

Abstract
Metabolic labeling of proteins with the methionine (1) surrogate azidonorleucine (2) can be targeted exclusively to specified cells through expression of a mutant methionyl-tRNA synthetase (MetRS). In complex cellular mixtures, proteins made in cells that express the mutant synthetase can be tagged with affinity reagents (for detection or enrichment) or fluorescent dyes (for imaging). Proteins made in cells that do not express the mutant synthetase are neither labeled nor detected.
Time-dependent changes in cellular proteomes can be monitored via a variety of powerful electrophoretic and spectroscopic methods. Traditionally, radiolabeled amino acids have been used to label proteins synthesized during an amino acid ‘pulse’; labeled proteins can be distinguished from preexisting (unlabeled) proteins through electrophoretic separation followed by radiographic detection1. More recently, mass spectrometry has enabled the use of stable isotopes in amino acid pulse labeling2.
In 2006, Dieterich and co-workers introduced the BONCAT (bio-orthogonal non-canonical amino acid tagging) strategy for selective enrichment and identification of newly synthesized proteins in cells3,4. The BONCAT approach reduces sample complexity and permits direct analysis of the primary protein synthesis response to stimuli. Bio-orthogonal functional groups5 are introduced into proteins by pulse-labeling with reactive, non-canonical amino acids. Labeled proteins are selectively modified with affinity tags for enrichment6,7; removal of unlabeled proteins simplifies subsequent analysis and identification by mass spectrometry.
All of these methods suffer from limitations when experiments are performed in systems that contain multiple cell types. Because incorporation of amino acids is non-specific with respect to cell identity, proteins from all cell types are labeled. In studies of interactions between different cell types in a single organism, the origin of the identified proteins can be difficult to ascertain because the cells share a common genome. When interactions between cells of different genomes are studied, detection of low abundance proteins can be problematic. In infection studies, for example, the protein content of the larger host cells can overwhelm that of the pathogen8 and limit detection and identification of the proteins of primary interest. In complex bacterial communities where hundreds of organisms can occupy a common biological niche9, probing the proteome of a single species in its natural context is an even greater challenge.
To address these difficulties, we describe here a versatile method for cell-selective protein labeling in mixed cellular environments. To achieve selective labeling, we employ non-canonical amino acids that are excluded by the endogenous protein synthesis machinery (Fig. 1a). These amino acids face discrimination by the quality control mechanisms found at the level of aminoacyl-tRNA synthetases10; they are not charged to tRNA and are not used in protein synthesis. By screening libraries of methionyl-tRNA synthetase (MetRS) mutants from Escherichia coli11,12 we have identified a mutant synthetase (NLL-MetRS) (Supplementary Fig. 1 online) that efficiently appends azidonorleucine (2, Fig. 1b) to cognate tRNA. Cells bearing the mutant MetRS are able to utilize 2 as a surrogate for methionine (1) in protein synthesis. Wild-type cells are inert to 2; proteins made in these cells utilize only methionine and are not labeled (Fig. 1a). In co-culture, protein labeling is restricted to mutant cells.
Figure 1.
