

Abstract
We investigate the conformational dynamics and mechanical properties of guanylate kinase (GK) using a multiscale approach combining high-resolution atomistic molecular dynamics and low-resolution Brownian dynamics simulations. The GK enzyme is subject to large conformational changes, leading from an open to a closed form, which are further influenced by the presence of nucleotides. As suggested by recent work on simple coarse-grained models of apo-GK, we primarily focus on GK's closure mechanism with the aim to establish a detailed picture of the hierarchy and chronology of structural events essential for the enzymatic reaction. We have investigated open-versus-closed, apo-versus-holo, and substrate-versus-product-loaded forms of the GK enzyme. Bound ligands significantly modulate the mechanical and dynamical properties of GK and rigidity profiles of open and closed states hint at functionally important differences. Our data emphasizes the role of magnesium, highlights a water channel permitting active site hydration, and reveals a structural lock that stabilizes the closed form of the enzyme.
Introduction
Guanylate kinase (GK), also called GMP kinase, is an essential enzyme for the equilibration of nucleotide ratios in the cell. GK catalyzes the reversible transfer of the terminal phosphoryl group from an ATP to a guanosine 5′ monophosphate (GMP) substrate, subsequently releasing guanosine diphosphate (GDP) as product (1,2). This is a crucial intermediate step in RNA or DNA synthesis foregoing the formation of the key nucleotide guanosine triphosphate (GTP) or its deoxy form dGTP (3). Furthermore, the GK enzyme is of pharmacological importance, as it activates pathways of guanosine analog prodrugs such as mercaptopurine and thigunine used against cancer, or of the drug carbovir used against HIV (4–7). GK enzymes share 40% sequence homology with membrane-associated guanylate kinases (8), which represent a large group of scaffolding proteins. Recently, it has been shown that the GK domain of membrane-associated guanylate kinases plays a crucial role in controlling gating of calcium channels (9). Hence, GK can be considered as a model system for exploring allosteric transmission between GK and SH3 domains (8,10–13). These properties render GK specific with respect to the related and much studied adenylate kinase (AK).
GMP kinase comprises three structural domains organized within a central five-stranded parallel β-sheet flanked by α-helices (Fig. 1). The innermost CORE domain is enclosed on its two opposite sides by the LID and GMP-binding (GMP-BD) domains (14). The highly conserved Walker A motif (or P-loop) is part of the CORE region and forms the ATP binding pocket interacting with the β-phosphate. Several invariant residues on the LID and GMP-BD domains participate in specific recognition of ATP and GMP substrates (15,16).
Figure 1.
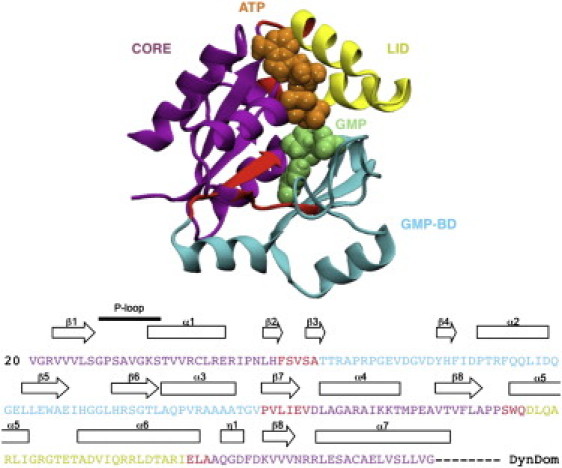
(Top) The three structural domains of GKMT as defined by the DynDom server (http://fizz.cmp.uea.ac.uk/dyndom/dyndomMain.do) are reported on a cartoon representation with hinge regions highlighted and each domain in a distinct shading: CORE, GMP-BD, and LID. (Bottom) The domain definition and secondary structure elements for GKMT are shown on the primary sequence. Fig. S3 provides an extensively annotated version of this figure.
GK's structure resembles a U-shape and is subject to large conformational changes during an enzymatic reaction, ranging from an open to a closed form, as suggested by the conformations captured in available crystal structures. In the closed form, the GMP-BD and LID domains enter into close contact (17). The allosteric spring probe (ASP) experiment on Mycobacterium tuberculosis (MT) guanylate kinase introduces an external mechanical perturbation interfering with the closure mechanism and leads to an enzyme activity loss (18,19). Using a reduced protein representation combined with Brownian dynamics simulations, we have recently shown that GK's anisotropic response to such mechanical stress is controlled by its main functional mode of motion and its mechanical properties (20). Such physics-based insight is very helpful to understand the link between enzymatic function and ductile properties built into the three-dimensional architecture of the protein.
Although molecular modeling of GK itself has only received little attention, several theoretical studies were carried out on the related AK enzyme (21–34). The dominant functional motion of both GK and AK is the opening and closing of their U-shaped structures.
In this study, we particularly focus on GK's closure mechanism with the aim to establish a detailed picture of the hierarchy and chronology of events that may be essential for the enzymatic reaction, using unbiased simulations without any assumptions about paths, transitions, and reaction coordinates, based on experimentally documented conformations. We compiled data from various GK structures among different organisms in order to build a range of functionally relevant coherent models for the M. tuberculosis GKMT form. We carry out a detailed exploration of the possible conformational states along GK's enzymatic reaction mechanism in order to identify key properties and dynamic effects. We characterize the open and closed, as well as substrate/product-loaded apo and holo forms of the MT GK system, probing the stability of these states and mapping their intrinsic mechanical properties. Such a comprehensive and consistent description for a single enzyme variant is largely complementary to previous experimental studies producing numerous valuable but incomplete data sets (15–17,35–37).
