

MetY is the first reported structure of an O-acetylhomoserine sulfhydrylase that utilizes a protein thiocarboxylate intermediate as the sulfur source in a novel methionine-biosynthetic pathway instead of catalyzing a direct sulfhydrylation reaction.
Keywords: Wolinella succinogenes, O-acetylhomoserine sulfhydrylases, pyridoxal 5′-phosphate, γ-elimination, direct sulfhydrylation, Cys/Met metabolism, O-acetylhomoserine, protein thiocarboxylate
Abstract
O-Acetylhomoserine sulfhydrylase (OAHS) is a pyridoxal 5′-phosphate (PLP) dependent sulfide-utilizing enzyme in the l-cysteine and l-methionine biosynthetic pathways of various enteric bacteria and fungi. OAHS catalyzes the conversion of O-acetylhomoserine to homocysteine using sulfide in a process known as direct sulfhydrylation. However, the source of the sulfur has not been identified and no structures of OAHS have been reported in the literature. Here, the crystal structure of Wolinella succinogenes OAHS (MetY) determined at 2.2 Å resolution is reported. MetY crystallized in space group C2 with two monomers in the asymmetric unit. Size-exclusion chromatography, dynamic light scattering and crystal packing indicate that the biological unit is a tetramer in solution. This is further supported by the crystal structure, in which a tetramer is formed using a combination of noncrystallographic and crystallographic twofold axes. A search for structurally homologous proteins revealed that MetY has the same fold as cystathionine γ-lyase and methionine γ-lyase. The active sites of these enzymes, which are also PLP-dependent, share a high degree of structural similarity, suggesting that MetY belongs to the γ-elimination subclass of the Cys/Met metabolism PLP-dependent family of enzymes. The structure of MetY, together with biochemical data, provides insight into the mechanism of sulfur transfer to a small molecule via a protein thiocarboxylate intermediate.
1. Introduction
Wolinella succinogenes is a Gram-positive bacterium that was originally found in cattle rumen. A member of the Helicobacteraceae group of ∊-proteobacteria, W. succinogenes is a close relative of Helicobacter pylori and Campylobacter jejuni, which are known pathogens in humans and animals. Specifically, H. pylori has been linked to ulcers and gastric cancer and C. jejuni is known to cause Guillain–Barre syndrome (Mileni et al., 2006 ▶).
O-Acetylhomoserine sulfhydrylase (OAHS; EC 4.2.99.10) is a sulfide-utilizing enzyme in the l-cysteine and l-methionine biosynthetic pathways employed by various enteric bacteria and fungi. OAHS is known to catalyze the conversion of O-acetylhomoserine (OAH) to homocysteine (Hcy) using sulfide directly (Yamagata, 1989 ▶). Recent biochemical studies (Krishnamoorthy & Begley, 2011 ▶) have suggested that the likely sulfur source in the methionine-biosynthetic pathway of W. succinogenes is a protein thiocarboxylate (Fig. 1 ▶). The pathway was identified from a gene cluster encoding a putative sulfur-carrier protein (HcyS), a putative metalloprotease (HcyD), an adenylating enzyme (HcyF), sulfur-assimilating proteins and proteins for methionine biosynthesis found in this species. In this proposed pathway, sulfate is converted to adenosine 5′-phosphosulfate (APS), which is then phosphorylated at the 3′-hydroxyl group. The resulting 3′-phosphoadenosine 5′-phosphosulfate (PAPS) is reduced in two steps to sulfide, which is transferred to the C-terminal carboxylate of HcyS to form HcyS thiocarboxylate (HcyS-COSH). MetY then catalyzes the pyridoxal 5′-phosphate (PLP) dependent condensation of HcyS-COSH and OAH, followed by an S,N-acyl shift, to form HcyS-homocysteine (HcyS-Hcy), which is cleaved by HcyD to form homocysteine (Hcy). Finally, methylation of Hcy by MetE using 5-methyltetrahydrofolate (5MeTHF) as the cofactor produces methionine. Thus, the S atom of methionine originates from sulfate and is incorporated by MetY using the sulfur-carrier protein HcyS rather than directly from sulfide as is the case for other OAHSs (Yamagata, 1989 ▶).