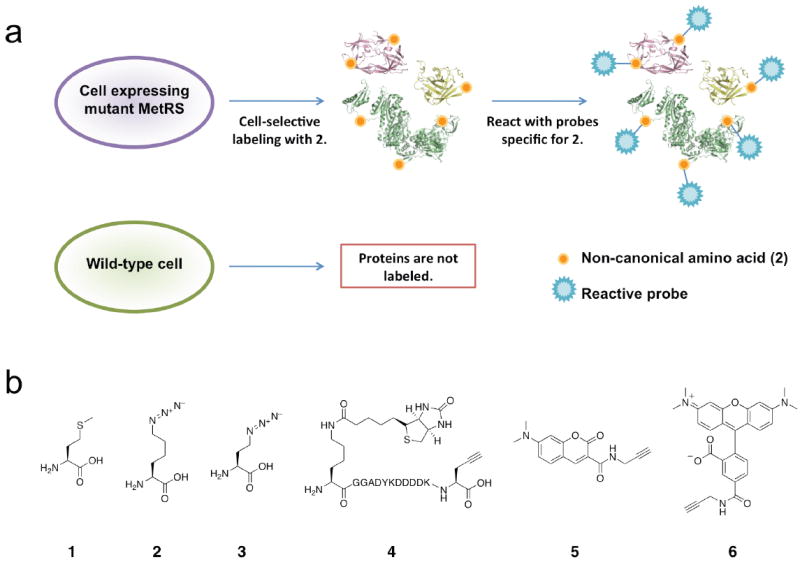
Cell-selective labeling of proteomes with azidonorleucine. (a) Schematic representation of incorporation of azidonorleucine exclusively in cells expressing NLL-MRS. (b) Structures of amino acids and probes used in this study: (1) methionine, (2) azidonorleucine, (3) azidohomoalanine, (4) biotin-FLAG-alkyne, (5) dimethylaminocoumarin-alkyne, (6) TAMRA-alkyne.
To validate this approach, we first confirmed that incorporation of 2 into newly synthesized proteins is dependent on expression of the mutant synthetase. An E. coli strain (DH10B/pJTN1) constitutively expressing a plasmid-borne copy of NLL-MetRS was pulse-labeled with 2 and compared to a control strain (DH10B/pQE-80L) that did not express the enzyme. Separate cultures of the two strains were grown in minimal medium containing the twenty canonical amino acids. When the cell density reached OD600 = 0.5, cells were pulse-labeled with 1 mM 2 for 10 m. Control cells were pulsed in the same fashion with 1, or incubated with the protein synthesis inhibitor chloramphenicol prior to labeling with 2. Cell lysates of each culture were probed for incorporation of 2 via Cu (I) catalyzed ligation13,14 to biotin-FLAG-alkyne (4) followed by Western blotting with protein detection by anti-FLAG antibody. The results of these experiments indicated that only proteins synthesized in cells constitutively expressing the NLL-MetRS (DH10B/pJTN1) were labeled with 2 and susceptible to ligation to 4 (Supplementary Fig. 2 online). A second control strain, in which the wild-type synthetase was over-expressed, was also inert to labeling (Supplementary Fig. 1 online).
The behavior observed in separate cultures was maintained when cells were incubated in co-culture to simulate a complex, mixed cellular environment. Two different heterologous proteins were employed as markers for the cells of origin to distinguish between NLL-expressing and wild-type E. coli. An E. coli strain (DH10B/pJTN2) expressing the NLL-MetRS was programmed to express green fluorescent protein (GFP) upon induction with isopropyl β-D-1-thiogalactopyranoside (IPTG). The control strain (DH10B/pJTN3) carried an IPTG-inducible gene for the marker protein DHFR. Both marker proteins were His-tagged to enable Ni-affinity purification and detection with Penta-His antibody. Individual cultures of these bacterial strains were grown to OD600 = 1.0 and a third culture was created by mixing cells in a volumetric ratio of 1:2 (DH10B/pJTN2:DH10B/pJTN3). To initiate labeling, 1 mM 2 was added to each of the three cultures and expression of marker proteins was induced with 1 mM IPTG for 3 h. Cell lysates from all three samples were subjected to Cu (I) catalyzed azide-alkyne ligation to 4, and marker proteins were isolated by Ni-affinity purification. Western analysis (Fig. 2a) of isolated proteins with Penta-His antibody revealed the expected expression patterns from the three cultures. In striking contrast, analysis of blots with streptavidin-HRP (for detection of conjugation to 4) revealed exclusive modification of GFP, the marker protein synthesized in cells expressing NLL-MetRS. DHFR isolated from control cells exhibited no such modification. N-terminal protein sequencing indicated 10-20% replacement of 1 by 2 in the GFP marker protein.