We point out possible origins for experimentally observed parameter variations related to ATP binding, GMP binding, and the overall catalytic rate under mechanical strain using the ASP technique. We further uncover a structural lock that stabilizes the closed form of the enzyme, hinting at residue pairs with major contributions to GK's mechanical properties. The study particularly highlights the crucial role of bound ligands in modulating the mechanical and dynamical properties of GK and emphasizes the role of magnesium. In this context, a water channel leading to the active site plays an important role.
Materials and Methods
System preparation
Fig. 2 illustrates the states along the enzymatic reaction pathway that were modeled and subsequently simulated using molecular dynamics (MD) as detailed in Table S1 in the Supporting Material.
Figure 2.
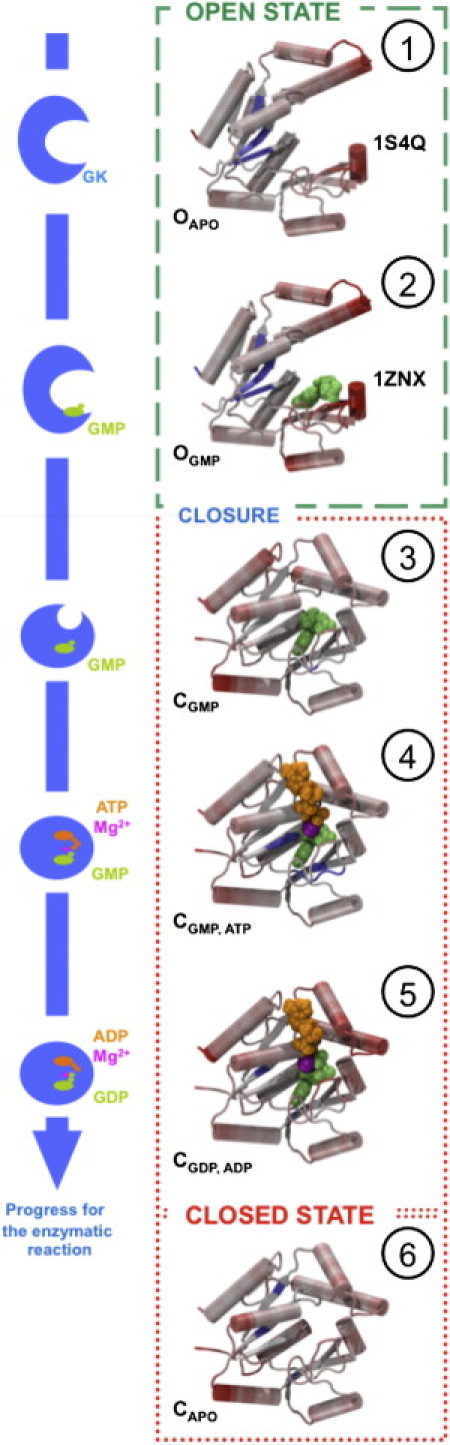
(Left side) Schematic representation of open (O, states 1 and 2) and closed (C, states 3–5) state structures describing GK's enzymatic reaction pathway. (Right side) Average RMS fluctuation measurements from 20-ns MD trajectories reported for each state (shading highlights flexible and rigid parts). GMP/GDP and ATP/ADP nucleotides as well as magnesium are shown both schematically and as van der Waals spheres. State 6 describes a putative stable form of the closed apo (CAPO) GK and is considered off the proposed enzymatic pathway.
Initial structures and homology modeling
Simulation of the OAPO and OGMP forms were, respectively, based on the experimental structures of apo GK and GMP⋅GK crystallized in their open conformations (Protein Data Bank (PDB) Nos. 1S4Q and 1ZNX) for M. tuberculosis. A single homology model was built for the closed state, based on an initial FASTA sequence alignment (37–43% sequence identity) inspired by the work of Hible et al. (15,16). Fig. 3 provides the sequence alignment used for modeling the closed form, highlighting the conservation of the active site. The model was generated with the ESyPred3d homology modeling server (based on the Modeler program by Sali and Blundell, as discussed in Lambert et al. (38)). The closed Mus musculus GK holo structure available in the Protein Data Bank (39) with PDB No. 1LVG (35) served as template for modeling the closed state of M. tuberculosis (see the Supporting Material for construction details and quality evaluation). In the loaded forms of the enzyme, the position and orientation of bound nucleotides were assigned by analogy to this structure as described in the following paragraph on nucleotide positioning. The apo model of the closed state, CAPO, was built from the GMP-bound closed form (CGMP) by removal of the substrate.
Figure 3.