Figure 1.

Proposed pathway for sulfur assimilation in W. succinogenes. MetY catalyses the PLP-dependent condensation of HcyS-COSH and OAH to form HcyS-Hcy. The S atom that originates from sulfate is highlighted in red throughout.
Sulfur-carrier proteins have been implicated in several biosynthetic pathways, including those for thiamin (ThiS; Dorrestein et al., 2004 ▶), molybdopterin (MoaD; Leimkühler et al., 2001 ▶), cysteine (CysO; Burns et al., 2005 ▶), thioquinolobactin (QbsE; Godert et al., 2007 ▶), 2-thioribothymidine (TtuB; Shigi et al., 2008 ▶) and 5-methoxycarbonylmethyl-2-thiouridine (Urm1p; Leidel et al., 2009 ▶). In general, sulfur-carrier proteins are of low molecular weight (<10 000 Da) with a Gly-Gly C-terminus and have a ubiquitin-like fold. In some cases, such as HcyS and QbsE, the Gly-Gly motif may be followed by one or more additional amino-acid residues which must be cleaved before activation. The C-terminus of the sulfur-carrier protein is activated by adenylation and subsequently converted to the thiocarboxylate, which serves as the sulfur source for subsequent reactions.
In this study, we report the 2.2 Å resolution structure of MetY, the first documented OAHS for which the S atom is provided by the thiocarboxylate of a sulfur-carrier protein. By comparing this structure with those of other proteins, we found that MetY has the same fold as Saccharomyces cerevisiae cystathionine γ-lyase (ScCGL; PDB entry 1n8p; Messerschmidt et al., 2003 ▶) and Pseudomonas putida methionine γ-lyase (PpMGL; PDB entry 2o7c; Kudou et al., 2007 ▶). The structural similarity and active-site conservation suggest that MetY belongs to the Cys/Met metabolism PLP-dependent family of enzymes (PF01053; Bateman et al., 2002 ▶), which catalyze a variety of reactions including γ-replacements, γ-eliminations and β-eliminations (Messerschmidt et al., 2003 ▶). The structure described in this study, together with published biochemical data (Krishnamoorthy & Begley, 2011 ▶), results in the assignment of MetY as a γ-elimination enzyme and allows us to propose a mechanism for the protein thiocarboxylate-dependent sulfur-transfer reaction.
2. Experimental procedures
The cloning, overexpression, purification and assay of MetY have been described previously (Krishnamoorthy & Begley, 2011 ▶).
2.1. Crystallization
The frozen protein was thawed at 277 K. Crystallization of MetY was carried out by the hanging-drop vapor-diffusion method at 295 K using sparse-matrix screening solutions from Hampton Research and Emerald BioSystems. 1.5 µl protein solution was combined with an equal volume of well solution. Extensive screenings led to one condition consisting of 25–30%(w/v) ethylene glycol with 5%(v/v) N-dodecyl-N,N-dimethylamine-N-oxide detergent as an additive. The crystals grew as small blocks ranging from 25 to 50 µm in two of the dimensions and from 50 to 75 µm in the third dimension. The crystals used for X-ray diffraction experiments took approximately three months to grow.
2.2. Data collection and processing
Because of the relatively high concentration of ethylene glycol (25–30%) in the crystallization condition, no additional cryoprotection was required prior to data collection. X-ray diffraction data were collected from crystals of MetY on the F1 beamline outfitted with capillary focusing optics at the Cornell High Energy Synchrotron Source (CHESS) using an Area Detector System Corporation Quantum 270 detector. The data were collected using a wavelength of 0.916 Å over 360° with a 1° oscillation range. The data set was indexed, integrated and scaled using the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). Data-collection statistics are shown in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Beamline | F1, CHESS |
| Wavelength (Å) | 0.916 |
| Resolution (Å) | 50–2.2 |
| Space group | C2 |
| Molecules per asymmetric unit | 2 |
| Unit-cell parameters | |
| a (Å) | 161.8 |
| b (Å) | 62.5 |
| c (Å) | 91.6 |
| α (°) | 90.0 |
| β (°) | 120.5 |
| γ (°) | 90.0 |
| Measured reflections | 89132 |
| Unique reflections | 37506 (1862) |
| 〈I/σ(I)〉 | 11.0 (2.9) |
| Multiplicity | 2.4 (2.3) |
| Completeness (%) | 96.6 (98.2) |
| Rmerge† (%) | 12.3 (40.1) |
R
merge = 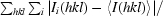
 , where 〈I(hkl)〉 is the mean intensity of the i reflections with intensities I
i(hkl) and common indices hkl.