Figure 2.
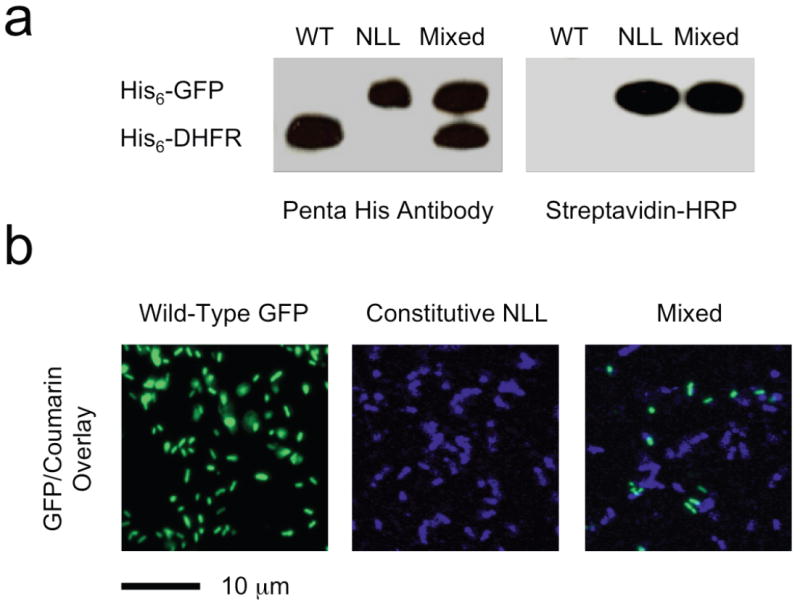
Cell-selective protein labeling in mixed populations. (a) Western blot detection of marker protein expression with Penta-His antibody (left) and with streptavidin-HRP (right). DHFR was made in cells lacking the NLL-MetRS (WT) and GFP in cells expressing the NLL-MetRS (NLL); both proteins contain multiple ATG (Met) codons (7 in DHFR and 8 in GFP). Azidonorleucine was added to all three cultures upon induction of protein synthesis. Samples from the mixed culture contain both proteins (as shown by Penta-His antibody detection), but only GFP is labeled with azidonorleucine and susceptible to labeling with 4. (b) Fluorescence images of cells expressing GFP but not NLL-MetRS (Wild-Type), cells expressing DHFR and NLL-MetRS (NLL), and a mixed culture of the two (Mixed). Azidonorleucine was added to all three cultures upon induction of protein synthesis. Cells from all three cultures were treated with 5, but only cells expressing the NLL-MetRS were labeled. Note that the marker proteins co-expressed with NLL-MetRS are different in the Western blotting and fluorescence imaging experiments.
To demonstrate further the utility of this approach, we used fluorescence microscopy to distinguish proteins made in cells expressing the NLL-MetRS from those made in cells that do not express the mutant synthetase. The control strain (DH10B/pJTN4) expressed an IPTG-inducible GFP while the strain (DH10B/pJTN5) constitutively expressing the NLL-MRS carried an IPTG-inducible DHFR. Labeling of individual and mixed cultures were performed as described earlier. After pulse-labeling with 2 and induction of protein synthesis, cells were collected by centrifugation and washed prior to Cu (I) catalyzed labeling of cells with dimethylaminocoumarin-alkyne (5). After washing with PBS to remove excess dye, cells were imaged by fluorescence microscopy. The results (Fig. 2b) were consistent with those of the Western analysis; the coumarin fluorescence was confined to cells that express NLL-MetRS. Control cells expressing GFP were inert with respect to labeling, as indicated by the absence of coumarin emission from these samples (Supplementary Fig. 3 online).
