

Abstract
K+-depolarization (KCl) of smooth muscle has long been known to cause Ca2+-dependent contraction, but only recently has this G protein-coupled receptor (GPCR)-independent stimulus been associated with rhoA kinase (ROCK)-dependent myosin light chain (MLC) phosphatase inhibition and Ca2+ sensitization. This study examined effects of ROCK inhibition on the concentration-response curves (CRCs) generated in femoral artery by incrementally adding increasing concentrations of KCl to intact tissues, and Ca2+ to tissues permeabilized with Triton X-100, β-escin and α-toxin. For a comparison, tissue responses were assessed also in the presence of protein kinase C (PKC) and MLC kinase inhibition. The ROCK inhibitor H-1152 induced a strong concentration-dependent inhibition of a KCl CRC. A relatively low GF-109203X concentration (1 μM) sufficient to inhibit conventional PKC isotypes also inhibited the KCl CRC but did not affect the maximum tension. ROCK inhibitors had no effect on the Ca2+ CRC induced in Triton X-100 or α-toxin permeabilized tissues, but depressed the maximum contraction induced in β-escin permeabilized tissue. GF-109203X at 1 μM depressed the maximum Ca2+-dependent contraction induced in α-toxin permeabilized tissue and had no effect on the Ca2+ CRC induced in Triton X-100 permeabilized tissue. The MLC kinase inhibitor wortmannin (1 μM) strongly depression the Ca2+ CRCs in tissues permeabilized with Triton X-100, α-toxin and β-escin. H-1152 inhibited contractions induced by a single exposure to a submaximum [Ca2+] (pCa 6) in both rabbit and mouse femoral arteries. These data indicate that β-escin permeabilized muscle preserves GPCR-independent, Ca2+- and ROCK-dependent, Ca2+ sensitization.
Keywords: vascular smooth muscle, permeabilization, contraction, Ca2+ signaling, signal transduction, Ca2+ sensitization
Introduction
The degree of isometric tension developed in smooth muscle involves mechanisms directly dependent on increases in cytosolic free Ca2+ ([Ca2+]i) and those that appear to be, if not entirely Ca2+-independent, at least independent of global increases in [Ca2+]i (reviewed by (Puetz, Lubomirov et al. 2009)). Influx of Ca2+ into smooth muscle activates Ca2+ and calmodulin-dependent myosin light chain (MLC) kinase (MLCK). MLCK phosphorylates the 20-kD regulatory MLC (MLC-p), which induces actomyosin crossbridge cycling and tension development. In addition, apparently Ca2+-independent protein cascades can inhibit MLC phosphatase (MLCP) activity, increasing the degree of MLC-p and concomitant tension despite constant or even decreasing [Ca2+]i. Early studies indicated that this Ca2+ sensitized tension production requires G protein-coupled receptor (GPCR) activation (Nishimura, Kolber et al. 1988; Sato, Ozaki et al. 1988; Fujiwara, Itoh et al. 1989; Kitazawa, Kobayashi et al. 1989). Effectors of Ca2+ sensitization include rhoA kinase (ROCK) and protein kinase C (PKC), which can phosphorylate the MLCP regulatory protein (targeting subunit), MYPT1, and the 17 kD PKC-dependent MLCP inhibitor, CPI-17, leading to inhibition of MLCP activity (Kitazawa, Masuo et al. 1991; Masuo, Reardon et al. 1994; Feng, Ito et al. 1999; Eto, Kitazawa et al. 2001; Velasco, Armstrong et al. 2002).
To study mechanisms regulating smooth muscle contraction, investigators often utilize high concentrations of extracellular K+ (KCl) as a stimulus to clamp the cell membrane potential above the threshold for voltage-operated calcium channel-dependent Ca2+ influx, increasing [Ca2+]i that directly activates Ca2+-calmodulin-dependent MLCK. This protocol was designed to circumvent the complex cell signaling cascades intrinsic to GPCR-mediated contraction (Somlyo and Somlyo 1968; Casteels, Kitamura et al. 1977; Somlyo and Himpens 1989). However, several studies have demonstrated that KCl-induced smooth muscle contraction also displays significant levels of Ca2+ sensitization (reviewed by (Ratz, Berg et al. 2005)). As described in an editorial in 2003 published in Circulation Research, “…it has become dogma that depolarization leads to only an increase in Ca2+ and a subsequent activation of MLCK, without the activation of additional signaling pathways. Challenging these generally accepted principles is recent evidence that ROCK inhibition almost completely eliminates the sustained portion of…contraction to…KCl depolarization…without affecting the Ca2+ transient (Brozovich 2003).” We recently showed that PKC also participates in KCl-induced Ca2+ sensitization (Ratz and Miner 2009). Thus, both ROCK- and PKC-dependent Ca2+ sensitizing systems seem to be “on” during a KCl-induced contraction.
Because KCl depolarizes the plasma membrane and opens Ca2+ channels, it can be proposed that membrane depolarization (Yanagisawa and Okada 1994), elevations in [Ca2+]i (Sakurada, Takuwa et al. 2003), or both mechanisms link the KCl stimulus with activation of cell signaling systems responsible for causing Ca2+ sensitizion of contraction. Precisely what links Ca2+ or membrane depolarization with activation of ROCK remains a matter of intensive investigation. Ca2+ may activate phosphoinositide 3-kinase (PI3K-C2α) or a pyk2-dependent PDZrhoGEF to induce elevations in active rhoA (Wang, Yoshioka et al. 2006; Yoshioka, Sugimoto et al. 2007; Ying, Giachini et al. 2009). Alternatively or concomitantly, Ca2+-dependent Ca2+ sensitization may reflect constitutive ROCK activity that basally inhibits MLCP resulting in basally elevated MLC-p (Alvarez, Miner et al. 2010). Indeed, basal levels of MYPT1-pT853 and MLC-p measured in resting artery and bladder smooth muscles are above zero (Ratz and Miner 2003; Porter, Evans et al. 2006; Poley, Dosier et al. 2008; Alvarez, Miner et al. 2010), and contraction does not occur in these tissues until MLC-p is elevated above a threshold level of ~15% (Rembold, Wardle et al. 2004).
Chemical permeabilization eliminates resting membrane potential and permits precise clamping of [Ca2+]i by using an EGTA-based Ca2+ buffering system (Cassidy, Hoar et al. 1979; Arner 1982; Nishimura, Kolber et al. 1988; Fujiwara, Itoh et al. 1989; Kitazawa, Kobayashi et al. 1989; Kobayashi, Kitazawa et al. 1989; Schubert 1996). Ca2+ sensitization was originally verified by using permeabilization procedures that protect plasma membrane GPCR signaling systems, and it is now generally accepted that contractile stimuli that activate certain GPCRs can cause Ca2+ sensitization. However, whether Ca2+ sensitization can be produced without activation of GPCRs in permeabilized tissues remains to be fully explored.
