

Abstract
High-speed countercurrent chromatography (HSCCC) and preparative high-performance liquid chromatography (prep-HPLC) were successively used for the separation of pogostone and four flavonoids from Pogostemon cablin (Blanco) Benth. An efficient HSCCC separation was achieved on a two-phase solvent system composed of n-hexane–ethyl acetate–methanol–water (11:5:11:5, v/v/v/v). Three well-separated peaks were obtained in the HSCCC chromatogram. The first and the second fractions each contained two flavonoids which were further separated by preparative HPLC. Consequently, the separation yielded 11.5 mg of 4′, 5-Dihydroxy-3′, 7-dimethoxyflavanone at a purity of 99%, 20.3 mg of 5- Hydroxy-7, 3′, 4′-trimethoxyflavanone at a purity of 98%, 18 mg of 5, 4′-Dihydroxy-3, 7, 3′-trimethoxyflavone at a purity of 96%, and 8 mg of 5-Hydroxy-3, 7, 4′-tetramethoxyflvone at a purity of 98%. The third HSCCC fraction yielded 18.5 mg of pogostone at a purity of 95%. The chemical structures of these compounds were identified by ESI-MSn, 1H-NMR, and 13C-NMR
Keywords: Pogostemon cablin (Blanco) Benth, Flavonoid, High-speed countercurrent chromatography, Preparative HPLC
INTRODUTION
Pogostemon cablin (Blanco) Benth native in Philippines has been cultivated extensively in Indonesia, Malaysia, China, and Brazil. The plant is a famous traditional Chinese medicine widely used in southern China. It has been used as traditional Chinese herbal medicine to remove dampness, relieve summer-heart, exterior syndrome, stop vomiting and stimulate the appetite[1]. The essential oil of P. cablin collected by steam distillation has been found to exhibit significant antifungal and antibacterial activities in vitro (MIC=0.08~1.0mg/L)[2]. Its alcoholic extract also showed some antifungal activities (MIC<2mg/L)[3]. The bioactive constituents of this herb are flavonoids, including 3, 5-Dihydroxy-7, 4′-dimethoxyflavone, 5-Hydroxy-7, 3′, 4′-trimethoxy-flavanone, pogostone, etc[3]. The chemical structures of these compounds are given in Fig. 1. These polyphenolic compounds have shown promising results for the treatment of coronary heart disease and used as anti-cancer, anti-aging, and anti-inflammatory agents [4–6]. In view of their pharmacological activities and broad applications, a large quantity of pure materials is urgently needed for further pharmacological studies. Therefore, development of an effective method for isolation and purification of flavonoids from P. cablin is warranted.
Fig.1.

HPLC chromatogram of n-hexane extract from P. cablin.. Conditions: column, reversed-phase Ultimate C18 (250 mm × 4.6 mm i.d., 5μm); mobile phase, methanol-0.1% aqueous acetic acid in gradient mode (methanol: 0–45min, 60–82%); flow rate, 1.0 ml/min; UV wavelength: 276 nm.
The separation and purification of flavonoids from P. cablin by classical methods require multiple chromatographic steps using silica gel, polyamide column, etc. [3, 6, 7], which are tedious, time consuming, and often cause loss of target compounds due to the highly adsorptive effect of the solid matrix. High-speed countercurrent chromatography (HSCCC) is a form of liquid–liquid partition chromatographic technique that uses no support solid matrix where the liquid stationary phase is retained by centrifugal force, hence eliminating irreversible adsorption of sample onto the solid matrix resulting in excellent sample recovery[8]. In addition the method permits introduction of crude samples into the column without extensive preparation. The method has been successfully applied to isolate and purify a number of natural products[8–11]. While HSCCC has many advantages as mentioned above, it also has some disadvantages. In our study, it was difficult to obtain the compound with the purity of over 90% by HSCCC in one step separation, especially when the system was complex and K values of target compounds are similar. While in many cases, preparative high-performance liquid chromatography (prep-HPLC) is a method needed to satisfy the purity specifications required on a routine basis, and it is also an important industrial separation process for the isolation and purification of pharmaceuticals and other valuable products[12]. The aim of the present study was to develop a method using HSCCC and prep-HPLC for purifying flavonoids from P. cablin.