Cell-selective protein labeling can also be accomplished in systems containing mixtures of bacterial and mammalian cells. Murine alveolar macrophages were infected with E. coli cells that constitutively express the NLL-MetRS (DH10B/pJTN1) or with control bacterial cells that express a GFP marker protein (DH10B/pJTN4). Prior to infection, 2 mM 2 was added to the macrophage medium; to initiate infection, bacteria were added to the culture medium and co-incubated for 35 m at 37 °C. Cells were fixed, permeabilized, and subjected to Cu (I) catalyzed conjugation to TAMRA-alkyne (6, Invitrogen). Bacteria were both bound and internalized by macrophages as confirmed by confocal microscopy and three-dimensional analysis (Supplementary Movie 1 online). Macrophage-associated bacterial cells that express the NLL-MetRS exhibited strong fluorescence emission from 6 (Fig. 3a). The control bacterial strain was bound and internalized by macrophages (as seen by detection of GFP, Supplementary Fig. 4 online) but exhibited no conjugation to 6 above background (Fig. 3a). To confirm protein synthesis by macrophages during the infection period, cells were treated with azidohomoalanine (3) in medium lacking 1 (Fig. 3b). Both 2 and 3 are susceptible to ligation to alkyne-functionalized probes; however, in contrast to 2, 3 is activated by wild-type MetRS and does not discriminate between cell types4. As shown in Fig 3, both bacterial cells and macrophages were labeled with 3, whereas labeling with 2 was observed only in bacterial cells that express the NLL-MetRS.
Figure 3.
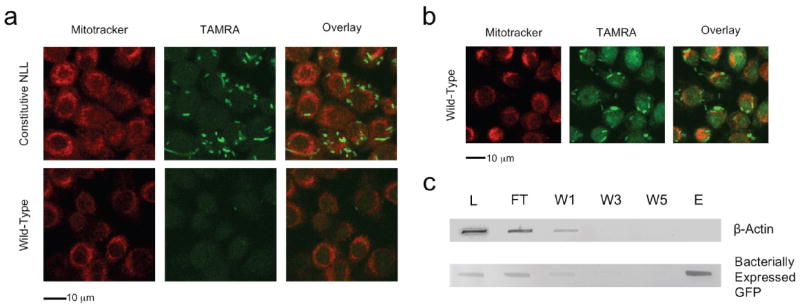
Cell-selective labeling in mixtures of bacterial and mammalian cells. (a) Fluorescence images of mixed cultures containing bacteria attached to or internalized by murine alveolar macrophages. Infection was performed in medium containing azidonorleucine. Bacterial cells constitutively expressing the NLL-MetRS were labeled by TAMRA-alkyne (Constitutive NLL) while cells lacking the NLL-MetRS (Wild type GFP) are visible only in the GFP channel (Supplementary Fig. 4, online). Macrophages were labeled with Mitotracker Deep Red (Invitrogen) and exhibited very low TAMRA background emission. In all cases, conjugation of TAMRA-alkyne was confined to bacterial cells expressing the NLL-MetRS. (b) Fluorescence images of macrophage infection with wild-type bacteria performed in the presence of azidohomoalanine. Protein synthesis by macrophages is indicated by strong TAMRA-alkyne emission from both bacterial cells and macrophages. (c) Macrophages were infected with bacterial cells that express GFP under induction with IPTG and constitutively express the NLL-MetRS. Infection was performed in medium containing IPTG and azidonorleucine to facilitate bacterial synthesis and labeling of GFP. Total cell lysate from the infection was subjected to conjugation with alkyne-functionalized biotin; labeled proteins were enriched with streptavidin avidity. Bacterially expressed GFP and mammalian β-actin were followed by immunoblot. Analysis of the lysate (L), unbound flow-through (FT), washes (W1, W3, W5), and eluent (E) reveal a separation of bacterial and mammalian representative proteins. Bacterially expressed GFP was labeled with azidonorleucine and thus subject to conjugation to biotin and enrichment with streptavidin. Protein originating from macrophages, including β-actin, were not labeled with 2 and therefore not conjugated to alkyne-functionalized biotin.
