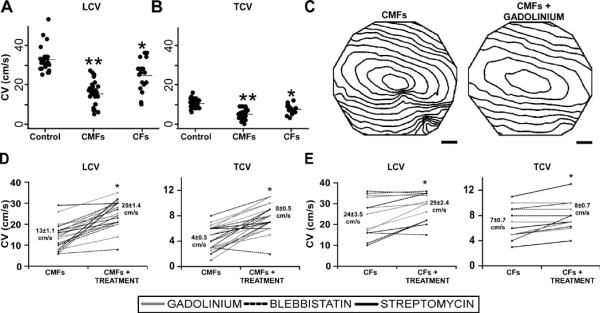
Drug effects on supplemented fibrotic model. LCV (A) and TCV (B) summary dot plots show that both the addition of CFs (n=17) and CMFs (n=26) to NRVC monolayers decreased CV compared with control monolayers (n=26), and that CV was significantly lower in CMF-supplemented monolayers as compared with CF-supplemented monolayers. *denotes significant difference from control monolayers, P<0.001; **denotes significant difference from control and CF monolayers, P<0.001. (C) Isochrone maps (10ms spacing) comparing CMF-supplemented monolayers before and after treatment with gadolinium at 2Hz pacing. Summary LCV and TCV line graphs of drug treatments for both CMF (D) and CF-supplemented monolayers (E). Blebbistatin (n=6) and MSC blockers (n=12) significantly increased LCV and TCV in CMF-supplemented monolayers (D), and to a lesser extent, in CF-supplemented monolayers (n=3 and 10, respectively) (E). * denotes significance after treatment, P<0.05.
Blebbistatin, gadolinium and streptomycin were again tested on the fibroblast-supplemented monolayers. Isochrone maps (shown for blebbistatin, Figure 5C) and line plots (Figure 5D) show that all treatments significantly increased LCV and TCV in CMF-supplemented monolayers and, to a lesser extent, in CF-supplemented monolayers (Figure 5E). Further, all three drugs significantly shortened APD (from 187±7ms to 166±6ms, n=12, p=10−3) and increased dV/dtmax (from 3.6±0.2 to 4.3±0.1, n=8, p=10−3) in CMF-supplemented monolayers but not in CF-supplemented monolayers (only changing from 150±5ms to 155±6ms, n=7, and from 3.9±0.3 to 4.1±0.1, n=8, respectively). Taken together, our results support the notion that the addition of TGF-β increases the contractile activity of CMFs, subsequently producing a greater impact on the electrophysiological function of NRVC monolayers.
Cx43 shRNA myofibroblasts retain slowing effects on NRVC monolayers
To assure a minimal degree of electrical coupling between myofibroblasts and myocytes, Cx43 was knocked down in myofibroblasts prior to supplementation, as confirmed with Western blots (Figure 6C). LCV of NRVC monolayers (27±1.7 cm/s, n=10) decreased to similar levels with the addition of scrambled shRNA CMFs (11±1.3 cm/s, n=11, p=10−7) or Cx43 shRNA CMFs (11±1.3 cm/s, n=8, p=10−6). Decreases in TCV were also similar (6±0.4 cm/s for control monolayers, 3±0.3 cm/s, p=10−6 with addition of scrambled shRNA CMFs, and 3±0.3 cm/s, p=10−5 with addition of Cx43 shRNA CMFs).
Figure 6.

Drug effects on monolayers supplemented with Cx43 shRNA myofibroblasts. Summary LCV (A) and TCV (B) line graphs for NRVC monolayers supplemented with myofibroblasts transduced with scrambled shRNA show significant increases in LCV and TCV with blebbistatin (n=2) and MSC blockers (n=6). A representative western blot (C) shows that purified fibroblasts transduced with Cx43 shRNA lentiviral particles have significantly less Cx43 expression compared with control fibroblasts (n=4, P<0.05). Summary LCV (D) and TCV (E) line graphs of drug treatments of NRVC monolayers supplemented with myofibroblasts transduced with Cx43 shRNA show that LCV and TCV again increased significantly with blebbistatin (n=2) and MSC blockers (n=6). Cx43 shRNA-transduced myofibroblasts contain SMA stress fibers (green), but insubstantial Cx43 (red) between the cellular contacts (denoted by white arrow) (F). * denotes significance after treatment, P <0. 05.