In the present studies, the two-phase solvent system composed of n-hexane-ethyl acetate-methanol-water(11:5:11:5, v/v/v/v) was used for preparative separation and purification of the five flavonoids in HSCCC. The partially purified fractions were further separated by reversed-phase prep-HPLC.
EXPERIMENTAL
Apparatus
The HSCCC instrument employed in present study is a GS10A-2 high-speed counter-current chromatograph (Beijing Institute of New Technology Application, Beijing, China), consisting of a type-J coil planet centrifuge equipped with a polytetrafluoroethylene multilayer coil separation column with a total capacity of 230 ml. The β value of this preparative column varied from 0.5 at the internal terminal to 0.7 at the external terminal (β = r/R, where r is the rotation radius or the distance from the coil to the holder shaft, and R is the revolution radius or the distances between the holder axis and central axis of the centrifuge). The revolution speed of the apparatus can be regulated from 0 to 1000 rpm. The system was also equipped with one NS-1007 constant flow pump, a Model 8823A-UV monitor operating at 254 nm, and a Yakogawa 3057 recorder.
The analytical HPLC system used in this study consisted of a Shimadzu LC-10ATvp Multisolvent Delivery System, a Shimadzu SPD-M10Avp UV detector, an injection valve (Model 7726) with a 20 μL loop, a system controller (SCL-10AVP) and a Shimadzu LC Solution Workstation (Shimadzu, Kyoto, Japan). The column applied in this work was a Ultimate C18 column (250 mm × 4.6 mm i.d., 5 μm, Welch Materials, Shanghai, china).
The semi-preparative HPLC system consisted of a Waters 600 pump, a 2487 detector (Waters, USA), a 5 ml sample loop, the model HSV4.0+ workstation (Empower, Hangzhou, China) and an YMC C18 column (250 mm×10.0 mm, i.d. 5μm, YMC Co. Ltd. Japan)
The nuclear magnetic resonance (NMR) spectrometer used here was a Mercury Plus 400 NMR system (Varian Inc., USA)., and the electro-spray mass spectrometer was an LCQ Deca XP Max system (Finnigan, USA).
Reagents
All organic solvents used for preparation of crude samples and HSCCC separation were of analytical grade (Damao Chemical Reagent Factory, Tianjin, China). Methanol used for HPLC analysis was of chromatographic grade and was purchased from Guangzhou Dikma Co., Ltd.(Guangzhou, China). P. cablin. was purchased from Guangzhou Traditional Chinese Medicine, Ltd. (Guangzhou, China) and indentified by Dr. Qingzhen Ran (Guangzhou University of Chinese Medicine, Guangzhou, China).
Preparation of Crude Sample
One kilogram of P. cablin was shattered to powder (about 30 mesh) and extracted with 3500 ml volume of 80% ethanol by reflux extraction for three times at 80 °C. The extraction duration was 3, 2 and 1 h, respectively. After filtration, the extracts were combined and evaporated to dryness under reduced pressure at 60°C, and the residue was redissolved in 20% ethanol (total volume 600 ml). The enriched extract was then extracted with n-hexane three times (600 ml for each time). All the n-hexane extracts were combined and evaporated to dryness under reduced pressure at 60°C, yielding 10.8 g of crude sample. It was stored in a refrigerator at 4°C for subsequent HSCCC separation.
Selection of Two-Phase Solvent System
n-Hexane-ethyl acetate-methanol-water was used as the two-phase solvent system of HSCCC. The composition of the two-phase solvent system was selected according to the partition coefficient (K) of each target component. The K values were determined by HPLC as follows: a suitable amount of crude extract was dissolved in the lower phase of the two phase solvent system. The solution was then analyzed by HPLC. The peak area was recorded as A1. An equal volume of the upper phase was then added to the solution and mixed thoroughly. After partition equilibration was reached, the lower phase solution was determined by HPLC again, and the peak area was recorded as A2. The K-values were calculated according to the following equation: K= (A1 − A2)/A2.