, where 〈I(hkl)〉 is the mean intensity of the i reflections with intensities I
i(hkl) and common indices hkl.
2.3. Structure determination
The MetY structure was determined by molecular replacement using MOLREP (Vagin & Teplyakov, 2000 ▶) with the Thermus thermophilus O-acetylhomoserine sulfhydrylase (TtOAHS) structure (PDB entry 2cb1; T. Imagawa, H. Utsunomiya, Y. Agari, S. Satoh & H. Tsuge, unpublished work) as the search model. CHAINSAW (Schwarzenbacher et al., 2004 ▶) was used to remove atoms that were not common to the two sequences. Refinement was performed using REFMAC (Murshudov et al., 1999 ▶) and alternated with successive manual model building using Coot (Emsley & Cowtan, 2004 ▶) guided by an F o − F c map, a 2F o − F c map and composite OMIT electron-density maps generated using CNS and CCP4 (Brünger et al., 1998 ▶; Winn et al., 2011 ▶). Water molecules were added after the R factor and R free converged. The geometry of MetY was validated using PROCHECK (Laskowski et al., 1993 ▶). Refinement statistics are summarized in Table 2 ▶.
Table 2. Data-refinement statistics.
| Resolution (Å) | 79–2.2 |
| No. of protein atoms | 5266 |
| No. of water atoms | 183 |
| Reflections in working set | 38264 |
| Reflections in test set | 2017 |
| R factor† (%) | 22.5 |
| Rfree‡ (%) | 25.1 |
| R.m.s.d. from ideals | |
| Bond lengths (Å) | 0.024 |
| Angles (°) | 1.942 |
| Average B factor (Å2) | 23.6 |
| Ramachandran plot | |
| Most favored (%) | 98.4 |
| Additionally allowed (%) | 1.6 |
| Generously allowed (%) | 0.0 |
| Disallowed (%) | 0.0 |
R factor = 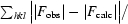
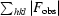 , where F
obs and F
calc are observed and calculated structure factors, respectively.
, where F
obs and F
calc are observed and calculated structure factors, respectively.
For R free, the sum is extended over a subset of reflections (5%) excluded from all stages of refinement.
2.4. Figure production
All figures were produced using PyMOL (DeLano, 2002 ▶), ALSCRIPT (Barton, 1993 ▶) and ChemBioDraw (CambridgeSoft).
3. Results
3.1. Structure of the MetY monomer
The Matthews coefficient (Matthews, 1968 ▶) for the MetY crystals was calculated to be 2.23 Å3 Da−1, which assuming two monomers per asymmetric unit corresponds to a solvent content of 45%. The final model (Fig. 2 ▶ a), including two monomers and 183 water molecules, had an R factor and R free of 22.5% and 25.1%, respectively. Of 407 possible amino-acid residues, monomer A contains residues 2–12, 57–367 and 377–407 and monomer B contains residues 2–11, 57–207, 211–364 and 381–406. Each monomer contains one active site. The monomer adopts an α/β fold and consists of a large central seven-stranded β-sheet, a two-stranded β-sheet and a three-stranded β-sheet. The β-sheets are flanked by a total of 12 α-helices and two 310-helices (Figs. 2 ▶ b and 2 ▶ c).
Figure 2.

Structure of MetY. (a) Dimer observed in the asymmetric unit. (b) Monomer with secondary-structure elements labelled. (c) Topology diagram with the same labels as in (b). The numbers for the first and last amino-acid residue in each secondary-structure element are shown. (d) The biological tetramer. PLP is represented as spheres. The letters N and C in the monomer and topology diagrams denote the N- and C-termini, respectively.