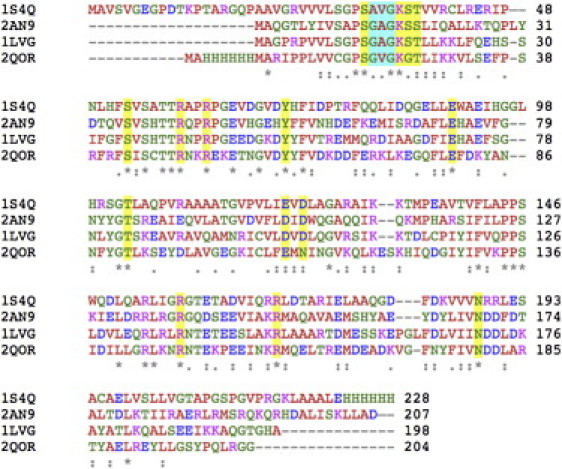
Primary sequence alignment of the experimental GK structures from different organisms used for the construction of the closed homology model: 1S4Q for M. tuberculosis, 2AN9 from E. coli, 1LVG from M. musculus, and 2QOR from P. vivax. Conservation of the catalytic site is highlighted for residues bound to substrates via their side chain (and eventually backbone) and for residues bound to substrates only via their backbone. Amino-acid letters are shaded depending on residue type.
Nucleotide positioning
The nucleotides were placed in the closed state GKMT models by least-squares fitting to the 1LVG GK structure of M. musculus comprising GMP and ADP substrates. The binding pockets are highly conserved (Fig. 3, and see Fig. S1 and Fig. S2 in the Supporting Material) and provide a very constrained accessible volume. Hence, nucleotide placement is straightforward. The ATP substrate position was obtained from ADP by adding an extra phosphate group taken from the Plasmodium vivax GK structure with PDB-id 2QOR to mimic Pγ. A single Mg2+ ion has been initially placed at the K+ position observed in the semiclosed GMP/GDP-bound structure from Escherichia coli GK (PDB-id 2AN9), i.e., close to the N7 atom of GMP. A spontaneous 6.5 Å displacement of the cation toward the nucleotide phosphate groups occurred during the equilibration run. The location of this magnesium ion binding site was qualitatively confirmed by an independent assessment using our MyPal software (40).
Simulations
All six GK models were simulated for 20 ns by molecular dynamics in explicit solvent. Low-resolution Brownian dynamics using the ProPhet program (41) were run on open and closed states. Full methods on molecular dynamics and Brownian dynamics simulation setups and analyses are described in the Supporting Material.
Results
Identification of architectural domains and structural hinges based on flexibility
Previously established GK domain definitions will serve as a basis to describe the structure of guanylate kinase throughout this work (15–17,35). A mobility-based domain characterization of GK is complementary to common structure-based descriptions. We used the DynDom (42) program to perform such an analysis. The DynDom domain definition shown in Fig. 1 highlights potential structural hinges. The LID domain is well established as a continuous stretch of residues between Asp147 and Ala175, involving the α5 and α6 helices. The GMP binding domain (GMP-BD) comprises residues Val52 to Leu117, whereas the remainder composes the CORE domain. Assignment of the α3 helix is ambiguous, as it can be part of the GMP-BD domain or of the CORE domain, supporting the idea that it forms a structural hinge ((16,17) and see Fig. S3). The residues linking sheets β2 and β3 are clearly identified as hinges.
Structural models of Mycobacterium tuberculosis guanylate kinase
We studied two open OAPO and OGMP states and four closed CGMP, CGMP, ATP, CGDP, ADP, and CAPO states along GK's enzymatic pathway (Fig. 2). Open states are based on crystal structures, whereas the closed states are based on a single homology model described in the subsequent paragraph. Each state was equilibrated using MD simulations (Table S1) and the structural drift was quantified using root mean-square deviations (RMSDs; Table S2). The RMSD is below 1.8 Å in between the two open or the four closed state structures, respectively, but is above 5 Å between open and closed states. The average residue flexibility, measured as RMS fluctuations, is reported on the structures in the right part of Fig. 2. The radii of gyration of the structures describing each state of the enzymatic pathway (Fig. S4) indicate that
-
1.
the OAPO state presents a more extended conformation than the OGMP state;
-
2.
the CGMP and CGMP, ATP states are well stabilized in their closed conformations; and
-
3.
the CGDP, ADP state is subject to structural fluctuations toward a partial opening of the enzyme.
Closed form of the enzyme
The CAPO model was derived from a M. musculus template with 43% sequence identity and highly conserved catalytic site residues (see Fig. 3 for definition and alignment). The quality of the homology model was assessed using a previously established evaluation protocol (43) and was found to be excellent, as described in detail in Fig. S1. The physico-chemical surface properties of the exposed ATP binding site are also well conserved (see Fig. S2). In addition, the model can be validated by comparison to closed structures from two other organisms, E. coli (PDB No. 2ANB) or P. vivax (PDB No. 2QOR), exhibiting a Cα RMSD of 2.9 and 1.3 Å, respectively. Essential contacts are very well conserved in the experimental structures and our model. For example, closure of the enzyme generates an interface between the GMP-BD and LID domains.
Contacts in this interface involve the β5-loop-β6/α6 and α5-loop-α6/β3-loop-β4 elements, with crucial residues such as His93 (GMP-BD) or Arg165-Arg166. Hydrophobic side-chain interactions form between the β5 sheet and the α4 helix defining an important interface between the CORE and GMP-BD domains. The CORE/LID domain interface is topologically identical to both experimental structures, with the P-loop being held in place by both the α5 and α6 helices. Compared to the closed structures of the aforementioned E. coli and P. vivax organisms, our closed state model features a stronger contact between the α4 and α6 helices, thus stabilizing the η1 turn. This interaction can be quantified by a 25% decrease of solvent-accessible surface area for the corresponding domains compared to the open state, representing a global measure of the increased contact between structural elements.