Blebbistatin, gadolinium and streptomycin were again tested on these monolayers. Figure 6 shows that all treatments significantly and similarly increased LCV and TCV in both scrambled and Cx43 shRNA–transduced CMF-supplemented monolayers. Further, compared with control, Cx43 shRNA CMF-supplemented monolayers had significantly lower dV/dtmax of propagating APs at 2Hz pacing, which was restored to near control levels following incubation with the aforementioned treatments (increasing from 2.6±0.1 to 3.5±0.2, n=8, p=0.01). Taken together, these results strengthen our findings that the contractile activity of CMFs is responsible for the electrophysiological consequences imparted by CMFs upon the NRVC monolayers.
Drug effects on the stress fiber organization and contractile force of myofibroblasts
Traction forces of individual cardiac fibroblasts were analyzed using elastic micropost arrays. Incubation with 5ng/ml TGF-β for 48–72 hours increased the cells' contractility by 50%, as measured by the total strain energy per cell imparted to the posts (E=183±12 fJ, n=100, Figure 7A), although untreated fibroblasts also had substantial contractility (E=120±8 fJ, n=93, p=10−5, Figure 7B). Further, for any given strain energy, TGF-β treatment increased the fraction of cells exerting at least that energy (Figure 7C). Myofibroblasts were also treated with blebbistatin, gadolinium or streptomycin for 20–30 minutes, and either immunostained for SMA to determine whether the drugs altered stress fiber organization, or analyzed for traction force changes using micropost arrays. Some of the CMFs incubated with blebbistatin experienced actin destabilization (Supplemental Figure 1C), likely through the inhibition of myosin II-actin interactions, and total strain energy decreased by 74% with blebbistatin (Figure 7E). On the other hand, CMFs treated with gadolinium had no apparent change in the SMA stress fiber organization (Supplemental Figure 1D), and similarly, no change was observed with streptomycin (not shown). Total strain energy did not significantly change with MSC blockers (Figure 7D). Finally, Cx43 shRNA transduced-CMFs showed no decrease in contractility compared with control CMFs (E=251±25 fJ, n=17, and 228±23 fJ, n=20 respectively, p=0.5) (Figure 7F), confirming the presence of contractile myofibroblasts in the supplemented Cx43 knockout model.
Figure 7.

Drug effects on contraction in cardiac myofibroblasts. Vector plots of traction forces at each post generated from an untreated cardiac fibroblast (A) and a TGF-β treated cardiac myofibroblast (B). Cumulative distribution from the same culture of fibroblasts (C) shows that at any given strain energy per cell, TGF-β treatment increases the proportion of cells imparting at least that energy to the posts (n=36 for untreated fibroblasts and n=42 for TGF-β-induced myofibroblasts). MSC blockers, streptomycin (n=9) or gadolinium (n=10), had no significant effect on the average myofibroblast strain energy (D), while blebbistatin (n=10) significantly reduced the average strain energy by 74% (E). Cumulative distribution shows that at any given strain energy per cell, control myofibroblasts and Cx43 shRNA myofibroblasts have a similar proportion of cells imparting at least that energy to the posts (F). * denotes significance after treatment, P<0.05.
Contraction and MSC inhibitors reduce spiral wave vulnerability in fibrotic monolayers
Because conduction velocity increased in fibrotic monolayers treated with blebbistatin, gadolinium, or streptomycin, we investigated the drug effects on susceptibility to rapid pacing-induced spiral waves (Figure 8A,B). The occurrence of spiral waves decreased in fibrotic monolayers after treatment (Figure 8C), despite the fact that it was possible to stimulate the treated monolayers at significantly faster capture rates (MCL decreased by 90ms, n=12, Figure 8D), which normally facilitates wave breaks. The pacing cycle length required to induce spiral waves in the treated monolayers also shortened significantly by 80ms (n=13, Figure 8C). Therefore, our results suggest that contraction or MSC inhibitors reduce susceptibility to arrhythmias in fibrotic NRVC monolayers.
Figure 8.