The electron density for two large segments was absent in both chains in the asymmetric unit. The longer segment is located at the N-terminus and consists of 44 residues (13–56) in monomer A and 45 residues (12–56) in monomer B, while the smaller segment is near the C-terminus and consists of nine residues (368–376) in monomer A and 16 residues (365–380) in monomer B. For both segments, electron density for the residues adjacent to the disordered loops (including the N-terminal helix α1) was clear, demonstrating that the protein was intact and not truncated. This is consistent with SDS–PAGE analysis, which showed that the molecular weight of the protein used for crystallization was near the expected value. The longer segment is also predicted to be involved in PLP binding (see below) and is most likely to be disordered because PLP is not bound in the crystal structure.
The monomer is composed of two domains. The large domain contains the PLP-binding site and the seven-stranded β-sheet, which is flanked by α2, α3, α4, α5, α6, α7, α8, η1, η2 and part of α9. The seven-stranded β-sheet consists of mostly parallel strands with a strand order of β1↑β9↓β6↑β5↑β4↑β2↑β3↑. The smaller domain, or the capping domain, resides near the C-terminal end and contains a slightly twisted antiparallel three-stranded β-sheet which is flanked by α9, α10, α11 and α12. β7 and β8 are continuous with β6 and β9, respectively, and extend to form another small β-sheet. Helix α9 is unusually long, consisting of 34 residues and spanning the two domains. The compact monomer is achieved by the tight packing between the two domains.
3.2. Structure of the MetY tetramer
The two monomers in the asymmetric unit are related by noncrystallographic twofold symmetry (Fig. 2 ▶ a) and a homotetramer with 222 symmetry is generated by the addition of a crystallographic twofold axis (Fig. 2 ▶ d). This tetramer was confirmed as the biological unit of MetY based on size-exclusion chromatography and dynamic light scattering (data not shown). The dimensions of the monomer are approximately 50 × 50 × 70 Å and those of the tetramer are approximately 75 × 85 × 95 Å. The monomers are arranged such that the two active sites of any pair of adjacent monomers face in opposite directions. In this arrangement, adjacent monomers are closely packed with 1018 Å2 of buried surface area per monomer as calculated by the PISA server (Krissinel & Henrick, 2005 ▶); however, diagonal monomers of the tetramer do not interact. Tightly packed monomers interact primarily via a long loop, α3, α4 and α8. The active sites of the two closest monomers are about 20 Å apart and the active sites of the farthest monomers are 37 Å apart.
3.3. MetY active site
Each monomer contains one PLP-binding site; however, the cofactor was not bound in the crystallized MetY. The putative active site was easily identified based on structural homology to PpMGL (PDB entry 2o7c; Kudou et al., 2007 ▶) and ScCGL (PDB entry 1n8p; Messerschmidt et al., 2003 ▶) and PLP was modeled into the active site. A stereo figure and schematic illustration of the active site are shown in Fig. 3 ▶. The active site appears as a relatively large cavity open to solvent. The active site is located near the N-terminus of α3. The PLP-binding site is occupied by water molecules in the absence of the cofactor. On the basis of the model, the phosphate group of PLP makes hydrogen bonds to Thr204, Ser202 and the amide N atoms of Gly83 and Met84. Asp180 is within salt-bridge distance of the N1 atom of PLP. Leu353, Met183, Ile87 and Met84 interact with the hydrophobic region of PLP. Glu151 does not appear to interact directly with PLP, but could form a hydrogen bond to Phe108, which could stabilize the pyridine ring of PLP by a stacking interaction. Thr182 and Ser202 are about 3 Å directly below the plane of the pyridine ring of PLP according to the orientation shown in Fig. 3 ▶. In addition, Thr182 is also within hydrogen-bonding distance of Asp180. No residues in the active site appear to interact with the methyl group of PLP. The hydroxyl group of PLP could hydrogen bond to an ordered water molecule as seen in the structures of the homologs. With the exception of two PLP-binding residues from the disordered N-terminal loop, all of the residues that potentially interact with PLP originate from the large PLP-binding domain.