An interesting feature of our closed GKMT model is the presence of a water channel (Fig. 4 and see Fig. S5) (44) constituted by a triple interface between GMP-BD (β5-loop-β6), CORE (α4) and LID (α6) domains. The water channel is located on the back side of the enzyme, with respect to the substrate-binding pockets on the front. The channel can be further characterized by radial distribution functions (RDFs), as described in the next paragraph.
Figure 4.
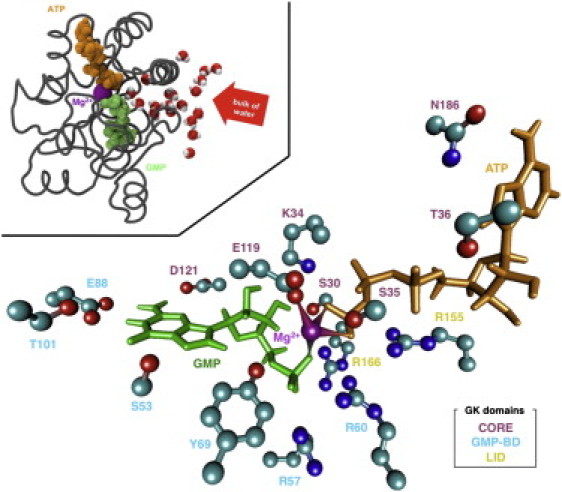
Main active site residue contacts with the substrates or cofactors during the GK OGMP, ATP simulation (see Tables 1–3). Hydrogen atoms are hidden for clarity. Catalytic residue annotations are shaded according to their domain (CORE, GMP-BD, and LID). GMP, ATP, and magnesium are represented as in Fig. 2. The HyperBalls representation was used to highlight magnesium coordination (d ≤ 2.9 Å) (44). (Top left) A channel is shown that could enable water access to the binding site in order to complete the catalytic reaction.
Substrate-bound enzyme
Tables 1 and 2 summarize the interactions between the enzyme and the nucleotides. Key interaction patterns for GMP-binding are described in Fig. 4 and Fig. S6, A and B. The Ser53, Glu88, Thr101, and Asp121 residues form specific contacts with GMP's guanine base. Residue Lys34 binds to the GMP sugar moiety. This nonspecific electrostatic interaction is also effective toward other nucleotides. The magnesium cation spontaneously formed a bridge between Glu119 and the GMP(Pα) and ATP(Pγ) phosphate groups. Water molecules completed the magnesium coordination sphere, forming a mostly regular hexacoordinated geometry around the ion (Table 3 and see Fig. S7). ATP-specific binding involves Asn186 via direct interactions with the nucleobase.
Table 1.
Summary of major direct protein-GMP/GDP substrate interactions
| OGMP |
CGMP |
CGMP, ATP |
CGDP,ADP |
|||
|---|---|---|---|---|---|---|
| Protein residue | Domain | GMP/GDP | (GMP binding) | (Closure) | (ATP binding) | (P transfer) |
| Strong interactions | ||||||
| Glu88 (OE1,2) | GMP-BD | GMP/GDP (N1,2) | >100% | >100% | >100% | >100% |
| Arg57 (NE, NH1) | GMP-BD | GMP/GDP (Pα,β) | >100% | 46% | >100% | >100% |
| Ser53 (OG) | GMP-BD | GMP/GDP (O6, N7) | 87% | 66% | 95% | 96% |
| Tyr69 (OH) | GMP-BD | GMP/GDP (Pα) | 97% | >100% | >100% | 48% |
| Arg166 (NH1,2) | LID | GMP/GDP (Pα) | — | >100% | >100% | — |
| LID | GDP (Pβ) | >100% | ||||
| Arg155 (NH1,2) | LID | GDP (Pβ) | >100% | |||
| Ser30 (OG) | CORE | GDP (Pβ) | 100% | |||
| Weaker interactions | ||||||
| Arg60 (NH1,2) | GMP-BD | GMP/GDP (Pα) | 60% | >100% | 28% | >100% |
| Thr101 (OG1) | GMP-BD | GMP/GDP (O6) | <5% | 71% | <5% | <5% |
| Asp121 (OD2) | CORE | GMP/GDP (N2) | — | 57% | 15% | 40% |
| Lys34 (NZ) | CORE | GMP/GDP (O3′,Pα) | — | 53% | 43% | — |
| CORE | GDP (Pβ) | 86% | ||||
Hydrogen-bonding and salt-bridge contact frequency in the catalytic site during the MD simulations of enzymatic substates 2–5 (see Fig. 2). Contacts are sorted from the strongest to the weakest interaction. Atoms or groups involved in the interaction are indicated in parentheses. Frequencies over 70% are shown in boldface. Percentages over 100% take into account multiple interactions from different groups of the same residue (such as bidentate contacts).
Table 2.