Spiral wave vulnerability decreases with contraction and MSC inhibitors. Voltage maps in specified time increments in a fibrotic monolayer before (A) and after (B) blebbistatin. The color bar indicates the normalized transmembrane potential: red represents peak depolarized potential and blue represents resting potential. Before treatment, a spiral wave was initiated during 3Hz pacing, due to wave breaks around two large regions of block (indicated by the pair of double lines at 110ms). After treatment, one region of block was eliminated and the other region was much smaller; subsequently, conduction was able to propagate across the monolayer without initiating reentry. Overall, the occurrence of spiral waves decreased in fibrotic monolayers after treatment with blebbistatin, gadolinium, or streptomycin (n=16, p=0.07) (C). Both the pacing cycle length needed to initiate spiral waves, as denoted by the numbers above the bar graphs in (C), and the minimum cycle length before loss of 1:1 capture (D) decreased in fibrotic monolayers after treatment. * denotes significant difference after treatment, P<0.05.
Discussion
Following an acute injury to the heart, as in myocardial infarction, fibroblasts participate in the wound healing process. Because of the limited capability of cardiomyocytes to regenerate, wound healing concludes with loss of ventricular muscle and formation of a stable scar, ultimately leading to a fibrotic, arrhythmogenic substrate.9 The myofibroblast repairs the injured region through the formation of granulation tissue,5,18 using contraction to reduce the total scar volume.19 It is well known that mechanoelectric coupling among cardiac cells, the process in which mechanical perturbations alter electrical activity, exists in the intact heart20 and in cardiac cell cultures.21 Cardiomyocytes also possess MSCs whose open probability increases with stretch22 and whose mechanical sensitivity increases following myocardial infarction.23 Thus, we tested the hypothesis that increased vulnerability to arrhythmias following cardiac injury could be related to a tonic tugging force that myofibroblasts exert on the cardiomyocyte membrane through mechanical coupling and, lead to conduction slowing and block through the action of MSCs.24
We used TGF-β in our fibrotic models to induce fibroblast differentiation to contractile myofibroblasts. Although fibroblasts in culture transition to proto-myofibroblasts that express some stress fibers,18 incubation with TGF-β increases complete differentiation to myofibroblasts,25 expression of SMA,5 and generation of strong contractile forces (Figure 7). Our results demonstrate that co-cultures of cardiomyocytes and untreated fibroblasts have a significant reduction in conduction velocity compared with NRVC control monolayers, similar to previous reports in the literature for co-cultures of cardiomyocytes with non-TGF-β treated fibroblasts.11 However, our experiments involving the supplementation of NRVC monolayers with TGF-β treated myofibroblasts suggest that conduction slowing is exacerbated well beyond that obtained with supplementation using untreated fibroblasts (Figure 5A,B), without a concordant increase in Cx43 expression (Figure 2F,G).
The recent perspective that fibroblasts may have an active electrical influence on cardiac electrophysiology has been bolstered by discoveries of in vitro and in situ gap junctional coupling between cardiac fibroblasts and cardiomyocytes,3,26 and by characterization of multiple fibroblast ion channels,27 including K channels,28 Na+-Ca2+ exchanger,19 and mechanosensitive channels.29 Numerous studies have focused on the potential electrophysiological consequences of electrical coupling between fibroblasts and cardiomyocytes,30–32 and the general dogma of these studies is that through electrical coupling, fibroblasts actively depolarize resting cardiomyocytes because of their more positive resting membrane potential.