Figure 3.

Model of the active site of MetY. (a) Stereo diagram. (b) Schematic representation. The PLP cofactor shown in cyan and red in (a) and (b), respectively, is manually positioned in the active site based on superposition of structural homologs. Predicted interactions are indicated by the dashed lines. ‘W’ denotes a water molecule.
4. Discussion
4.1. Comparison of MetY to homologous enzymes
Although the asymmetric unit contains a dimer, the tetramer can be generated from the dimer using a crystallographic twofold axis. The tetrameric structure (Fig. 2 ▶) was supported by a PISA server analysis (Krissinel & Henrick, 2005 ▶) and experimentally by gel-filtration and dynamic light-scattering analysis. Furthermore, OAHS from Schizosaccharomyces pombe has been biochemically demonstrated to be tetrameric (Yamagata, 1984 ▶). The γ-lyases PpMGL and ScCGL also function biologically as tetramers (Kudou et al., 2007 ▶; Messerschmidt et al., 2003 ▶; Motoshima et al., 2000 ▶; Nikulin et al., 2008 ▶). In addition, the MetY tetramer is structurally homologous to those of PpMGL, ScCGL and TtOAHS.
The monomer contains two domains: a large PLP-binding domain and a small C-terminal domain. A search using the DALI server identified several structural homologs. The top representative hits are shown in Table 3 ▶ (Holm & Sander, 1998 ▶) and a superposition of the monomers from each structure with MetY is shown in Fig. 4 ▶. TtOAHS, which was the search model for the molecular-replacement structure determination of MetY, was the top hit, with a Z score of 45.4 and an aligned sequence identity of 41%. The next two hits, ScCGL and PpMGL, with Z scores of 41.9, belong to the Cys/Met metabolism PLP-dependent family of enzymes (PF01053; Bateman et al., 2002 ▶). This family of enzymes utilizes PLP to carry out β-eliminations, γ-eliminations and γ-replacements. Therefore, the DALI search results suggest that MetY may belong to the γ-elimination subfamily of the Cys/Met metabolism PLP-dependent family of enzymes.
Table 3. Enzymes structurally similar to MetY from DALI .
Figure 4.

Structural superposition of the MetY monomer with homologous enzymes. Color coding is as follows: MetY, green; TtOAHS (PDB entry 2cb1), blue; ScCGL (PDB entry 1n8p), magenta; PpMGL (PDB entry 2o7c), gray. The PLP molecule is shown as cyan spheres.
The disordered N-terminal loop is located between helices α1 and α2. Both of these helices have well defined electron density in both monomers of MetY. In the structures of TtOAHS, PpMGL and ScCGL the corresponding loop is located at the monomer–monomer interface (Kudou et al., 2007 ▶). Two absolutely conserved PLP-binding residues, corresponding to Tyr53 and Arg55 in MetY, are located near the end of the loop. These residues cap the PLP portion of the active site of the adjacent monomer. In the other structures both residues form hydrogen bonds to the PLP phosphate. Therefore, the absence of PLP in the MetY crystal structure is most likely to cause the loop to be disordered. The shorter disordered loop is located near the surface at an opening into the active site. In the structures of TtOAHS, PpMGL and ScCGL this region forms a short α-helix and caps the active site. It is likely that in MetY this region becomes ordered when substrate binds or that it mediates protein–protein interactions in the complex with HcyS-COSH.