Three detergents and one pore-forming molecule are generally used to chemically permeabilize smooth muscle tissue (Van Heijst, Blange et al. 2000). Saponin forms a complex with cholesterol in membrane leaflets to form ~9 nm pores (Bangham, Horne et al. 1962) and uncouples receptors involved in physiological activation of smooth muscle (Itoh, Kuriyama et al. 1983). Compared to saponin, the specific saponin ester, β-escin, has a milder action on the plasma membrane than saponin, is a more potent modifier of sarcoplasmic reticulum function (Launikonis and Stephenson 1999) and retains GPCR-contraction coupling mechanisms (Kobayashi, Kitazawa et al. 1989). Staphylococcus aureus α-toxin forms ~3 nm pores permeable to molecules of up to ~4 kD (Fussle, Bhakdi et al. 1981; Lind, Ahnert-Hilger et al. 1987) and, like β-escin, retains GPCR-contraction coupling mechanisms (Kitazawa, Kobayashi et al. 1989). Triton X-100 leads to an extensively permeabilized cell in which all membrane functions are eliminated including the loss of GPCR-contraction coupling mechanisms, sarcoplasmic reticulum function, and likely because of loss of CPI-17, responsiveness to activators of PKC (Kitazawa, Takizawa et al. 1999). By utilizing Triton X-100, α-toxin and β-escin permeabilized rabbit femoral artery, this study tests the hypothesis that Ca2+-dependent contraction is dependent on ROCK activity.
Recent studies strongly suggest that aberrant levels of Ca2+ sensitization participate in causing certain vascular smooth muscle hyper-contractile disorders. Increased ROCK activity has been shown to contribute to hypertension and coronary artery spasm (Uehata, Ishizaki et al. 1997; Masumoto, Hirooka et al. 2001; Masumoto, Mohri et al. 2002). Moreover, direct inhibition of ROCK has proven to be a therapeutic treatment for both hypertension and vasospasm (Liao, Seto et al. 2007). Thus, a more thorough understanding of the mechanisms participating in ROCK activation should permit the identification of novel cellular and molecular targets for clinical treatment of vascular hyper-contractile disorders.
Methods
Tissue Preparation and Isometric Tension
Tissues were prepared and contractions were measured as previously described (Ratz, 1993). All animal care and experimental protocols complied with the appropriate animal welfare regulations and guidelines of the US Public Health Service as approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee. Femoral arteries isolated from adult male and female New Zealand White rabbits were cleaned of adhering tissue, mechanically denuded of endothelium and cut into 3 mm wide rings under an Olympus SZX12 binocular dissecting microscope (Olympus America, Inc., Center Valley, PA), and used for all studies except that shown in Figs 5D–5F. Femoral arteries from female C57BL/J mice were isolated, cleaned of adhering tissue, mechanically denuded of endothelium, cut into 2 mm wide rings, and used for the study shown in Figs 5D–5F. Throughout storage and experimentation, tissues were maintained in a modified physiological salt solution (PSS; in mM: 140 NaCl, 4.7 KCl, 2.0 morpholino-propanesulfonic acid (MOPS), 0.02 ethylenediaminetetraacetic acid (EDTA) to chelate heavy metals, 1.2 Na2HPO4·7H2O, 1.2 MgCl2, 1.6 CaCl2, 5.6 α-D-glucose and adjusted to pH 7.4 at 37°C with 5N NaOH). Each artery artery ring was secured between two stainless steel pins (or the tungsten wires of two Mulvany clips form mouse arteries) attached to a temperature-controlled myograph tissue chamber (for rabbit arteries: Model 610M, DMT-USA, Inc., Atlanta, GA, USA; for mouse arteries: M-series, Radnoti LLC, Monrovia, Ca 91016, USA). Artery rings were allowed to equilibrate for one hour in aerated PSS at 37°C. One pin (or Mulvany clip) was connected to a micrometer for muscle length adjustments and the other pin (or Mulvany clip) was connected to an isometric tension transducer. For each tissue, equilibration was followed by an abbreviated length-tension curve requiring approximately two hours to identify optimum tension (To) and length (Lo). Muscle contraction was induced by replacement of the PSS tissue bathing solution with a solution containing 110 mM KCl substituted isosmotically for NaCl (high KCl). Each contraction induced by high KCl was maintained for ~5 min, at which time tension and intracellular free Ca2+ in rabbit muscular arteries achieve steady-state levels (Ratz, Miner et al. 2009). Voltage signals from each myograph tension transducer were digitized (National Instruments, Austin, TX, USA) and visualized on a computer screen as tension (T), with units in grams for convenience. The digital data were recorded and analyzed using DASYLab (National Instruments, Austin, TX, USA)
Fig 5.
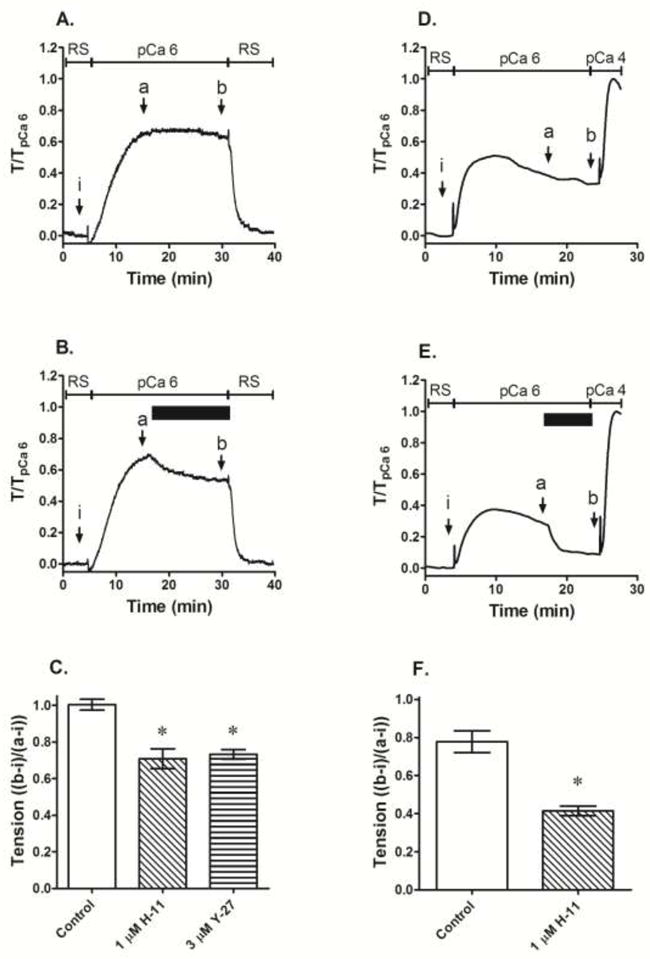
Effect of ROCK inhibitors (B, C, E and F) compared to control (A and B) when added at steady-state contraction (B and E, solid horizontal bar) induced by a single submaximum Ca2+ concentration in rabbit (A–C) and mouse (D–E) femoral arteries permeabilized with β-escin. Data in A, B, D and E are representative tension tracings; H-11 = H-1152, Y-27 = Y-27632, RS = relaxing solution, solid bar indicates duration of exposure to ROCK inhibitor i, a and b indicate where data were taken to calculate tension values shown in C and F. Data in C and F are means ± S.E., n=5 (Control), 4 (H-11) and 3 (Y-27 and Wort), * p < 0.05 compared to Control.