However, in our studies, immunolabeling for Cx43 and pan-cadherin of fibroblast-only cultures suggests a different mechanism by which myofibroblasts and cardiomyocytes can interact. We found that SMA-negative fibroblasts expressed abundant Cx43, whereas SMA-positive myofibroblasts had little Cx43 expression but enhanced pan-cadherin expression (Figure 2C–E), suggesting that myofibroblasts form strong mechanical rather than electrical junctions. However, TGF-β treated myofibroblasts did have significantly more soluble Cx43 (Figure 2G), indicating that Cx43 is still present in the cytoplasm. This finding is consistent with recent evidence demonstrating that Cx43 is necessary for fibroblast differentiation into myofibroblasts.33 In heterocellular contacts between adjoining cardiomyocytes and myofibroblasts, we also found that adherens junction expression dominated over gap junction expression (Figure 2B). Our key finding that myofibroblast-induced slowing of conduction could be restored with contraction or MSC blockers (Figures 3, 5) was unchanged with connexin43 silencing in the myofibroblasts (Figure 6). Further, given that changes in myofibroblast contractile force (Figure 7) paralleled the changes in conduction velocity brought about by both TGF-β and blebbistatin (Figure 1,3,5), and considering the plethora of work demonstrating the significance of myofibroblast contraction in other tissues,5 we believe that mechanical coupling between myofibroblasts and cardiomyocytes is an important factor contributing to conduction slowing in co-cultures of these two cell types. We suggest that this form of mechanical signaling can activate MSCs that depolarize the cardiomyocyte membrane,34 thereby inactivating the Na+ channels that drive conduction. Further, heterogeneity in the distribution of myofibroblasts can accentuate spatial gradients in conduction and action potential duration, leading to increased propensity to reentrant arrhythmias that again, may be suppressed by MSC or contraction blockers (Figure 8). As a caveat, we cannot rule out the possibility of paracrine signaling between myofibroblasts and cardiomyocytes as a means of modulating cardiac conduction, although the continuous exchange of solution in our experimental setup suggests that paracrine factors do not play a dominant role.
We believe that our in vitro model, consisting of longitudinally patterned myocytes and adjacent contractile SMA myofibroblasts, retains several key aspects of the environment in a healing infarct. Previous studies have shown that both SMA positive myofibroblasts35 and TGF-β are markedly expressed36 during infarction, and can remain elevated for several months to years in the infarct border zone.37,38 Further, although numerous studies have shown that Cx43 is downregulated and redistributed post infarction,39,40 cadherin expression levels and distribution remain unaffected or are downregulated to a lesser degree compared with Cx43.40 On the other hand, our results are specific to in vitro conditions, and it remains to be seen whether they extrapolate to the in vivo heart in light of known differences between the two environments. First, our cells are maintained under culture conditions (culture media, 2D rigid substrate, lack of hemodynamic loading), which can affect cell shape, protein expression and cell function.21 Second, the spatial distribution of heterocellular gap junctions and adherens junctions in cell monolayers may differ significantly from that found in vivo. Although myofibroblast-myocyte junctions have not been characterized in the intact heart, in normal myocardium, myocyte-myocyte junctions occur primarily within intercalated disks, in contrast to their distribution around the cell perimeter in cell culture,21 although in infarcted myocardium the distribution also tends to occur around the cell perimeter.40 If future studies confirm our findings in infarcted regions of the heart in vivo, it is plausible that mechanosensitive channel blockers or fibroblast-specific contraction inhibitors may provide a means to reduce the incidence of arrhythmias.
To conclude, our data demonstrate that impaired conduction in an in vitro fibrotic model is mechanically dependent. Furthermore, our findings support the current view that myofibroblasts are capable of actively decreasing conduction among cardiomyocytes, and suggest that mechanical coupling between myofibroblasts and cardiomyocytes can play a more prominent role in this regard than electrical coupling. Finally, we propose a novel mechanism in which myofibroblasts may impair cardiomyocyte electrophysiological function through the application of contractile force to the cardiomyocyte membrane and activation of mechanosensitive channels.
Supplementary Material
Clinical perspective.
Myocardial infarction engages a fibrotic process in which myofibroblasts secrete extracellular matrix proteins to replace the injured tissue. The contractile properties of myofibroblasts help to ensure a smaller and stronger scar area and to preserve mechanical function. However, in the infarct border zone, arrhythmias are prone to initiate owing to slowed and heterogeneous conduction. Though fibrosis has classically been considered to be arrhythmogenic because of the creation of an unexcitable region, together with zigzag conduction in the border zone, we tested the hypothesis that myofibroblasts can actively influence electrophysiological function through mechanoelectric coupling to the cardiomyocytes. In this study, impaired electrical conduction in co-cultured monolayers of myofibroblasts and cardiomyocytes can be dramatically improved by applying an excitation-contraction inhibitor or mechanosensitive channel blockers. Our findings advocate a novel mechanism whereby cardiac myofibroblasts exert tension on the myocyte membrane, which leads to slowed and heterogeneous electrical conduction through the action of mechanosensitive ion channels. Provided that these in vitro results are corroborated in the intact heart, inhibition of this form of mechanoelectric interaction in the heart may be a way to decrease susceptibility to arrhythmias.