Preparation of Two-Phase Solvent System
In the present study, n-hexane–ethyl acetate–methanol–water (11:5:11:5, v/v) was used as the two-phase solvent system for HSCCC separation. It was prepared by adding the solvents to a separation funnel according to their volume ratios followed by thorough equilibration by repeated shaking. Then, the two phases were separated and degassed by sonication for 30 min shortly before use. The sample solution was prepared by dissolving 300 mg of the crude extract in 10 ml a mixture of lower and upper phases (1:1, v/v) of the solvent system used for HSCCC separation.
HSCCC Separation Procedure
HSCCC was performed as follows. The multilayer coiled column was first entirely filled with the upper phase as a stationary phase. The lower aqueous mobile phase was then pumped into the head end of the column inlet at a flow rate of l.5 ml/min, while the apparatus was run at a revolution speed of 800 rpm. After hydrodynamic equilibrium was established, as indicated by a clear mobile phase eluting from the tail outlet, the sample solution was injected through the injection valve. The effluent from the tail end of the column was continuously monitored with a UV detector at 254 nm and the chromatogram was recorded. Each peak fraction was collected according to the elution profile and analyzed by HPLC. After the separation was completed, retention of the stationary phase was measured by collecting the column contents by forcing them out of the column with pressurized nitrogen gas.
Preparative HPLC Purification Procedure
The partially purified fractions of HSCCC (fractions “I ”and “II”) was evaporated to dryness under reduced pressure at 60°C and each dissolved in the mobile phase at a concentration of about 30 mg/ml. The separation was perform with a YMC C18 column (250 mm × 10.0 mm, i. d., 5μm, YMC Co., Ltd., Japan) at a column temperature of 30°C. After 4 ml of sample solution was injected, the mobile phase composed of methanol-0.1% aqueous acetic acid (70:30, v/v, for fraction “I”) and (75:25, v/v, for fraction “II”) was isocratically eluted at a flow-rate of 5 ml/min. The effluent was monitored at 276 nm and peak fraction was collected according to the elution profile.
HPLC Analysis and Identification of HSCCC Peak Fractions and Preparative HPLC Fractions
The n-hexane extract from the P. calbin and peak fraction obtained by HSCCC and preparative HPLC were analyzed by HPLC. The analyses were performed with a Ultimate C18 column (250 mm × 4.6 mm i.d., 5 μm, Welch Materials, Shanghai, china). The mobile phase, composed of methanol-0.1% aqueous acetic acid was eluted with linear gradient elution (methanol: 0–45 min, 60–82%). The total flow rate was 1.0 ml/min and the effluent was monitored by a Shimadzu SPD10Avp UV detector.
Identification of both HSCCC peak fractions and prep-HPLC peak fractions was carried out by ESI-MS, 1H-NMR and 13C-NMR.
RESULTS AND DISCUSSION
Optimization of HPLC Separation Condition
The crude extract and the fractions obtained by HSCCC and prep-HPLC were analyzed by HPLC. In order to achieve a resolution for each target comound, several mobile phases such as methanol–water and methanol–aqueous acetic acid with different concentrations of acetic acid were tested with various flow rates and eluted modes.
The results indicated that the best separations would be achieved using methanol–0.1% aqueous acetic acid in a gradient mode (methanol: 0–45min, 60–82%) at a flow rate of 1.0 ml/min with a column temperature of 30°C and an effluent monitoring wavelength of 276 nm. Under the above conditions, a satisfactory separation of targeted compounds was obtained (Fig. 1). The crude n-hexane extract and peak fractions separated by HSCCC and preparative HPLC were analyzed by HPLC under the above optimum conditions.
Selection of Two-Phase Solvent System of HSCCC
A successful separation of the target compounds using HSCCC requires a suitable two-phase solvent system using the following considerations[10,13]: (1) the setting time of the solvent system should be shorter than 30 s for satisfactory retention of the stationary phase; and (2) the partition coefficient (K) should be close to 1. In general, small K values usually result in poor peak resolution, while large K values tend to produce excessive sample band broadening.