Tissue Permeabilization
Each artery ring at Lo was incubated at 30° C in a Ca2+-free “relaxing solution” (in mM: 74.1 potassium methanesulfonate, 4.0 magnesium methanesulfonate, 4.0 Na2ATP, 4.0 EGTA, 5.0 creatine phosphate, 30.0 PIPES, adjusted to pH 7.1 with 1N KOH and ionic strength 180 with additional 0.5 M potassium methanesulfonate). α-toxin (EMD Bioscience, (formerly Calbiochem), San Diego, CA, USA) permeabilization was achieved by incubating each tissue for 50 minutes with 20 μg/ml α-toxin dissolved in Ca2+-free relaxing solution. Triton X-100 (Pierce, Rockford, IL, USA) permeabilization was achieved by incubating each tissue for 15 minutes with 1% Triton X-100. α-toxin was reused because of cost and slowly lost potency over a period of 1–2 months, resulting in a small rightward shift in the [Ca2+]-response curve. To compensate for this shift, for each day that the effect of a drug was measured, a control response was also measured. For rabbit artery, β-escin (Sigma-Aldrich, St. Louis, MO, USA) permeabilization was achieved by incubating tissues with 100 μM β-escin dissolved in Ca2+-free relaxing solution at 4°C for 45 minutes then at 30°C for 15 minutes. The low temperature incubation of tissues in β-escin facilitates the slow penetration and/or binding of detergent molecules to the smooth muscle surface (Masuo, Reardon et al. 1994). Mouse arteries were permeabilized with β-escin dissolved in Ca2+-free relaxing solution at 30°C for 7 minutes. Following permeabilization each tissue was washed thoroughly with Ca2+-free relaxing solution to remove the permeabilizing agent. To deplete sarcoplasmic reticular calcium, 10 μM A23187 (a calcium ionophore) was used in all permeabilized tissue. In Triton X-100 and β-escin permeabilized tissue, 1unit/ml of calmodulin (Sigma Aldrich, St. Louis, MO) was added to compensate for loss during permeabilization. All permeabilized tissues and solutions were maintained at 30°C. Each tissue underwent an internal control contraction after permeabilization where the [Ca2+] was clamped for 10 min at 1 μM (pCa 6) in a “contracting solution” made by adding the appropriate amount of 1 M CaCl2 stock (Fluka Chemicals, Sigma-Aldrich, St. Louis, MO, USA) to Ca2+-free relaxing solution that had not yet been pH- and ionic strength-adjusted, followed by pH and ionic strength adjustments, as determined by the software program WEBMAXC developed by Chris Patton (maxchelator.stanford.edu). pCa is defined as the negative logarithm of the free [Ca2+] in moles/L. All subsequent contractions for each tissue was normalized to its internal control contraction and reported as a fraction of the initial pCa 6 contraction. Subsequent contractions were often slightly weaker than the initial contraction because of “run-down” commonly seen in permeabilized smooth muscle tissues (Masuo, Reardon et al. 1994). In the experiment shown in Figs 6D–6F, intact tissues were first contracted with 110 mM KCl in the presence of 0.4 mM CaCl2 (pCa 3.4), and subsequent contractions were normalized to this contraction.
Fig 6.
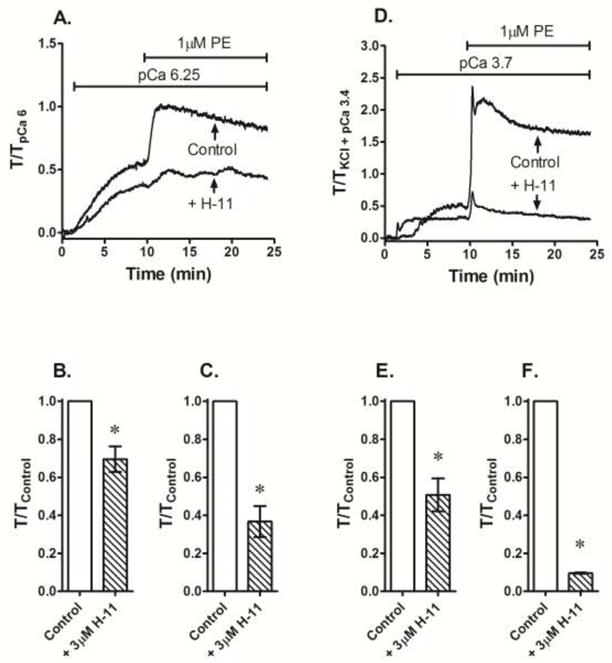
Effect of 3 μM H-1152 (H-11) compared to no drug addition (Control) added prior to stimulation with a single submaximum Ca2+ concentration and 1 μM phenylephrine (PE) on steady-state tension achieved by these contractile agents in β-escin permeabilized (A–C) and KCl-depolarized intact (D–F) femoral artery. Data in A and D are representative tension tracings. Data in B, C, E and F are means ± S.E., n=3, * p < 0.05 compared to Control.
Concentration-Response Curves (CRCs)
Following the internal control contraction, permeabilized tissues were incubated for 15 –20 minutes with specific kinase inhibitors. Control tissues received either no inhibitor or the vehicle used to dissolve the inhibitor. A Ca2+ CRC was constructed using seven to eleven contracting solutions made of incrementally increasing [Ca2+]s from 3.16 ×10−8 (pCa 7.5) to 10−5 (pCa 5). For each [Ca2+], the resulting steady-state tension response was measured. Each of the seven to eleven contracting solutions was made separately by adding the appropriate amount of 1 M CaCl2 to a fixed amount of pre-adjusted relaxing solution followed by pH and ionic strength adjustments. Each contracting solution was added to the tissue for 5–10 min which permitted tension to reach a steady-state value. For a comparison, intact (not permeabilized) tissues were subjected to a KCl CRC. Intact tissues incubated in PSS were contracted by adding KCl from 10 mM to 60 mM in 10 mM increments over a period of ~30 – 45 min. Steady-state tension values produced for each added [KCl] were normalized to To. The KCl-induced contractions were not due to changes in osmolarity because adding sucrose (20 mM to 120 mM incrementally in steps of 20 mM) or LiCl2 (10 mM to 60 mM incrementally in steps of 10 mM) had no effect on tension (n = 4, data not shown). For a comparison with the KCl CRC, some intact tissues were depolarized with high KCl and subjected to a Ca2+ CRC. Phentolamine (1 μM) was added to inhibit the potential activation of -adrenergic receptors induced by periarterial nerve stimulation in response to the depolarizing stimuli. Tissues pre-incubated with a specific kinase inhibitor prior to the Ca2+ and KCl CRCs were exposed to the same kinase inhibitor concentration throughout the CRCs.
Drugs and Solutions
GF-109203X (2-[1-(3-dimethylaminopropyl)indol-3-yl]-3-(indol-3-yl) maleimide), H-1152 ((S)-(+)-2-Methyl-1-[(4-methyl-5-isoquinolinyl)sulfonyl]-hexahydro-1H-1,4-diazepine dihydrochloride), HA-1077 (fasudil), KT-5720 ((9R, 10S, 12S)-2,3,9,10,11,12-hexahydro-10hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-k]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid) and Y-27632 (trans-4-[(1R)-1-aminoethyl]-N-4-pyridinylcyclohexanecar boxamide dihydrochloride) were from EMD Biosciences (formerly Calbiochem), San Diego, CA, USA. β-escin was from Sigma-Aldrich Corporation, St. Louis, MO, USA. Wortmannin was from Alexis Biochemicals, San Diego, CA, USA. A23187 (Free acid, Streptomyces chartreusensis, Calcimycin) was from Fisher Scientific, Pittsburg, PA, USA. GF-109203X, wortmannin, and A23187 were dissolved in DMSO, while all other compounds were dissolved in de-ionized water. DMSO was added at final concentrations no more than 0.1%.
Statistics
The null hypotheses were examined using Students’ unpaired t-test or a one-way analysis of variance (ANOVA). Differences between experimental groups were analyzed using the Neuman-Keuls post-hoc test. The null hypothesis was rejected in all cases where p<0.05.