Acknowledgements
We wish to thank Seth Weinberg for helpful discussions and Christopher S. Chen for providing expertise and materials for fabricating micropost arrays.
Funding Sources Funding for this work was provided by NIH grant R01-HL066239 (L.T.), a Grant-in-Aid from the Mid-Atlantic Affiliate of the American Heart Association (L.T.) and National Science Foundation Integrative Graduate Education and Research Traineeship (S.A.T.,C.R.C.).
Conflict of Interest Disclosures Funding for this work was provided by NIH grant R01-HL066239 (L.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann N Y Acad Sci. 2006;1080:76–84. doi: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- 2.Souders CA, Bowers SLK, Baudino TA. Cardiac Fibroblast: The Renaissance Cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Brown RD, Ambler SK, Mitchell MD, Long CS. The Cardiac Fibroblast: Therapeutic Target in Myocardial Remodeling and Failure. Annual Review of Pharmacology and Toxicology. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 5.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat M-L, Gabbiani G. The Myofibroblast: One Function, Multiple Origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 7.Branton MH, Kopp JB. TGF-β and fibrosis. Microbes and Infection. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 8.Drobic V, Cunnington RH, Bedosky KM, Raizman JE, Elimban VV, Rattan SG, Dixon IMC. Differential and combined effects of cardiotrophin-1 and TGF-β1 on cardiac myofibroblast proliferation and contraction. Am J Physiol Heart Circ Physiol. 2007;293:H1053–1064. doi: 10.1152/ajpheart.00935.2006. [DOI] [PubMed] [Google Scholar]
- 9.van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 10.Xie Y, Garfinkel A, Camelliti P, Kohl P, Weiss JN, Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Heart Rhythm. 2009;6:1641–1649. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohr S. Myofibroblasts in diseased hearts: New players in cardiac arrhythmias? Heart Rhythm. 2009;6(6):848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 12.Kohl P, Camelliti P, Burton FL, Smith GL. Electrical coupling of fibroblasts and myocytes: relevance for cardiac propagation. Journal of Electrocardiology. 2005;38(Supplement 1):45–50. doi: 10.1016/j.jelectrocard.2005.06.096. [DOI] [PubMed] [Google Scholar]
- 13.Pedrotty DM, Klinger RY, Badie N, Hinds S, Kardashian A, Bursac N. Structural coupling of cardiomyocytes and noncardiomyocytes: quantitative comparisons using a novel micropatterned cell pair assay. Am J Physiol Heart Circ Physiol. 2008;295:H390–400. doi: 10.1152/ajpheart.91531.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wipff P-J, Hinz B. Myofibroblasts work best under stress. Journal of Bodywork and Movement Therapies. 2009;13:121–127. doi: 10.1016/j.jbmt.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 15.Follonier L, Schaub S, Meister J-J, Hinz B. Myofibroblast communication is controlled by intercellular mechanical coupling. J Cell Sci. 2008;121:3305–3316. doi: 10.1242/jcs.024521. [DOI] [PubMed] [Google Scholar]
- 16.Sniadecki NJ, Lamb CM, Liu Y, Chen CS, Reich DH. Magnetic microposts for mechanical stimulation of biological cells: fabrication, characterization, and analysis. Rev Sci Instrum. 2008;79 doi: 10.1063/1.2906228. 044302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, van Grunsven LA, Van Rossen E, Schroyen B, Timmermans JP, Geerts A, Reynaert H. Blebbistatin inhibits contraction and accelerates migration in mouse hepatic stellate cells. Br J Pharmacol. 2010;159:304–315. doi: 10.1111/j.1476-5381.2009.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. The Journal of Pathology. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 19.Raizman JE, Komljenovic J, Chang R, Deng C, Bedosky KM, Rattan SG, Cunnington RH, Freed DH, Dixon IM. The participation of the Na+-Ca2+ exchanger in primary cardiac myofibroblast migration, contraction, and proliferation. J Cell Physiol. 2007;213:540–551. doi: 10.1002/jcp.21134. [DOI] [PubMed] [Google Scholar]
- 20.Franz MR. Mechano-electrical feedback in ventricular myocardium. Cardiovascular Research. 1996;32:15–24. [PubMed] [Google Scholar]
- 21.Tung L, Thompson S. Advantages and pitfalls of cell cultures as model systems to study cardiac mechanoelectric coupling. In: Kohl P, Sachs F, Franz MR, editors. Cardiac Mechano-Electric Coupling and Arrhythmias. 2nd ed. Oxford University Press; Oxford, U.K.: In press: chap 20. [Google Scholar]