In this work, a simple method was used to select two-phase solvent systems. If the sample is an extract of plant material, the search may be started at any point according to the polarity of the extracts; if the sample is moderate polarity, the search may begin at n-hexane–ethyl acetate–methanol–water (1:1:1:1, v/v/v/v), whereas if the sample is a strong polarity, the search may be started at 1-butanol–ethyl acetate–water (4:1:4, v/v/v), suggested by Ito[14]. The mixing ratio of the solvents may be optimized according to the K values and the solubility of sample. In our research, the sample was n-hexane extract which is probably identified as moderately hydrophobic compounds. Then n-hexane–ethyl acetate–methanol–water was selected as a two-phase solvent system which has been used to separate a broad range of compounds based on their hydrophobicity by modifying the volume ratio of the four components. The K values of target compounds obtained from n-hexane–ethyl acetate–methanol–water at various volume ratios ranging from 4:5:4:5 to 3:1:3:1 (v/v) were tested to isolate the crude extract as shown in Table 1.
Table 1.
K-values of the target compounds in several two-phase solvent systems
| Solvent system (n-hexane–ethyl acetate–methanol –water) (v/v) |
K-value of the peaks
|
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| 4:5:4:5 | 9.43 | 16.02 | 8.92 | 24.83 | 24.83 |
| 1:1:1:1 | 3.85 | 7.50 | 3.44 | 18.15 | 18.15 |
| 3:2:3:2 | 1.05 | 2.48 | 0.98 | 7.90 | 7.90 |
| 2:1:2:1 | 0.25 | 0.87 | 0.25 | 3.79 | 3.79 |
| 11:5:11:5 | 0.22 | 0.72 | 0.22 | 3.38 | 3.38 |
| 13:5:13:5 | 0.14 | 0.60 | 0.14 | 2.86 | 2.86 |
| 3:1:3:1 | 0.11 | 0.45 | 0.11 | 2.35 | 2.35 |
The result indicated that the solvent system composed of n-hexane–ethyl acetate–methanol–water at the volume ratios of 1:1:1:1, 4:5:4:5 and 3:2:3:2 (v/v) had too large K values, while the volume ratios of 13:5:13:5 and 3:1:3:1 (v/v) produced too small K value. The appropriate K values are obtained at two volume ratios of 2:1:2:1(v/v) and 11:5:11:5 (v/v). Since K value of the compound 4 at 11:5:11:5 (v/v) is smaller than that of 2:1:2:1 (v/v), the two-phase solvent system composed of n-hexane–ethyl acetate–methanol–water at a volume ratio of 11:5:11:5 was selected to separate the target compounds in the present study. A preliminary HSCCC experiment at a constant flow rate of 1.5 ml/min yielded three well resolved fractions from 300 mg crude extract in 4.5 h and at high retention of the stationary phase of 78% (Fig. 2).
Fig. 2.
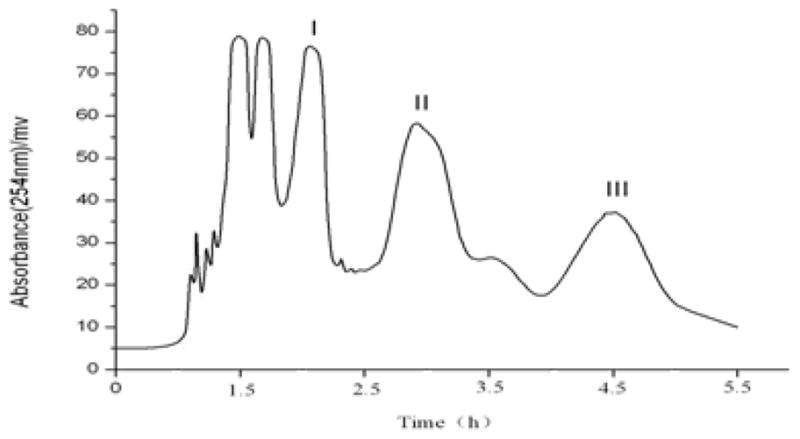
Preparative HSCCC chromatogram of the n-hexane extract from P. cablin.; solvent system: n-hexane-ethyl acetate–methanol–water (11:5:11:5, v/v/v/v); stationary phase: upper phase; mobile phase: lower phase; flow rate: 1.5 ml/min; revolution speed: 800 rpm; sample: 300 mg dissolved in 10 ml mixture solution of each phase (1:1, v/v).