Results
Effect of PKC and ROCK inhibitors on KCl CRC in depolarized intact artery
Intact tissues contracted by incremental additions of 10 mM KCl in the absence of a protein kinase inhibitor produced a concentration-dependent increase in tension (Fig 1A, DMSO) with a maximum contraction of ~90 % To. GF-109203X at 1 μM reduced the strength of contraction at all KCl concentrations except the highest concentration used (i.e., GF-109203X did not significantly reduce the maximum tension; Fig 1A, 1 μM GF-109). It is generally agreed that 1 μM GF109203X can inhibit α/β and δ PKC isotypes and cannot inhibit the atypical ζ/γ PKC isotype or MLCK or ROCK (Toullec, Pianetti et al. 1991; Martiny-Baron, Kazanietz et al. 1993; Gailly, Gong et al. 1997; Davies, Reddy et al. 2000). These data suggest that PKCα/β or δ participates in the regulation of a KCl-induced contraction, but that strong membrane depolarization obscures this contribution.
Figure 1.

Inhibition of a KCl CRC generated in intact femoral artery by the ROCK inhibitor H-1152 (0.3 μM H-11 and 1 μM H-11) and a relatively low concentration (1 μM) of the PKC inhibitor GF-109203X (1 GF) compared to DMSO control (A), and lack of effect of 1 μM KT-5720 on a KCl CRC (B). Data are means ± S.E., n = 6–8 for panel A and 4 for panel B, * p < 0.05 for all data points except 10 mM (log [KCl] = −2.0) compared to DMSO control. ψ p < 0.05 for all data points except 10 mM and 60 mM (the highest [KCl] used) compared to DMSO control.
The ROCK inhibitor H-1152 at 0.3 μM and 1 μM strongly suppressed the strength of KCl-induced contractions at all KCl concentrations tested (Fig 1A, 0.3 μM H-11 and 1 μM H-11). Although H-1152 and Y-27632 have been used for several years as selective ROCK inhibitors, control agents should be applied whenever possible to test for potential inhibition of other ser/thr kinases. Based on an extensive study comparing the specificities of 65 inhibitors of protein kinases against a panel of 70–80 protein kinases (Bain, Plater et al. 2007), we chose KT-5720 to serve as a control for potential non-selective activity of H-1152 and Y-27632 in our tissue. Specifically, 1 μM KT-5720, 1 μM H-1152 and 10 μM Y-27632 inhibit two protein kinases that may alter smooth muscle contraction, protein kinase C-related kinase-2 (PRK2) and 3-phosphoinositide-dependent protein kinase (PDK1). Importantly, 1 μM KT-5720 is a poor inhibitor of ROCK whereas 1 μM H-1152 and 10 μM Y-27632 are strong ROCK inhibitors. In rabbit femoral artery, a KCl CRC in the presence of 1 μM KT-5720 was not different than a KCl CRC in the presence of the drug vehicle (Fig 1B). These data suggest that the inhibitory effect of H-1152 and Y-27632 on a KCl CRC was most likely due to attenuation of ROCK activity.
Effect of selected kinase inhibitors on Ca2+ CRC in permeabilized artery
H-1152 at a concentration that attenuated a maximum KCl-induced contraction by nearly 50% (0.3 μM, see Fig 1A) had no effect on a Ca2+ CRC induced in Triton X-100 permeabilized muscle (Fig 2A, 0.3 H-11). A 10-fold higher concentration likewise did not cause inhibition (Fig 2A, 3 H-11). At 10 μM, two other ROCK inhibitors, Y-27632 and HA-1077, likewise did not diminish the strength of a Ca2+ CRCs in tissues permeabilized with Triton X-100 (Fig 2B). GF-109203X at a concentration that caused a rightward shift in the KCl CRC (1 μM, see Fig 1A) had no effect on a Ca2+ CRC induced in Triton X-100 permeabilized muscle (Fig 2C, 1 GF-109). At 3 μM, GF-109203X shifted the curve rightward and did not inhibit the maximum response, and at 10 μM, GF-109203X also inhibited the maximum contraction (Fig 2C, 3 GF-109 and 10 GF-109). The MLC kinase inhibitor wortmannin, at 1 μM, caused a rightward shift of the Ca2+ CRC and depressed of the maximum response (Fig 2D, 1 Wort). These data support the notion that in Triton X-100 permeabilized muscle, conventional PKC- and ROCK-dependent contractile signaling systems were not retained, and that MLC kinase is the primary signaling molecular responsible for Ca2+-dependent contraction.
Fig 2.
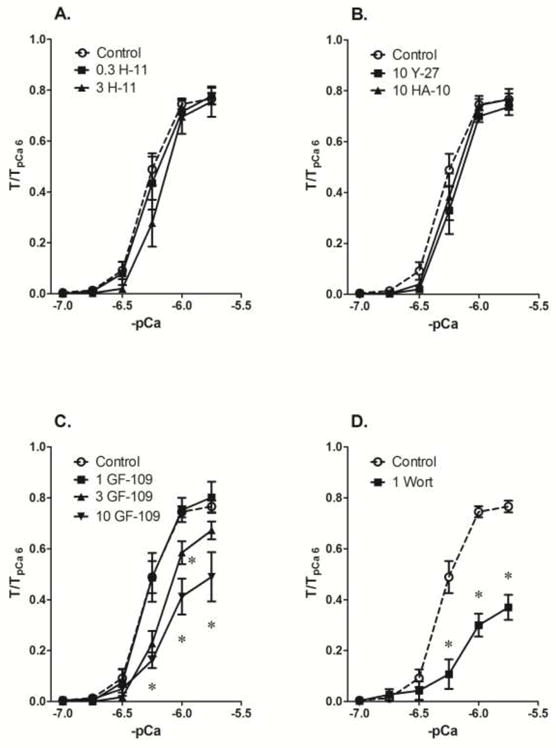
Inhibition of a Ca2+ CRC generated in Triton X-100 permeabilized femoral artery by 1 μM of the MLCK inhibitor wortmannin (D, 1 Wort) but not by 0.3 μM and 3 μM of the ROCK inhibitor H-1152 (A, 0.3 H-11 and 3 H), 10 mM of the ROCK inhibitors Y-27632 and HA-1077 (B, 10 Y-27 and HA-10), nor a relatively low concentration (1 μM) of the PKC inhibitor GF-109203X (1 GF) compared to DMSO control (Control). Data are means ± S.E., n=11 (Control), 4 (H-11, GF and Wort) and 3 (Y-27 and HA-10), * p < 0.05 compared to Control.
The control Ca2+ CRC induced in α-toxin permeabilized artery rings (Fig 3, Control) was leftward shifted compared to that induced in Triton X-100. Moreover, contraction of α-toxin permeabilized artery did not display any degradation because the contraction induced by pCa 6 at the end of the Ca2+ CRC reached a level of at least 1-fold that of the initial contraction induced by exposure to pCa 6 prior to the Ca2+ CRC. By comparison, the pCa 6-induced contraction induced at the end of the Ca2+ CRC in tissues permeabilized by Triton X-100 was degraded by ~20% (see Fig 2, Control). Thus, in our hands, Ca2+ is a more potent stimulus in α-toxin permeabilized compared to Triton X-100 permeabilized tissue (compare Control curves in Figs 2A and 3A). As in tissues permeabilized with Triton X-100, 3 μM H-1152, 10 μM Y-27632 and 10 μM H-1077 had no effect on the Ca2+ CRC in α-toxin permeabilized muscle (Fig 3A). Also like that seen in Triton X-100 permeablized muscle 1 μM wortmannin caused a rightward shift and depression of the maximum in α-toxin permeabiilzed muscle (Fig 3C, 1 Wort). Unlike that seen in Triton X-100 permeabilized muscle, 1 μM GF-109203X significantly inhibited contractions induced at pCa 6.5 and above in α-toxin permeabiilzed muscle (Fig 3B).