Newly synthesized bacterial proteins can be enriched from such cultures by affinity chromatography. Using an E. coli strain that expresses the NLL-MetRS constitutively and GFP under induction with IPTG, we infected macrophages in medium containing 2 mM 2. Immediately upon infection, IPTG was added to initiate bacterial synthesis of GFP. After 35 m at 37 °C, the total cell mixture was collected by centrifugation and lysed. Proteins were subjected to Cu (I) catalyzed azide-alkyne ligation with alkyne-functionalized biotin. Biotinylated proteins were selectively enriched by collection on neutravidin-agarose beads. After five washes, proteins were eluted from the resin with 2 mM free biotin and 2% SDS. To examine the extent of enrichment, the lysate, resin flow-through, washes and eluent were analyzed by immunoblot (Fig. 3c). The mammalian protein β-actin was detected with anti-β-actin and served as a representative macrophage protein. The bacterial marker GFP was detected with anti-Penta-His antibody. No actin was detected in the eluent, indicating at least 50-fold depletion of the mammalian marker. In contrast, comparison of the GFP band intensities in the eluent and lysate confirmed good recovery of the affinity-tagged bacterial protein.
The results described here illustrate the use of mutant aminoacyl-tRNA synthetases to enable cell-specific protein labeling with non-canonical amino acids. In mixed cellular systems, newly synthesized proteins in selected cells can be labeled with affinity reagents or fluorescent dyes for enrichment, identification, and visualization. This approach will enable unambiguous determination of the cellular origins of proteins made in complex multicellular systems and will provide new insight into intercellular communication. We are expanding the studies described here by engineering new amino acid/synthetase pairs and by using the azidonorleucine/NLL-MetRS pair to examine a variety of intercellular interactions.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institute of Health R21 DA020589, National Institutes of Health R01 GM62523, and the Army Research Office Institute for Collaborative Biotechnologies for support of this work. We thank K. Boulware for assistance in generating the three-dimensional movie of infected macrophages. E.M. Schuman is an investigator of the Howard Hughes Medical Institute.
References
- 1.Beynon RJ, Pratt JM. Mol Cell Proteomics. 2005;4:857–872. doi: 10.1074/mcp.R400010-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Mann M. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 3.Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Proc Natl Acad Sci USA. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dieterich DC, et al. Nat Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- 5.Prescher JA, Bertozzi CR. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 6.Beatty KE, Xie F, Wang Q, Tirrell DA. J Am Chem Soc. 2005;127:14150–14151. doi: 10.1021/ja054643w. [DOI] [PubMed] [Google Scholar]
- 7.Beatty KE, Liu JC, Xie F, Dieterich DC, Schuman EM, Wang Q, Tirrell DA. Angew Chem Int Ed Engl. 2006;13:7364–7367. doi: 10.1002/anie.200602114. [DOI] [PubMed] [Google Scholar]
- 8.Zhang GC, Chromy BA, McCutchen-Maloney SL. Expert Rev Proteomic. 2005;2:187–202. doi: 10.1586/14789450.2.2.187. [DOI] [PubMed] [Google Scholar]
- 9.Macpherson AJ, Harris NL. Nat Rev Immunol. 2004;4:478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 10.Ibba M, Söll D. Science. 1999;286:1893–1897. doi: 10.1126/science.286.5446.1893. [DOI] [PubMed] [Google Scholar]
- 11.Link AJ, Vink MK, Agard NJ, Prescher JA, Bertozzi CR, Tirrell DA. Proc Natl Acad Sci USA. 2006;103:10180–10185. doi: 10.1073/pnas.0601167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo TH, Tirrell DA. Angew Chem Int Ed. 2006;46:5340–5346. doi: 10.1002/anie.200700779. [DOI] [PubMed] [Google Scholar]
- 13.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 14.Lewis WG, et al. Angew Chem Int Ed. 2002;41:1053–51057. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.