Since the K value of compound “1” is equal to that of compound “3”, these two compounds cannot be separated by HSCCC. On the other hand the compounds “4” and “5” are not resolved in HPLC and therefore their K values are not accurately determined by analytical HPLC. In fact, it turned out that the K value of compound “2” is similar to compound “4” and it was difficult to separate them by HSCCC as well. However, HSCCC can easily separate compounds “4” and “5” which cannot be resolved by HPLC. As shown in Fig. 3, the HPLC analysis of each HSCCC fraction revealed that two mixtures and one pure flavonoid were obtained from the crude extract, i.e., fraction “I” (76.8 mg) which mainly contained compounds “1” and “3”, fraction “II” (52.5 mg) which mainly contained compounds “2” and “4”, and fraction “III” which contained compound “5” (18.5 mg) at a purity of 95%.
Fig.3.

HPLC analyses of partially purified fractions of HSCCC. Conditions: the same as showed in Fig. 3. (a): HSCCC fraction I (b): HSCCC fraction II and (c): HSCCC fraction III (the purity = 95%).
Optimization of Prep-HPLC Separation Conditions
As showed in Fig. 3, fraction “I” and fraction “II” from HSCCC were mixtures each containing two target compounds. In order to obtain pure materials, these fractions were subjected to prep-HPLC for further purification. The prep-HPLC mobile phase was selected based on the polarity of the two compounds and the analytical HPLC conditions. Various concentrations of methanol (60%, 65%, 70%, 75%, and 80%) were tested at flow rates of 3 ml, 4 ml, and 5 ml. The results indicated that the best separation conditions were achieved using methanol 0.1% aqueous acetic acid with the isocratic elution mode (0–25 min, 70:30, fraction “I”) and (0–20 min, 75:25, fraction “II”), respectively, at a flow rate of 5.0 ml/min with a column temperature of 30°C and an effluent monitoring wavelength of 276 nm (Fig. 4). Under the above conditions, a satisfactory separation of each targeted compound was achieved. The effluent of each target compound (in peaks “1”, “2”, “3” and “4”) was collected and analyzed by HPLC (Fig. 5). The results indicated that the purities of fraction “1” (4′, 5-Dihydroxy-3′, 7 dimethoxyflavanone, 11.5 mg), fraction “2” (5-Hydroxy-7, 3′, 4′-trimethoxyflavanone, 20.3 mg), fraction “3” (5, 4′-Dihydroxy-3, 7, 3′-trimethoxyflavone, 18 mg) and fraction “4” (5-Hydroxy-3, 7, 3′, 4′-tetramethoxyflvone, 8 mg) were 99%, 98%, 96% and 98%, respectively.
Fig. 4.


Prep-HPLC chromatograms of peak I and peak II in HSCCC. Conditions: reversed phase C18 column (250×10 mm I.D.,5μm, YMC-Pack). Mobile phase: (a) fraction “I”: methanol–0.1% aqueous acetic acid (70: 30, v/v), (b) fraction “II”: methanol–0.1% aqueous acetic acid (75: 25, v/v); flow rate,5 ml/min; detection wavelength, 276 nm.
Fig. 5.


HPLC analyses of Prep- HPLC fractions. HPLC conditions: the same as those in Fig. 1. (a) peak 1 (b) peak 2 (c) peak 3, and (d) peak 4
Identification of Chemical Structures of Target Compounds
The structure of each target compound was identified by MS, 1H-NMR and 13C-NMR as shown in Fig. 6.
Fig. 6.