Fig 3.
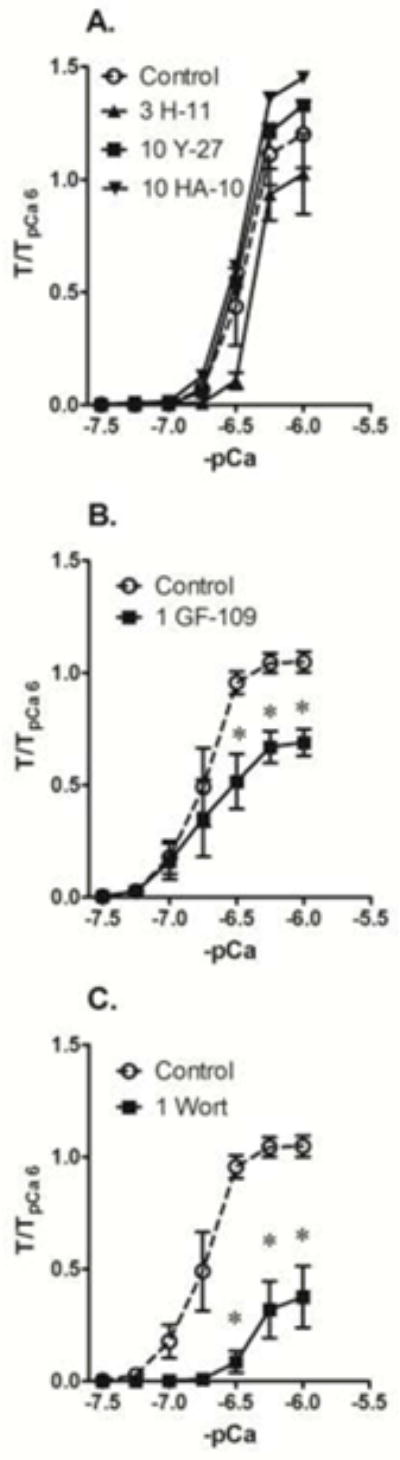
Inhibition of a Ca2+ CRC generated in α-toxin permeabilized femoral by 1 μM of the MLCK inhibitor wortmannin (C, Wort) and a relatively low concentration (1 μM) of the PKC inhibitor GF-109203X (B, 1 GF-109) but not by the ROCK inhibitors H-1152 (3 μM, H-11), Y-27632 (10 μM, Y-27) and HA-1077 (10 μM, 10-HA) compared to DMSO control (Control). Data are means ± S.E., n=5 (Control) and 3 for ROCK, PKC and MLCK inhibitors, * p < 0.05 compared to Control.
Like α-toxin permeabilized muscle, the average control Ca2+ CRC induced in β-escin permeabilized artery rings displayed little degradation, but the control curve was rightward-shifted compared to that induced in α-toxin permeabililzed muscle (compare Control curves in Figs 3 and 4). Unlike that seen in Triton X-100 and α-toxin permeabilized tissues, the chemically distinct ROCK inhibitors H-1152 (3 μM) and Y-27632 (10 μM) reduced the maximum Ca2+ CRC tension compared to control in β-escin permeabilized tissues (Fig 4A). Like that seen in Triton X-100 and α-toxin permeabilized tissues, 1 μM wortmannin caused a rightward shift in the Ca2+ CRC and reduced the maximum tension response in β-escin permeabilized tissues (Fig 4C). As in α-toxin permeabilized tissues, 1 μM GF-109203X appeared to strongly inhibit contraction in β-escin permeabilized tissues (Fig 4B, n=2). The general ser/thr kinase inhibitor staurosporine at 1 μM completely prevented the development of a Ca2+ CRC in β-escin permeabilized artery (data not shown).
Fig 4.
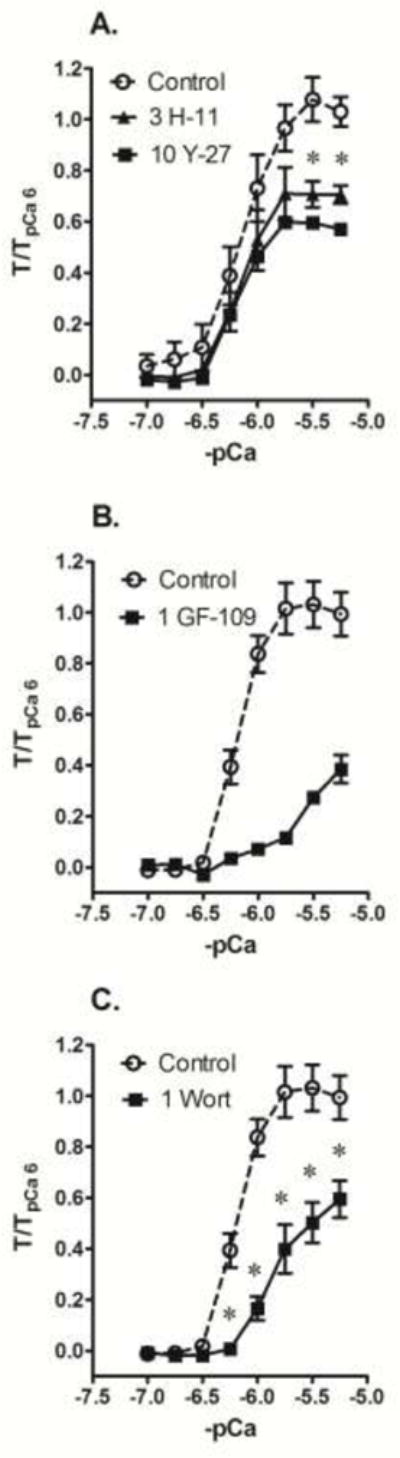
Inhibition of maximum tension induced during a Ca2+ CRC generated in β-escin permeabilized femoral artery by the ROCK inhibitors H-1152 (A, 3 μM, 3 H-11) and Y-27632 (B, 10 μM, 10 Y-27), and strong inhibition of the Ca2+ CRC by the MLCK inhibitor wortmannin (C, 1 μM, 1 Wort), compared to DMSO control (Control). Data are means ± S.E., n=5 (Control), 4 (H-11) and 3 (Y-27 and Wort), * p < 0.05 compared to Control. A relatively low concentration (1 μM) of the PKC inhibitor GF-109203X (B, 1 GF-109) also appeared to cause strong inhibition of a Ca2+ CRC (n=2).
Effect of ROCK inhibition on a submaximum Ca2+-induced contraction in β-escin permeabilized muscle
Although the average values of contraction produced by increases in Ca2+ during a Ca2+ CRC in β-escin permeabilized artery exposed to H-1152 were less than the average values for control tissues, this apparent difference was significant only at the highest Ca2+ concentrations (see Fig 4A). To determine whether H-1152 can inhibit a contraction induced by a submaximum Ca2+ concentration, tissues permeabilized with β-escin were stimulated with Ca2+ at pCa 6, a stimulus producing an ~70% maximum contraction (see Fig 4A) and at steady-state, challenged with 1 μM H-1152. Compared to control tissues (Fig 5A), H-1152 caused a slowly-developing inhibition (Fig 5B) achieving ~30% inhibition within 15 min (Fig 5C). Y-27632 at 3 μM produced inhibition of a similar degree (Fig 5C). To determine whether this effect was species specific, a similar protocol was applied to mouse femoral artery. In this tissue, 1 μM H-1152 inhibited steady-state pCa 6-induced contraction by ~50% compared to control (Figs 5D–5F). Tissues washed to remove H-1152 and stimulated with Ca2+ at pCa 4 displayed a much stronger contraction compared to that induced by pCa 6, confirming that pCa 6 was a submaximum stimulus in mouse femoral artery permeabilized with β-escin (Figs 5D and 5E).