Summary of major direct protein-ATP/ADP substrate interactions
| CGMP, ATP |
CGDP,ADP |
|||
|---|---|---|---|---|
| Protein residue | Domain | ATP/ADP | (ATP binding) | (P transfer) |
| Strong interactions | ||||
| Ala31 (N) | CORE | ATP/ADP (Pβ) | >100% | >100% |
| Lys34 (N, NZ) | CORE | ATP/ADP (Pβ,γ) | >100% | >100% |
| Ser35 (N, OG) | CORE | ATP/ADP (Pβ) | >100% | >100% |
| Thr36 (N, OG1) | CORE | ATP/ADP (Pα) | >100% | >100% |
| Arg155 (NH1,2) | LID | ATP/ADP (Pα,β,γ) | >100% | 97% |
| Gly33 (N) | CORE | ATP/ADP (Pα) | >100% | — |
| CORE | ATP/ADP (Pβ) | — | >100% | |
| Ser30 (OG) | CORE | ATP (Pγ) | >100% | |
| Arg60 (NH2) | GMP-BD | ATP (Pγ) | 97% | |
| Weaker interactions | ||||
| Val32 (N) | CORE | ATP/ADP (Pβ) | 44% | 56% |
| Asn186 (OD1, ND2) | CORE | ATP/ADP (N6, N7) | 42% | 64% |
| Arg166 (NH1) | LID | ATP (Pγ) | 44% | |
Hydrogen-bonding and salt-bridge contact frequency in the catalytic site during the MD simulations of enzymatic substates 4 and 5 (see Fig. 2). Contacts are sorted from the strongest to the weakest interactions. Atoms or groups involved in the interaction are indicated in parentheses. Frequencies over 70% are shown in boldface. Percentages over 100% take into account multiple interactions from different groups of the same residue (such as bidentate contacts).
Table 3.
Coordination numbers for the magnesium ion in the CGMP, ATP and CGDP, ADP substates derived from radial distribution functions (see Fig. S5)
| Oxygen carrier | C(GMP, ATP) | C(GDP, ADP) |
|---|---|---|
| GMP/GDP | 1.5 | 0.5 |
| ATP/ADP | 2 | 1 |
| Protein | 1 | 2 |
| Water | 1.5 | 2.5 |
Oxygens from GMP/GDP, ATP/ADP, protein, and water in the coordination sphere of Mg2+ were taken into account up to a cutoff radius of 2.9 Å.
The three phosphate groups of ATP form a dense network with CORE, LID, and GMP-BD domain residues. Water RDFs calculated for all MD trajectories clearly show that GMP/GDP is less water accessible than ATP/ADP, except in the open substate OGMP (Fig. 5). In the closed states, nucleotides in CGMP, ATP, a state before the enzymatic reaction, are more protected from solvent than in CGMP, and even more than in CGDP, ADP, a state that would occur just after phosphate transfer. By considering the deeply buried position of both GMP and the Mg2+ ion, given the compactness of the catalytic site and the presence of water below a distance of 15 Å, the mean gyration radius observed for the closed states (Fig. S4) is an additional indication of the persistence of the water channel in all closed state simulations.
Figure 5.
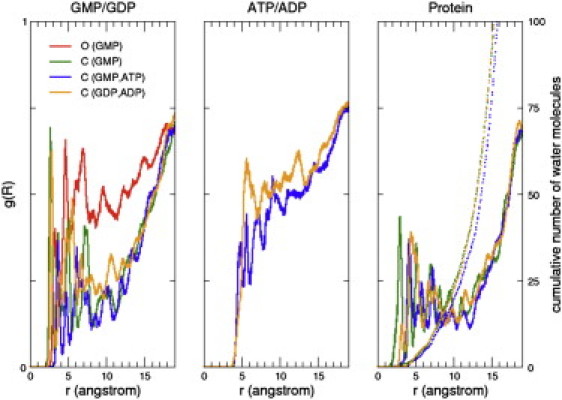
Radial distribution function g(R) of water oxygens around GMP/GDP (left panel), ATP/ADP (middle panel), and protein (right panel). OGMP, CGMP, CGMP, ATP, and CGDP, ADP simulation data is shown. The distance (in Å) shown on the x axis is measured between the center of mass of the molecule of interest (substrate, product, or protein) and water oxygen atoms. (Dotted lines on the right panel) Cumulative number of water molecules.
Interactions at the domain interfaces
The open apo enzyme mainly exhibits strong structuring of delimited individual domains. Upon GMP binding, part of the GMP-BD domain swings upward to stabilize its substrate and forms a new GMP-BD/LID interface with a surface area of ≈150 Å2. At the same time, the α6 helix of the LID domain moves and the CORE/LID contact surface shows a 20% increase. Interestingly, no significant change of the CORE/GMP-BD contact surface is observed despite a sliding motion of the α4 helix along the β5 sheet after GK closure (Fig. S8) and an internal reorganization within the core. Helix α7 participates in this reorganization. Upon docking of ATP:Mg2+, only the contacts in the β6-α3 region increase. An important loss of intraproteic contacts is observed for the product-bound closed structure.
Principal components analysis (see Fig. S9) clearly illustrates how modulations in the domain interfaces impact the intrinsic dynamics of GK's substates. For instance, the first principal modes of motion for OAPO and OGMP favor closure of the structure by modulating GMP-BD and LID domains close to the ATP binding platform (P-loop). In CGMP the opening/closing motion involves the hinge and GMP-BD regions; a light contribution of the vicinity of the P-loop should also be noticed. Except for its terminal regions, the CGMP, ATP state is very rigid, as one would expect for preorganizing the phosphate transfer. The CGDP, ADP state shows a strong tendency for opening, which may help the escape of the products of the reaction.