4.2. Comparison of active sites for enzymes similar to MetY
Because MetY, TtOAHS, PpMGL and ScCGL are members of the same superfamily and share the same fold, we compared their active sites. The superposition of the active sites (Fig. 5 ▶, Table 4 ▶) of these homologs and MetY confirmed the location of the active site of MetY and showed highly conserved amino-acid residues. In addition to the biochemical data, which show that MetY requires PLP for activity, MetY appeared yellow throughout the purification procedure, suggesting the presence of PLP, despite its absence in the crystal structure. By comparing the active-site residues of MetY with those of TtOAHS (PDB entry 2cb1; T. Imagawa, H. Utsunomiya, Y. Agari, S. Satoh & H. Tsuge, unpublished work), PpMGL (Kudou et al., 2007 ▶) and ScCGL (Messerschmidt et al., 2003 ▶), the critical active-site residues were identified. A sequence alignment with these residues identified is shown in Fig. 6 ▶. The equivalent residues to Phe108 of MetY in the other three structures form π-stacking interactions with the pyridine ring of PLP (Clausen et al., 1998 ▶; Hayashi et al., 1990 ▶), suggesting that Phe108 plays the same role in MetY. The conserved Glu151 does not interact directly with PLP, but is positioned to form a hydrogen bond to the backbone amide of the active-site residue Phe108. In the absence of PLP, Phe108 is rotated slightly away from where PLP is postulated to bind. The conserved residues Thr182 and Ser202 may form van der Waals interactions with the pyridine ring of PLP as well as forming electrostatic interactions with the π-electron ring. Together with Phe108, Thr182 and Ser202 sandwich PLP in the active site. Thr182 also hydrogen bonds to the amide H atom of the conserved Asp180 and positions it for a salt bridge to N1 of PLP. Lys205 is conserved within the superfamily and forms a Schiff-base linkage with PLP prior to substrate binding. In the MetY active site, Thr204 and Ser202 are predicted to form hydrogen bonds to the PLP phosphate group and are conserved. On the basis of comparison with the homologous structures, Phe108, Thr204 and Lys205 move away from the active site in the absence of PLP. The active site is accessible to solvent in the absence of PLP; however, ordering of the shorter disordered loop region would reduce active-site accessibility. The backbone amide groups of Met84 and the conserved Gly83 are in position to hydrogen bond to the phosphate group of PLP. The conserved Arg387 does not appear to make any contacts with PLP, while its equivalents in PpMGL and ScCGL are known to bind to the carboxylate group of the incoming substrate (Kudou et al., 2007 ▶; Messerschmidt et al., 2003 ▶). Ile353, Met183, Ile87 and Met84 are in position to interact with the hydrophobic region of PLP.
Figure 5.

Superposition of the active sites of MetY with those of other γ-lyases. The enzymes and coloring schemes are the same as in Fig. 4 ▶, except that the color of PLP is the same as its corresponding enzyme. Residues marked with an asterisk are from a neighboring monomer.
Table 4. Comparison of active-site residues of different structural homologs of MetY.
| PpMGL | ScCGL | MetY | TtOAHS | Predicted role in MetY |
|---|---|---|---|---|
| Gly89 | Gly79 | Gly83 | Gly80 | Amide hydrogen interacts with the phosphate group of PLP |
| Met90 | Ser80 | Met84 | Gln81 | Interacts hydrophobically with PLP ring and its amide hydrogen makes a hydrogen bond with the phosphate group of PLP |
| Ile93 | Thr83 | Ile87 | Thr84 | Interacts with the hydrophobic part of PLP ring |
| Tyr114 | Tyr103 | Phe108 | Phe105 | Stabilizes the pyridine ring of PLP by π-stacking interaction |
| Glu157 | Glu145 | Glu151 | Glu147 | Positions Phe108 in the active site |
| Asp186 | Asp178 | Asp180 | Asp176 | Forms a salt bridge with the N1H atom of PLP |
| Thr188 | Thr180 | Thr182 | Thr178 | Makes van der Waals interaction and a hydrogen bond with the π-electron ring of PLP |
| Tyr189 | Phe181 | Met183 | Phe179 | Interacts with the hydrophobic part of PLP ring |
| Ser208 | Ser200 | Ser202 | Ser199 | Makes van der Waals interaction with the π-electron ring and a hydrogen bond with the phosphate group of PLP |
| Thr210 | Thr202 | Thr204 | Thr201 | Hydrogen bonds with the phosphate group of PLP |
| Lys211 | Lys203 | Lys205 | Lys202 | Makes a Schiff-base linkage with PLP |
| Leu341 | Leu335 | Ile353 | Leu356 | Interacts with the hydrophobic part of the PLP ring |
| Arg375 | Arg369 | Arg387 | Arg390 | Interacts with the carboxylate group of O-acetylhomoserine |
| Tyr59† | Tyr48† | Tyr50† | Tyr53† | Hydrogen bonds with the phosphate group of PLP |
| Arg61† | Arg51† | Arg52† | Arg55† | Hydrogen bonds with the phosphate group of PLP |
Residues from the adjacent dimer of the active monomer.