To determine whether H-1152 added prior to stimulation with a single submaximum Ca2+ concentration can also cause inhibition of steady-state tension, rings of rabbit femoral artery permeabilized with β-escin were exposed to 3 μM H-1152 for 15 min, then stimulated with Ca2+ at pCa 6.25. When normalized to the response produced by control tissues that were not exposed to H-1152, pCa 6.25-induced contractions were inhibited by ~30% (Fig 6A and 6B). To confirm that the β-escin permeabilized muscle could support a GPCR-induced contraction, and that H-1152 could inhibit this contraction, tissues were then exposed to 1 μM phenylephrine (PE, Fig 6A). As expected, PE induced a strong contraction that was inhibited by H-1152 (Figs 6A and 6C). A similar protocol was applied to intact tissues. Tissues were depolarized with KCl in a nominally Ca2+-free solution (PSS absent CaCl2), then exposed to pCa 3.7 (0.2 mM) to cause a submaximum contraction, and at the steady-state of Ca2+-induced contraction, were stimulated further with 1 μM PE (Fig 6D). Compared to control tissues, those incubated with 3 μM H-1152 contracted weakly to both Ca2+ (Figs 6D and 6E) and PE (Figs 6D and 6F).
Discussion
Results from this study support the hypothesis that ROCK and conventional PKC isotypes participate in the regulation of contraction of tissues in which the sole applied stimulus is Ca2+. Components of this GPCR-independent, Ca2+-, ROCK- and PKC-dependent contractile signaling system were preserved in β-escin and α-toxin, but not Triton X-100, permeabilized muscle; the least “harsh” permeabilizing agent, α-toxin, preserved the PKC- but not ROCK-dependent regulatory system, and β-escin preserved the ROCK- and PKC-dependent regulatory systems. Recent evidence reveals that ROCK and conventional PKC isotypes appear to be constitutively active in intact, resting (not stimulated to contract) smooth muscles (Navedo, Amberg et al. 2006; Porter, Evans et al. 2006; Poley, Dosier et al. 2008; Ratz and Miner 2009; Wang, Kendig et al. 2009; Alvarez, Miner et al. 2010). Thus, these data together support the hypothesis that the Ca2+ sensitization observed in KCl-stimulated smooth muscle (reviewed by (Ratz, Berg et al. 2005)) is due to activation of ROCK and PKC by increases in [Ca2+]i or to the constitutive activities of ROCK and PKC.
Wortmannin at 1 μM inhibited maximum tension in permeabilized muscle by up to ~65%, supporting the generally accepted hypothesis that MLCK plays a significant role in Ca2+-activated contraction (He, Peng et al. 2008). Notably, wortmannin not only shifted the Ca2+ CRC rightward, but also reduced the Ca2+ CRC maximum tension. This was the anticipated response because wortmannin should diminish MLCK activity at each [Ca2+] added to the tissue, and this should result in a rightward shifted Ca2+ CRC with a depressed maximum tension. Although wortmannin at 1 μM abolishes MLCK activity in vitro (Nakanishi, Kakia et al. 1992; Davies, Reddy et al. 2000) and tension induced by the Ca2+ channel agonist Bay k 8644 in intact rabbit femoral artery (Alvarez, Miner et al. 2010), this concentration did not abolish contraction induced in permeabilized femoral artery in the present study. At 10 μM, wortmannin inhibits α-toxin permeabilized rat caudal artery contraction by ~50%. This might suggest that additional mechanisms participate in regulation of a Ca2+ CRC in permeabilized muscle (Wilson, Sutherland et al. 2005), or that ATP concentrations in permeabilized muscle are slightly higher than in intact tissue (wortmannin competes with ATP at the MLCK catalytic domain (Nakanishi, Kakia et al. 1992)). However, the general ser/thr kinase inhibitor staurosporine (Ruegg and Burgess 1989) also acts by competing with ATP, and at 1 μM, abolished the ability of Ca2+ to induce a contraction in β-escin permeabilized muscle in the present study. These data suggest that signaling events involving ser/thr phosphorylation are necessary for the development of Ca2+-dependent tension.
KCl causes increases in [Ca2+]i, active rhoA, MYPT1-pT853, MLC-p and tension, and ROCK inhibition attenuates MYPT1-pT853, MLC-p and tension without reducing [Ca2+]i (Mita, Yanagihara et al. 2002; Sakamoto, Hori et al. 2003; Sakurada, Takuwa et al. 2003; Urban, Berg et al. 2003; Janssen, Tazzeo et al. 2004; Ratz and Miner 2009). These data suggest that KCl causes ROCK activation leading to Ca2+ sensitization because rhoA activates ROCK, MYPT1-pT853 is a substrate for ROCK, phosphorylation of MYPT1 at T853 causes MLCP inhibition and MLCP inhibition would be expected to elevate MLC-p for a given level of [Ca2+]i (for review, see (Puetz, Lubomirov et al. 2009)). In addition, PKC inhibition attenuates KCl-induced increases in MYPT1-pT853 and tension (Ratz and Miner 2009), suggesting that KCl may also cause PKC activation leading to Ca2+ sensitization. Although the precise link between KCl and Ca2+ sensitization of contractions remains to be determined, one proposal is that KCl-dependent increases in [Ca2+]i cause ROCK activation (Sakurada, Takuwa et al. 2003; Urban, Berg et al. 2003; Janssen, Tazzeo et al. 2004; Yoshioka, Sugimoto et al. 2007). We recently showed that the Ca2+ channel activator Bay k 8644 causes a strong increase in [Ca2+]i, active rhoA, MLC-p and tension, but phosphorylation of the ROCK substrates, MYPT1-pT853 and MYPT1-pT696, did not increase above the basal level, suggesting that an increase in [Ca2+]i and rhoA does not necessarily cause ROCK activation (Alvarez, Miner et al. 2010). Despite showing no increase in MYPT1-pT853, ROCK inhibitors greatly attenuated basal MYPT1-pT853 and Bay k 8644-induced increases in tension and MLC-p, but not [Ca2+]i. These data lead to a paradoxical scenario in which a ROCK inhibitor can greatly attenuate a contraction induced by a stimulus that apparently does not increase ROCK activity. This paradox could be resolved if constitutively active ROCK causes basal MYPT1-pT853 and basal inhibition of MLCP, and that these basal activities participate in regulation of stimulated contraction. In this case, the tension increase induced by any agent that causes an increase in MLCK activity would be reduced by a ROCK inhibitor even if the stimulus does not itself induce a further increase in ROCK activity and MYPT1 phosphorylation above this basal levels. Alternatively, the balance between RhoGEF and RhoGAP activities may be regulated by Ca2+.