Mechanical properties and flexibility of enzymatic pathway intermediates
We investigated the mechanical properties for the open and closed states using coarse-grain Brownian dynamics. Simulations on the closed state of GKMT were carried out on the protein without substrates, because earlier studies (40,45) have shown that this choice enables the intrinsic mechanical properties of the enzyme to be studied independently of the nature and position of any bound ligand.
Rigidity profile of the apo enzyme in its open state
The rigidity profile of GKMT is represented in Fig. 6. Such force constants can actually be measured experimentally (see, e.g., work by Dietz et al. (46)) and are a direct physical property that is relevant for enzymatic function. Remarkably, most of the force constant peaks from the first-half of the protein sequence correspond to residues belonging to the ATP:Mg2+ and GMP binding sites. We note the rigidity peaks of Ser27 and Lys34, which surround a flexible P-loop, and the highly rigid area around Glu119. More generally both the GMP and the ATP:Mg2+ binding sites are quite rigid with respect to the rest of the protein. The average force constant of their constitutive residues (listed in Tables 1 and 2) is 36 kcal mol−1 Å−2, compared to 23 kcal mol−1 Å−2 for the complete molecule.
Figure 6.
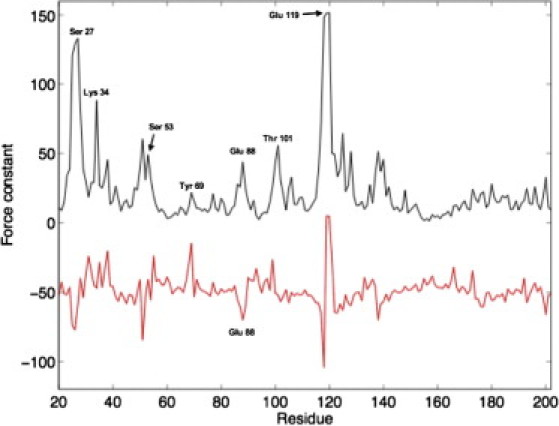
(Top line) Rigidity profile of GKMT in its open conformation (PDB code No. 1S4Q); the annotated residues belong either to the GMP or the ATP-binding site. (Bottom line) Variation of the residue force constants upon closure of the protein structure; the curve has been vertically shifted for visibility. All the force constants are in kcal.mol−1.Å−2.
Mechanical variations upon closure
It is genuinely difficult to predict force constant variations related to conformational changes, because structural rearrangements can result in both residue rigidity increases and decreases, as is also the case for GK. Simulations on the closed CAPO conformation (Fig. 6) show a nonmonotonous perturbation of the rigidity profile, yet on average the closed conformation is more rigid than the open one. This stiffening of the protein concerns the ATP:Mg2+ binding site, more affected than the GMP binding site, but globally all the residues from the ligand binding sites undergo an increase of their force constant upon closure, with the notable exception of Glu88 and Thr101. This agrees with the observation that closure leads to loss of interactions between these residues and the neighboring α4 helix at the atomistic level (Fig. S8). Overall, these measures indicate that closure of the enzyme profoundly alters the malleability of the structure, in particular of the ligand binding sites.
Mechanical resistance maps reveal a structural lock
Mechanical resistance maps provide further physics-based insight into such ductility changes and the evolution of mechanical properties induced by the closure of the structure. The histograms plotted in Fig. 7 A confirm that closure induces a shift of the force constant distribution toward larger values, with the disappearance of many residue pairs with low force constants (<1 N m−1). The evolution of the mechanical map shown in Fig. 7 B indicates that the increase of the directional force constant (DFC) values upon protein closure is restricted to a small number of residue pairs. By restricting this selection to residue pairs undergoing an increase of at least 600% of their DFC upon closure, four structural groups (Fig. 7 C) are detected. One group is on the P-loop, two are on the GMP-BD domain, and a fourth large group includes residues from the LID domain. These four groups come in close contact upon protein closure, which might explain the strong increase of the DFC. The residues form a dense set of interactions, resembling a structural lock that helps to maintain GK in its closed conformation.
Figure 7.
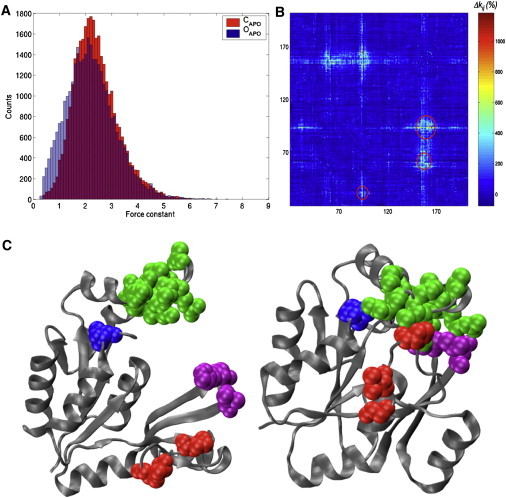
(A) Distribution of the directional force constants of GKMT over all possible pulling directions. Data is shown for the open conformation of GK (PDB code No. 1S4Q) without any ligand (OAPO), and the closed conformation of GKMT without any ligand (CAPO). (B) Relative variations of the DFCs upon closure of the GKMT protein structure. (Circles) Areas with important rigidity changes. (C) A cartoon representation of the open (left) and closed (right) conformations of GKMT with the residues forming rigid pairs upon closure plotted as van der Waals spheres: Val32 (leftmost); Pro61, Val64, and Gly66 (bottom); His93, Gly94, and Leu96 (rightmost); Leu152, Gly154, Arg155, Thr157, Ala 160, Ile163, Arg165, Arg166, and Thr169 (top).