Figure 6.

Sequence alignment of MetY with TtOAHS (PDB entry 2cb1), ScCGL (PDB entry 1n8p) and PpMGL (PDB entry 2o7c). Numbering and secondary-structure assignments are for MetY. Helices are indicated by blue cylinders, strands are indicated by green arrows and the two disordered loops are indicated by black dashed lines. Conserved residues are shown in boxes with white font and red shading. Conservative substitutions are shown in boxes with a red font. The conserved active-site residues are marked with asterisks and other active-site residues of MetY are marked with black circles. The two conserved residues from the disordered loop of an adjacent monomer, which are predicted to bind to the PLP phosphate group, are marked with red triangles.
4.3. Mechanistic implications
The primary sequence of MetY and its structural alignment with homologs suggest that MetY belongs to the Cys/Met metabolism PLP-dependent family of enzymes (PF01053; Bateman et al., 2002 ▶). This family also includes CysM, which catalyzes the transfer of sulfur from CysO thiocarboxylate (CysO-COSH) to O-phosphoserine (OPS; Jurgenson et al., 2008 ▶; O’Leary et al., 2008 ▶). Our biochemical and structural data suggest that MetY-catalyzed sulfur transfer from HcyS-COSH to OAH is similar to the CysM-catalyzed sulfur transfer from CysO-COSH to OPS. A mechanistic proposal is outlined in Fig. 7 ▶. In this proposal, a transimination between 1 and OAH gives imine 2. Deprotonation gives 3. A second deprotonation gives the stabilized carbanion 4, from which the acetyl group is eliminated to give 5. By reversal of these steps, HcyS-COSH gives 8. Product release by a transimination gives 9, which then undergoes a spontaneous N/S acyl shift to give the HcyS-Hyc 10. Our structure suggests that the active site of MetY is accessible to the C-terminus of HcyS-COSH, thus allowing the thiocarboylate to position itself for sulfur transfer.
Figure 7.

Mechanistic proposal for MetY-catalyzed sulfur transfer from HcyS-COSH to OAH. The S atom that originates from sulfate is highlighted in red.
4.4. Significance
The structural studies reported here demonstrate that MetY belongs to the γ-elimination subclass of the Cys/Met metabolism family of PLP-dependent enzymes (PF01053; Bateman et al., 2002 ▶). This suggests that MetY catalyzes a γ-elimination reaction to produce Hcy that is covalently attached to the HcyS sulfur-carrier protein. Therefore, MetY is likely to be mechanistically similar to OAHS except that the sulfur donor is a protein thiocarboxylate rather than a sulfide. MetY is a member of a growing family of proteins involved in sulfur transfer from protein thiocarboxylates. Eight species identified to date in the SEED database (Overbeek et al., 2005 ▶) contain a gene-clustering pattern in which a sulfur-carrier protein gene is clustered with genes for a sulfur-assimilation pathway and methionine biosynthesis.
Supplementary Material
PDB reference: MetY, 3ri6
Acknowledgments
We thank Dr Cynthia Kinsland of the Cornell University Protein Facility for cloning MetY and the MacCHESS staff, supported by NIH grant RR01646, at the Cornell High Energy Synchrotron Source for assistance during data collection. We thank Dr Michael Fenwick for performing the light-scattering experiments. We thank Ms Leslie Kinsland for her assistance during manuscript preparation. This work supported by NIH grant DK67081.