In smooth muscle, CPI-17 is entirely lost after Triton X-100 permeabilization, is reduced by 65% after β-escin permeabilization, and is preserved by α-toxin permeabilization (Kitazawa, Takizawa et al. 1999). Moreover, PKCα/β and ROCK, but not MYPT1 nor MLCK, are also lost from tissues permeabilized with Triton X-100 (Weber, Seto et al. 2000; Araki, Ito et al. 2001). Our data showed that the Ca2+-stimulated contraction induced in tissues permeabilized with α-toxin was inhibited by 1 μM GF-109203X, but that induced in Triton X-100 permeabilized tissues was not. In addition, ROCK inhibition had no effect on a Ca2+ CRC in tissues permeabilized by Triton X-100. Why the ROCK-dependent contraction was preserved in β-escin, and not in α-toxin, permeabilized muscle remains to be determined. This is especially puzzling given that arachidonic acid-induced activation of ROCK-dependent contraction is inhibited by Y-27632 in α-toxin permeabilized rabbit femoral artery (Fu, Gong et al. 1998; Araki, Ito et al. 2001). We speculate that α-toxin may interfere with a step in the Ca2+-dependent ROCK signaling cascade. Alternatively, the surprising difference between α-toxin and β-escin in response to ROCK inhibitors could reflect loss of RhoGAPs from β-escin treated muscles.
In summary, data from the present study combined with recently published studies (Ratz and Miner 2009; Ratz, Miner et al. 2009; Alvarez, Miner et al. 2010) are consistent with a model in which constitutively active ROCK maintains MLCP activity at a basally low level, and the low basal MLCP activity is responsible for the basal level of MLC-p that is measured in smooth muscle tissues (Rembold, Wardle et al. 2004). Any stimulus that leads to an increase in [Ca2+]i would stimulate contraction by increasing the activity of MLCK, leading to elevations in the degree of MLC-p above the basal level. Inhibition of the constitutive activity of ROCK would lead to activation of MLCP and a reduction in the basal level of MLC-p. Any contractile stimulus added to a muscle in which basal MLCP activity is elevated because of inhibition of constitutive ROCK activity should induce a weaker contraction than in tissues in which basal MLCP activity is not elevated. This would be true even if the stimulus acts only to elevate [Ca2+]i and does not itself activate ROCK. In other words, the ability of ROCK inhibitors to attenuate KCl and Ca2+-induced smooth muscle contractions may reflect an inhibitory action of these agents on constitutive rather than, or in addtition to, stimulated ROCK activity. This model does not exclude the possibility that membrane depolarization and Ca2+ may lead to additional activation of key signaling systems responsible for enhancing the degree of MLC-p.
Acknowledgments
This work was supported by a grant from the National Institutes of Health R01-HL61320.
References
- Alvarez SM, Miner AS, Browne BM, Ratz PH. Failure of Bay K 8644 to induce RhoA kinase-dependent calcium sensitization in rabbit blood vessels. Br J Pharmacol. 2010;160(6):1326–1337. doi: 10.1111/j.1476-5381.2010.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki S, Ito M, Kureishi Y, Feng J, Machida H, Isaka N, Amano M, Kaibuchi K, Hartshorne DJ, Nakano T. Arachidonic acid-induced Ca2+ sensitization of smooth muscle contraction through activation of Rho-kinase. Pflugers Arch. 2001;441(5):596–603. doi: 10.1007/s004240000462. [DOI] [PubMed] [Google Scholar]
- Arner A. Mechanical characteristics of chemically skinned guinea-pig taenia coli. Pflugers Arch. 1982;395(4):277–284. doi: 10.1007/BF00580790. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangham AD, Horne RW, Glauert AM, Dingle JT, Lucy JA. Action of saponin on biological cell membranes. Nature. 1962;196:952–955. doi: 10.1038/196952a0. [DOI] [PubMed] [Google Scholar]
- Brozovich FV. Rho signaling: agonist stimulation and depolarization come together. Circ Res. 2003;93(6):481–483. doi: 10.1161/01.RES.0000093183.00556.D5. [DOI] [PubMed] [Google Scholar]
- Cassidy P, Hoar PE, Kerrick WG. Irreversible thiophosphorylation and activation of tension in functionally skinned rabbit ileum strips by [35S]ATP gamma S. J Biol Chem. 1979;254(21):11148–11153. [PubMed] [Google Scholar]
- Casteels R, Kitamura K, Kuriyama H, Suzuki H. Excitation-contraction coupling in the smooth muscle cells of the rabbit main pulmonary artery. J Physiol. 1977;271(1):63–79. doi: 10.1113/jphysiol.1977.sp011990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Kitazawa T, Yazawa M, Mukai H, Ono Y, Brautigan DL. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C alpha and delta isoforms. J Biol Chem. 2001;276(31):29072–29078. doi: 10.1074/jbc.M103206200. [DOI] [PubMed] [Google Scholar]
- Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, Nakano T. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Itoh T, Kubota Y, Kuriyama H. Effects of guanosine nucleotides on skinned smooth muscle tissue of the rabbit mesenteric artery. J Physiol. 1989;408:535–547. doi: 10.1113/jphysiol.1989.sp017474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussle R, Bhakdi S, Sziegoleit A, Tranum-Jensen J, Kranz T, Wellensiek HJ. On the mechanism of membrane damage by Staphylococcus aureus alpha-toxin. J Cell Biol. 1981;91(1):83–94. doi: 10.1083/jcb.91.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. J Physiol. 1997;500(Pt 1):95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WQ, Peng YJ, Zhang WC, Lv N, Tang J, Chen C, Zhang CH, Gao S, Chen HQ, Zhi G, Feil R, Kamm KE, Stull JT, Gao X, Zhu MS. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology. 2008;135(2):610–620. doi: 10.1053/j.gastro.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Kuriyama H, Suzuki H. Differences and similarities in the noradrenaline- and caffeine-induced mechanical responses in the rabbit mesenteric artery. J Physiol. 1983;337:609–629. doi: 10.1113/jphysiol.1983.sp014645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LJ, Tazzeo T, Zuo J, Pertens E, Keshavjee S. KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L852–858. doi: 10.1152/ajplung.00130.2004. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ J Biol Chem. 1989;264(10):5339–5342. [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc Natl Acad Sci U S A. 1991;88(20):9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Takizawa N, Ikebe M, Eto M. Reconstitution of protein kinase C-induced contractile Ca2+ sensitization in triton X-100-demembranated rabbit arterial smooth muscle. J Physiol 520 Pt. 1999;1:139–152. doi: 10.1111/j.1469-7793.1999.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Kitazawa T, Somlyo AV, Somlyo AP. Cytosolic heparin inhibits muscarinic and alpha-adrenergic Ca2+ release in smooth muscle. Physiological role of inositol 1,4,5-trisphosphate in pharmacomechanical coupling. J Biol Chem. 1989;264(30):17997–18004. [PubMed] [Google Scholar]
- Launikonis BS, Stephenson DG. Effects of beta-escin and saponin on the transverse-tubular system and sarcoplasmic reticulum membranes of rat and toad skeletal muscle. Pflugers Arch. 1999;437(6):955–965. doi: 10.1007/s004240050867. 94370955.424 [pii] [DOI] [PubMed] [Google Scholar]
- Liao JK, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol. 2007;50(1):17–24. doi: 10.1097/FJC.0b013e318070d1bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind I, Ahnert-Hilger G, Fuchs G, Gratzl M. Purification of alpha-toxin from Staphylococcus aureus and application to cell permeabilization. Anal Biochem. 1987;164(1):84–89. doi: 10.1016/0003-2697(87)90371-x. 0003-2697(87)90371-X [pii] [DOI] [PubMed] [Google Scholar]