Discussion
We present a structural study of guanylate kinase in a variety of conformational and nucleotide-bound forms along its enzymatic pathway, using physics-based descriptors to gain insight into functionally relevant properties. Whereas previous analyses were based on comparisons between homologous structures from different species (15–17,35–37), we provide insight into the role of specific interactions and mechanical variations relevant for the enzymatic cycle of a unique enzyme variant.
Specific binding of GMP, ATP, and Mg2+ to guanylate kinase
GMP- and ATP-binding patterns in our models are in excellent agreement with experimental structures from other organisms such as M. musculus (35) and E. coli (15). Mutagenesis experiments suggest a critical role of the Glu119 residue in transition state stabilization (17). The Glu119 residue has been initially proposed to bind the sugar ring of GMP (35), leading to specificity of GMP binding toward dGMP such as measured experimentally (47,48). Glu119 is part of a patch of three CORE domain residues interacting with GMP in the mouse and Saccharomyces cerevisiae crystals. This interaction is not seen in the MT crystal structure of the enzyme (16).
The all-atom MD simulations show that Glu119 is not directly involved in specific binding to GMP's sugar moiety. Instead, Lys34 and Ser30 interact specifically with the O3′ atom and may discriminate dGMP with respect to GMP. Selective binding to GMP's base may arise from the recognition of its amino group by Asp121 and Glu88, consistent with features of the experimental structures and similar to previous observations made on other GMP-specific systems (49).
Lys34 and Asp121 also participate in interdomain CORE/GMP-BD bridges. These interactions stabilize the closed form of the enzyme, which is further sustained by the presence of the magnesium ion. Mg2+ bridges both nucleotides and is strongly bound to Glu119 (Fig. 4 and Fig. S7). The Glu119 residue has previously been predicted to contribute to magnesium coordination (16) and it has been established that the presence of both substrates enhances magnesium binding (50). Furthermore, Glu119 belongs to the most rigid part of the protein as seen in the rigidity profile (Fig. 6), a common signature of residues belonging to enzymatic catalytic sites (45,51).
To our knowledge, our models are the first to provide such insights into the possible role of magnesium coordination. Our simulations of the MT form clearly show that Glu119 interacts with the nucleotides, albeit indirectly via the Mg2+ ion. This yields a coherent description for all GK variants.
Dynamic rearrangement of the substrates within the active site
Our simulations—in agreement with recent experimental data (48)—show that GMP binding induces important structural changes. Tables 1 and 2 summarize the variations observed in the specific interactions of enzyme and nucleotides. We observe a tight fit of the enzyme to its ligands in the closed GK structure, hence limiting the conformational freedom of the bound nucleotides. After completion of the enzymatic reaction, during which the Ser30 and Arg60 residues maintain their interactions with the phosphate groups, they bridge the GDP-Pα and -Pβ phosphates (Fig. S10). Furthermore, four other residues rearrange their interactions with the phosphates: Gly33, Lys34, Arg155, and Arg166. The coordination of the magnesium ion is also modified during the enzymatic reaction. GMP and ATP substrates coordinate more strongly in the CGMP, ATP model, yet lose some of their interactions in favor of the protein (Glu119 and Ser36) and of water molecules in the CGDP, ADP model (Table 3 and Fig. S6 B and Fig. S7). All these rearrangements taken together induce flexibility in the lock region, specifically in helices α5 and α6 (Figs. 2 and 7 C), favoring the reopening of the structure. The tendency for reopening is further confirmed by the principal components analysis (Fig. S9) and the observed RDF (Fig. 5) and g(R) (Fig. S4) variations of the CGDP, ADP state.
A structural lock secures the closed state
Analyzing physics-based mechanical resistance properties allowed us to establish a set of specific residues in the enzyme structure. Amino acids involved in the GMP-BD/LID interface contribute to this structural lock detected in the BD simulations (Fig. 7 C). Most of these residues are close to Arg60 (GMP-BD) and Glu158 (LID). This observation highlights the importance of electrostatic interactions emanating from these two residues in the closed GK state, stabilizing the domain interfaces in addition to prevalent interactions between the complementary hydrophobic surfaces of GMP-BD or LID domains. Interestingly, the positions of the structural lock residues on the enzyme's structure are very similar to the set of coevolved residues forming anticorrelated pairs that were detected by Armenta-Medina et al. (34) in adenylate kinase. This observation suggests that the important role of these amino acids for the protein's enzymatic activity is conserved throughout the whole functional family.
Further work on the role played by the residues involved in this structural lock of the closed GK form with respect to enzymatic activity could be pursued experimentally, by studying specific GKMT mutants such as Val64Ser, Leu96Ser, Ala160Ser, and Ile163Ser.