Chemical structures of target compounds
Fraction “1”: λmax UV (MeOH): 216, 287 nm; ESI-MS m/z: 315 (M-H)−; 1H-NMR (400MHz, CDCl3) δ: 12.02 (1H, s, 5-OH), 8.00 (1H, s, H-2′), 6.96 (1H, d, H-6′), 6.93 (1H, d, H-5′), 6.05 (2H, d, H-6, 8), 5.70 (1H, s, H-4′), 5.35 (1H, dd, H-2), 3.92 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.09 (1H, dd, Hax-3), 2.74 (1H, dd, Heq-3); δ: 13C-NMR (400MHz, CDCl3): δ: 196.0 (C-4, C=O), 167.9 (C-7, C), 164.1 (C-5, C), 162.8 (C-9, C), 146.7 (C-3′, CH), 146.2 (C-4′, C), 130.2 (C-1′, C), 119.6 (C-5′, CH), 114.5 (C-2′, CH), 108.7 (C-10, C), 103.1 (C-6′, CH), 95.1 (C-6, CH), 94.2 (C-8, CH), 79.3 (C-2, CH), 55.7 (C-3, CH2), 43.4 (C-3, CH2). Compared with the data given in[14], it was identified as 4′, 5-Dihydroxy-3′, 7 dimethoxyflavanone.
Fraction “2”: yellow needles; λmax UV (MeOH): 254, 356 nm; ESI-MS m/z: 343 (M-H)−; 1H-NMR (400MHz, CDCl3) δ: 12.62 (1H, s, 5-OH), 7.67 (2H, dd, H-2′, 6′), 7.03 (1H, d, H-5′), 6.43 (1H, s, H-8), 6.34 (1H, s, H-6), 6.00 (1H, s, 4′-OH), 3.97 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.84 (3H, s, OCH3); 13C-NMR (400MHz, CDCl3) δ: 178.7 (C=O), 165.4 (C-7), 162.0 (C-9), 156.7 (C-5), 155.9 (C-2), 148.3 (C-4′), 146.3 (C-3′), 138.8 (C-3), 122.6 (C-6′), 122.4 (C-1′), 114.5 (C-5′), 110.9 (C-2′), 106.0 (C-10), 97.8 (C-6), 92.2 (C-8), 60.1, 56.1, 55.8 (3, 7, 3′-OCH3). Compared with the data given in the related report[3], it was comfirmed as 5, 4′-Dihydroxy-3, 7, 3′-trimethoxyflavanone.
Fraction “3”: colorless needles; λmax UV (MeOH): 205, 286 nm; ESI-MS m/z: 329(M-H)−; 1H-NMR (400MHz, CDCl3) δ: 12.01 (1H, s, 5-OH), 6.97 (2H, d, H-2′, 5′), 6.90 (1H, d, H-6′), 6.05 (2H, dd, H-6,8), 5.36 (1H, dd, H-2), 3.92 (3H, s, OCH3), 3.90 (3H, s, OCH3), 3.81 (3H, s, OCH3), 3.11 (1H, dd, Hax-3), 2.81 (1H, dd, Heq-3); 13C-NMR (400MHz, CDCl3) δ: 195.0 (C=O), 168.0 (C-7), 164.2 (C-5), 162.8 (C-9), 149.6 (C-4′), 149.3 (C-3′), 130.8 (C-1′), 118.8 (C-6′), 111.2 (C-2′), 109.4 C-3 (C-5′), 103.2 (C-10), 95.1 (C-6), 94.3 (C-8), 79.2 (C-2), 56.0 (3′, 4′-OCH3), 55.7 (7-OCH3), 43.4 (C-3). According to the data reported [3], it was identified as 5-Hydroxy-7, 3′, 4′-trimethoxyflavanone.
Fraction “4”: yellow needles; λmax UV (MeOH): 253, 353 nm; ESI-MS m/z : 357 (M-H)−; 1H-NMR (400MHz, CDCl3) δ: 12.63 (1H, s, 5-OH), 7.72 (1H, dd, H-6′), 7.68(1H, d, H-2′), 6.98 (1H, d, H-5′), 6.44 (1H, d, H-8), 6.35 (1H, d, H-6), 3.98 (3H, s, OCH3), 3.97 (3H, s, OCH3), 3.88 (3H, s, OCH3), 3.87 (3H, s, OCH3). According to the data reported [3], it was identified as 5-Hydroxy-3, 7, 3′, 4′-tetrmethoxyflavanone.