- Mackay D. An analysis of functional antagonism and synergism. Br J Pharmacol. 1981;73(1):127–134. doi: 10.1111/j.1476-5381.1981.tb16781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension. 2001;38(6):1307–1310. doi: 10.1161/hy1201.096541. [DOI] [PubMed] [Google Scholar]
- Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation. 2002;105(13):1545–1547. doi: 10.1161/hc1002.105938. [DOI] [PubMed] [Google Scholar]
- Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca(2+)-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. J Gen Physiol. 1994;104(2):265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita M, Yanagihara H, Hishinuma S, Saito M, Walsh MP. Membrane depolarization-induced contraction of rat caudal arterial smooth muscle involves Rho-associated kinase. Biochem J. 2002;364(Pt 2):431–440. doi: 10.1042/BJ20020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S, Kakia S, Takahashi I, Kawahara K, Tsukuda E, Sano T, Yamada K, Yoshida M, Kase H, Matsuda Y, Hashimoto Y, Nonomura Y. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem. 1992;267(4):2157–2163. [PubMed] [Google Scholar]
- Navedo MF, Amberg GC, Nieves M, Molkentin JD, Santana LF. Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle. J Gen Physiol. 2006;127(6):611–622. doi: 10.1085/jgp.200609519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura J, Kolber M, van Breeman C. Norepinephrine and GTP-γ-S increase myofilament Ca2+ sensitivity in α-toxin permeabilized arterial muscle. Biochem Biophys Res Commun. 1988;157:677–683. doi: 10.1016/s0006-291x(88)80303-6. [DOI] [PubMed] [Google Scholar]
- Poley RN, Dosier CR, Speich JE, Miner AS, Ratz PH. Stimulated calcium entry and constitutive RhoA kinase activity cause stretch-induced detrusor contraction. Eur J Pharmacol. 2008;599:137–145. doi: 10.1016/j.ejphar.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter M, Evans MC, Miner AS, Berg KM, Ward KR, Ratz PH. Convergence of Calcium Desensitizing Mechanisms Activated by Forskolin and Phenylephrine Pretreatment, But Not 8-bromo-cGMP. Am J Physiol Cell Physiol. 2006;290(6):C1552–1559. doi: 10.1152/ajpcell.00534.2005. [DOI] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 2009;24:342–356. doi: 10.1152/physiol.00023.2009. [DOI] [PubMed] [Google Scholar]
- Ratz PH, Berg KM, Urban NH, Miner AS. Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol. 2005;288(4):C769–783. doi: 10.1152/ajpcell.00529.2004. [DOI] [PubMed] [Google Scholar]
- Ratz PH, Miner AS. Length-dependent regulation of basal myosin phosphorylation and force in detrusor smooth muscle. Am J Physiol Regul Integr Comp Physiol. 2003;284(4):R1063–R1070. doi: 10.1152/ajpregu.00596.2002. [DOI] [PubMed] [Google Scholar]
- Ratz PH, Miner AS. Role of protein kinase Czeta and calcium entry in KCl-induced vascular smooth muscle calcium sensitization and feedback control of cellular calcium levels. J Pharmacol Exp Ther. 2009;328(2):399–408. doi: 10.1124/jpet.108.142422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz PH, Miner AS, Barbour SE. Calcium-independent phospholipase A2 participates in KCl-induced calcium sensitization of vascular smooth muscle. Cell Calcium. 2009;46(1):65–72. doi: 10.1016/j.ceca.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Wardle RL, Wingard CJ, Batts TW, Etter EF, Murphy RA. Cooperative attachment of cross bridges predicts regulation of smooth muscle force by myosin phosphorylation. Am J Physiol Cell Physiol. 2004;287(3):C594–602. doi: 10.1152/ajpcell.00082.2004. [DOI] [PubMed] [Google Scholar]
- Ruegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10(6):218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Hori M, Izumi M, Oka T, Kohama K, Ozaki H, Karaki H. Inhibition of high K+-induced contraction by the ROCKs inhibitor Y-27632 in vascular smooth muscle: possible involvement of ROCKs in a signal transduction pathway. J Pharmacol Sci. 2003;92(1):56–69. doi: 10.1254/jphs.92.56. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93(6):548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- Sato K, Ozaki H, Karaki H. Changes in cytosolic calcium level in vascular smooth muscle strip measured simultaneously with contraction using fluorescent calcium indicator fura 2. J Pharmacol Exp Ther. 1988;246(1):294–300. [PubMed] [Google Scholar]
- Schubert R. Multiple ligand-ion solutions: a guide for solution preparation and computer program understanding. J Vasc Res. 1996;33(1):86–98. doi: 10.1159/000159136. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Himpens B. Cell calcium and its regulation in smooth muscle. Faseb J. 1989;3(11):2266–2276. doi: 10.1096/fasebj.3.11.2506092. [DOI] [PubMed] [Google Scholar]
- Somlyo AV, Somlyo AP. Electromechanical and pharmacomechanical coupling in vascular smooth muscle. J Pharmacol Exp Ther. 1968;159(1):129–145. [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266(24):15771–15781. [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol. 2003;285(6):C1377–1385. doi: 10.1152/ajpcell.00501.2002. [DOI] [PubMed] [Google Scholar]
- Van Heijst BG, Blange T, Jongsma HJ, De Beer EL. The length dependency of calcium activated contractions in the femoral artery smooth muscle studied with different methods of skinning. J Muscle Res Cell Motil. 2000;21(1):59–66. doi: 10.1023/a:1005609319445. [DOI] [PubMed] [Google Scholar]
- Velasco G, Armstrong C, Morrice N, Frame S, Cohen P. Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr850 induces its dissociation from myosin. FEBS Letters. 2002;527(1–3):101–104. doi: 10.1016/s0014-5793(02)03175-7. [DOI] [PubMed] [Google Scholar]
- Wang T, Kendig DM, Smolock EM, Moreland RS. Carbachol-induced rabbit bladder smooth muscle contraction: roles of protein kinase C and Rho kinase. Am J Physiol Renal Physiol. 2009;297(6):F1534–1542. doi: 10.1152/ajprenal.00095.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yoshioka K, Azam MA, Takuwa N, Sakurada S, Kayaba Y, Sugimoto N, Inoki I, Kimura T, Kuwaki T, Takuwa Y. Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem J. 2006;394(Pt 3):581–592. doi: 10.1042/BJ20051471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber LP, Seto M, Sasaki Y, Sward K, Walsh MP. The involvement of protein kinase C in myosin phosphorylation and force development in rat tail arterial smooth muscle. Biochem J 352 Pt. 2000;2:573–582. [PMC free article] [PubMed] [Google Scholar]
- Wilson DP, Sutherland C, Borman MA, Deng JT, Macdonald JA, Walsh MP. Integrin-linked kinase is responsible for Ca 2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem J. 2005 doi: 10.1042/BJ20051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T, Okada Y. KCl depolarization increases Ca2+ sensitivity of contractile elements in coronary arterial smooth muscle. Am J Physiol. 1994;267(2 Pt 2):H614–H621. doi: 10.1152/ajpheart.1994.267.2.H614. [DOI] [PubMed] [Google Scholar]
- Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol. 2009;29(10):1657–1663. doi: 10.1161/ATVBAHA.109.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Sugimoto N, Takuwa N, Takuwa Y. Essential role for class II phosphoinositide 3-kinase alpha-isoform in Ca2+-induced, Rho- and Rho kinase-dependent regulation of myosin phosphatase and contraction in isolated vascular smooth muscle cells. Mol Pharmacol. 2007;71(3):912–920. doi: 10.1124/mol.106.032599. [DOI] [PubMed] [Google Scholar]