References
- Barton, G. J. (1993). Protein Eng. 6, 37–40. [DOI] [PubMed]
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002). Nucleic Acids Res. 30, 276–280. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Burns, K. E., Baumgart, S., Dorrestein, P. C., Zhai, H., McLafferty, F. W. & Begley, T. P. (2005). J. Am. Chem. Soc. 127, 11602–11603. [DOI] [PMC free article] [PubMed]
- Clausen, T., Huber, R., Prade, L., Wahl, M. C. & Messerschmidt, A. (1998). EMBO J. 17, 6827–6838. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Dorrestein, P. C., Zhai, H., McLafferty, F. W. & Begley, T. P. (2004). Chem. Biol. 11, 1373–1381. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Godert, A. M., Jin, M., McLafferty, F. W. & Begley, T. P. (2007). J. Bacteriol. 189, 2941–2944. [DOI] [PMC free article] [PubMed]
- Hayashi, H., Inoue, Y., Kuramitsu, S., Morino, Y. & Kagamiyama, H. (1990). Biochem. Biophys. Res. Commun. 167, 407–412. [DOI] [PubMed]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res. 26, 316–319. [DOI] [PMC free article] [PubMed]
- Jurgenson, C. T., Burns, K. E., Begley, T. P. & Ealick, S. E. (2008). Biochemistry, 47, 10354–10364. [DOI] [PMC free article] [PubMed]
- Krishnamoorthy, K. & Begley, T. P. (2011). J. Am. Chem. Soc. 133, 379–386. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2005). CompLife 2005, edited by M. R. Berthold, R. Glen, K. Diederichs, O. Kohlbacher & I. Fischer, pp. 163-174. Berlin, Heidelberg: Springer-Verlag.
- Kudou, D., Misaki, S., Yamashita, M., Tamura, T., Takakura, T., Yoshioka, T., Yagi, S., Hoffman, R. M., Takimoto, A., Esaki, N. & Inagaki, K. (2007). J. Biochem. 141, 535–544. [DOI] [PubMed]
- Laskowski, R. A., Moss, D. S. & Thornton, J. M. (1993). J. Mol. Biol. 231, 1049–1067. [DOI] [PubMed]
- Leidel, S., Pedrioli, P. G., Bucher, T., Brost, R., Costanzo, M., Schmidt, A., Aebersold, R., Boone, C., Hofmann, K. & Peter, M. (2009). Nature (London), 458, 228–232. [DOI] [PubMed]
- Leimkühler, S., Wuebbens, M. M. & Rajagopalan, K. V. (2001). J. Biol. Chem. 276, 34695–34701. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Messerschmidt, A., Worbs, M., Steegborn, C., Wahl, M. C., Huber, R., Laber, B. & Clausen, T. (2003). Biol. Chem. 384, 373–386. [DOI] [PubMed]
- Mileni, M., MacMillan, F., Tziatzios, C., Zwicker, K., Haas, A. H., Mäntele, W., Simon, J. & Lancaster, C. R. (2006). Biochem. J. 395, 191–201. [DOI] [PMC free article] [PubMed]
- Motoshima, H., Inagaki, K., Kumasaka, T., Furuichi, M., Inoue, H., Tamura, T., Esaki, N., Soda, K., Tanaka, N., Yamamoto, M. & Tanaka, H. (2000). J. Biochem. 128, 349–354. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Nikulin, A., Revtovich, S., Morozova, E., Nevskaya, N., Nikonov, S., Garber, M. & Demidkina, T. (2008). Acta Cryst. D64, 211–218. [DOI] [PubMed]
- O’Leary, S. E., Jurgenson, C. T., Ealick, S. E. & Begley, T. P. (2008). Biochemistry, 47, 11606–11615. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Overbeek, R. et al. (2005). Nucleic Acids Res. 33, 5691–5702. [DOI] [PMC free article] [PubMed]
- Schwarzenbacher, R., Godzik, A., Grzechnik, S. K. & Jaroszewski, L. (2004). Acta Cryst. D60, 1229–1236. [DOI] [PubMed]
- Shigi, N., Sakaguchi, Y., Asai, S., Suzuki, T. & Watanabe, K. (2008). EMBO J. 27, 3267–3278. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2000). Acta Cryst. D56, 1622–1624. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yamagata, S. (1984). J. Biochem. 96, 1511–1523. [DOI] [PubMed]
- Yamagata, S. (1989). Biochimie, 71, 1125–1143. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: MetY, 3ri6