- 22.Akay M, Craelius W. Mechanoelectrical feedback in cardiac myocytes from stretch-activated ion channels. IEEE Trans Biomed Eng. 1993;40:811–816. doi: 10.1109/10.238466. [DOI] [PubMed] [Google Scholar]
- 23.Kamkin A, Kiseleva I, Wagner K-D, Leiterer KP, Theres H, Scholz H, Günther J, Lab MJ. Mechano-Electric Feedback in Right Atrium After Left Ventricular Infarction in Rats. Journal of Molecular and Cellular Cardiology. 2000;32:465–477. doi: 10.1006/jmcc.1999.1091. [DOI] [PubMed] [Google Scholar]
- 24.Kuijpers NHL, ten Eikelder HMM, Bovendeerd PHM, Verheule S, Arts T, Hilbers PAJ. Mechanoelectric feedback leads to conduction slowing and block in acutely dilated atria: a modeling study of cardiac electromechanics. Am J Physiol Heart Circ Physiol. 2007;292:H2832–2853. doi: 10.1152/ajpheart.00923.2006. [DOI] [PubMed] [Google Scholar]
- 25.Vaughan MB, Howard EW, Tomasek JJ. Transforming Growth Factor-β1 Promotes the Morphological and Functional Differentiation of the Myofibroblast. Experimental Cell Research. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- 26.Rook MB, van Ginneken AC, de Jonge B, el Aoumari A, Gros D, Jongsma HJ. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am J Physiol Cell Physiol. 1992;263:C959–977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 27.Li G-R, Sun H-Y, Chen J-B, Zhou Y, Tse H-F, Lau C-P. Characterization of Multiple Ion Channels in Cultured Human Cardiac Fibroblasts. PLoS ONE. 2009;4:e7307. doi: 10.1371/journal.pone.0007307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benamer N, Moha Ou Maati H, Demolombe S, Cantereau A, Delwail A, Bois P, Bescond J, Faivre J-F. Molecular and functional characterization of a new potassium conductance in mouse ventricular fibroblasts. Journal of Molecular and Cellular Cardiology. 2009;46:508–517. doi: 10.1016/j.yjmcc.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Kamkin A, Kiseleva I, Isenberg G, Wagner K-D, Günther J, Theres H, Scholz H. Cardiac fibroblasts and the mechano-electric feedback mechanism in healthy and diseased hearts. Progress in Biophysics and Molecular Biology. 2003;82:111–120. doi: 10.1016/s0079-6107(03)00009-9. [DOI] [PubMed] [Google Scholar]
- 30.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of Cardiac Electrical Activity Over Extended Distances by Fibroblasts of Cardiac Origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 31.Miragoli M, Gaudesius G, Rohr S. Electrotonic Modulation of Cardiac Impulse Conduction by Myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 32.Jacquemet V, Henriquez CS. Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am J Physiol Heart Circ Physiol. 2008;294:H2040–2052. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asazuma-Nakamura Y, Dai P, Harada Y, Jiang Y, Hamaoka K, Takamatsu T. Cx43 contributes to TGF-β signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Exp Cell Res. 2009;315:1190–1199. doi: 10.1016/j.yexcr.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 34.Kamkin A, Kiseleva I, Isenberg G. Stretch-activated currents in ventricular myocytes: amplitude and arrhythmogenic effects increase with hypertrophy. Cardiovasc Res. 2000;48:409–420. doi: 10.1016/s0008-6363(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 35.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovascular Research. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 36.Bujak M, Frangogiannis NG. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovascular Research. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-β1 in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24. doi: 10.1111/j.1365-2567.2006.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 39.Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, Hirst-Jensen BJ, Duffy HS, Sorgen PL. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–1112. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noorman M, van der Heyden MAG, van Veen TAB, Cox MGPJ, Hauer RNW, de Bakker JMT, van Rijen HVM. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. Journal of Molecular and Cellular Cardiology. 2009;47:23–31. doi: 10.1016/j.yjmcc.2009.03.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.