Fraction “5” Pogostone(III): λmax UV (MeOH): 308 nm; ESI-MS m/z : 223 (M-H)−; 1H-NMR (400MHz, CD3OD) δ: 6.09 (1H, s, H-8), 4.79 (1H, q, H-9), 3.01 (2H, t, H-5), 2.26 (3H, d, H-12), 1.61 (1H, m, H-3), 1.52 (2H, m, H- 4), 0.92 (6H, d, H-1, 2), 5.46 (CH2Cl2 solvent residual peak); 13C-NMR (400MHz, CD3OD) δ: 206.8 (C-6, C=O), 179.9 (C-8), 168.8 (C-11, C=O), 160.6 (C-10), 100.0 (C-9), 98.1 (C-7), 38.8 (C-5), 31.8 (C-4), 26.8 (C-3), 20.6 (C-1, 2), 18.3 (C-12), 52.6 (CH2Cl2 solvent residual peak). According to the data reported [16], it was confirmed as Pogostone.
CONCLUSIONS
Using HSCCC and semi-preparative HPLC, five target compounds including 4′, 5-Dihydroxy-3′, 7-dimethoxyflavanone, 5, 4′-Dihydroxy-3, 7, 3′-trimethoxyflavanone, 5-Hydroxy-7, 3′, 4′-trimethoxyflavanone, 5-Hydroxy-3, 7, 3′, 4′-tetrmethoxyflavanone, pogostone, were successfully separated from an extract of P. Cablin. The method is simple, fast, and without the requirement of complex solvent systems. The present studies demonstrate that HSCCC is a powerful method for separating bioactive components from natural products. In general, the combined use of HSCCC and prep-HPLC is an effective separation mode for natural compounds due to the complementary action of these two methods.
Acknowledgments
This work was supported by the National Natural Science Foundation for Young Scholars of China (Grant No. 30801515).
References
- 1.Committee CP, editor. Phatmacopoeia of the People’s Republic of China. Beijing: Chemical Industry Press; 2000. [Google Scholar]
- 2.Su J, Zhang G, Li H, Zeng L, et al. Chinese Traditional and Herbal Drugs. 2001;32:204–205. [Google Scholar]
- 3.Zhang G, Feng X, Su J, Zeng L, et al. Chinese Traditional and Herbal Drugs. 2001;32:871–874. [Google Scholar]
- 4.Kimata M, Inagaki N, Nagai H. Planta Medica. 2002;66:25. doi: 10.1055/s-2000-11107. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Ren L. Chinese Traditional and Herbal Drugs. 1997;28:198–202. [Google Scholar]
- 6.Miyazawa M, Okuno Y, Nakamura S, Kosaka HC. Journal of Agricultural and Food Chemistry. 2000;48:642–647. doi: 10.1021/jf990160y. [DOI] [PubMed] [Google Scholar]
- 7.Guan L, Quan L, Xu L, Cong P. China Journal of Chinese Materia Medica. 1994;19:355–356. [PubMed] [Google Scholar]
- 8.Ito Y, Conway WD, editors. High-Speed Countercurrent Chromatography. Wiley-Interscience; 1996. [Google Scholar]
- 9.Liu R, Feng L, Sun A, Kong L. Journal of Chromatography A. 2004;1057:89–94. doi: 10.1016/j.chroma.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Peng J, Fan G, Wu Y. Journal of Chromatography A. 2005;1092:216–221. doi: 10.1016/j.chroma.2005.07.064. [DOI] [PubMed] [Google Scholar]
- 11.Peng J, Fan G, Chai Y, Wu Y. Journal of Chromatography A. 2006;1102:44–50. doi: 10.1016/j.chroma.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 12.Han X, Ma X, Zhang T, Zhang Y, et al. Journal of Chromatography A. 2007;1151:180–182. doi: 10.1016/j.chroma.2007.02.105. [DOI] [PubMed] [Google Scholar]
- 13.Shan Y, Seidel-Morgenstern A. Journal of Chromatography A. 2004;1041:53–56. doi: 10.1016/j.chroma.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 14.Ito Y. Journal of Chromatography A. 2005;1065:145–168. doi: 10.1016/j.chroma.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 15.Geissman T. Australian Journal of Chemistry. 1958;11:376–382. [Google Scholar]
- 16.Yang Z, Xie P. Chinese Science Bulletin. 1977;22:318–320. [Google Scholar]