Independence and interdependence of GMP and ATP binding
Previous structural studies show that simultaneous binding of both GMP and ATP substrates yields a catalytically active closed GK conformation similar among all nucleotide monophosphate kinases (14). Binding of GMP alone induces the closure of the GMP domain (17,36) and subsequent cobinding of ATP results in the fully closed conformation (35).
On the whole, our data sustain the idea that GMP binding alone should be sufficient to totally close GK, unlike what was observed by Hible et al. (16). Conversely, when full enzyme closure is hindered by mechanical tension applied in ASP experiments, the GMP binding constant is lowered (18). Long-range electrostatics centered on the P-loop (Fig. S2) would then drive ATP:Mg2+ into its selective binding pocket, with more subtle structural effects on the enzyme. The network of interactions enabled by the presence of ATP:Mg2+ is, however, essential to stabilize and lock the closed form. The enzymatic reaction can only be achieved if the GMP and ATP phosphates are well ordered with the help of the magnesium ion coordination. The closed GK state is weakened again upon phosphate transfer, with numerous rearrangements concerning the bound nucleotides. Given these intricate structural links, it appears unlikely that ATP binding and GMP binding are independent as suggested in Choi and Zocchi (18).
The interplay with the actual enzymatic reaction cannot be studied directly at the level of the classical description used in this work. For this purpose, mixed QM/MM methods would be more appropriate, which is beyond the scope of this study.
Detection of a water channel leading toward the active site
The water channel (Figs. 4 and 5 and see Fig. S5) identified in the simulations of the closed form of the enzyme might be functionally relevant by enabling selected water molecules to access the enzymatic site (14), which could be crucial for the mechanism of such phosphotransferases (52). Kandeel and Kitade (48) have also recently pointed out the importance of structured water molecules at the active site of Plasmodium falciparum GK. The observation of a stable water channel establishes what we believe to be a new hypothesis for the enzymatic activity of guanylate kinase.
Conclusions
Using a multiresolution simulation approach we unveil important features of the GK enzyme. Brownian dynamics applied to an elastic network coarse-grain model highlights catalytic residues, main binding mode modulations, and domain mobilities. All-atom MD simulations provide a detailed description of the substrate binding patterns and differentiate conformational and ligand-related mechanical properties. The generated complexes representing important states of the GK system form an essential basis for further work at a mixed quantum/classical level, taking into account the enzymatic reaction itself.
Based on the classical simulations presented here, we propose a sequence of structural events leading to enzyme closure, highlighting the importance of specific residues toward GK's enzymatic activity. Asp121 contributes to GMP specific recognition and initiates a swinging motion of the GMP-BD closing the structure. Our data emphasizes the pivotal role of magnesium coordination in the enzymatic reaction and as an additional factor controlling the stabilization of the closed state. A water channel, localized at the junction of three domains, permits hydration of the active site—which we perceive as a new avenue for a possible block of the enzyme's activity.
Bridging residues at domain interfaces participate in concert with the substrates to the overall mechanical stability of the system. We detected a structural lock that stabilizes the closed form of the enzyme, hinting at residue pairs with major contributions to GK's mechanical properties. Furthermore, our study clearly highlights the crucial role of bound ligands in modulating the mechanical and dynamical properties of GK.
Acknowledgments
We thank Dr. Brigitte Hartmann for critical reading of the manuscript and stimulating discussions.
This interdisciplinary work was funded by the French Agency for Research (grants No. ANR-06-PCVI-0025 and No. ANR-2010-BIOE-003-04). We thank IDRIS (CNRS's National Supercomputer Center in Orsay) for allocating computing time (project No. 071714).
Footnotes
Olivier Delalande's present address is Université de Rennes 1, Faculté des Sciences Pharmaceutiques et Biologiques, UMR CNRS 6026, Equipe RMN-ILP, CS 34317, 35043 Rennes Cedex, France.
Supporting Material
References
- 1.Oeschger M.P. Guanylate kinase from Escherichia coli B. Methods Enzymol. 1978;51:473–482. doi: 10.1016/s0076-6879(78)51065-3. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal K.C., Miech R.P., Parks R.E., Jr. Guanylate kinases from human erythrocytes, hog brain, and rat liver. Methods Enzymol. 1978;51:483–490. doi: 10.1016/s0076-6879(78)51066-5. [DOI] [PubMed] [Google Scholar]
- 3.Konrad M. Cloning and expression of the essential gene for guanylate kinase from yeast. J. Biol. Chem. 1992;267:25652–25655. [PubMed] [Google Scholar]
- 4.Miller W.H., Miller R.L. Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase. J. Biol. Chem. 1980;255:7204–7207. [PubMed] [Google Scholar]
- 5.Boehme R.E. Phosphorylation of the antiviral precursor 9-(1,3-dihydroxy-2-propoxymethyl)guanine monophosphate by guanylate kinase isozymes. J. Biol. Chem. 1984;259:12346–12349. [PubMed] [Google Scholar]
- 6.Miller W.H., Daluge S.M., Miller R.L. Phosphorylation of carbovir enantiomers by cellular enzymes determines the stereoselectivity of antiviral activity. J. Biol. Chem. 1992;267:21220–21224. [PubMed] [Google Scholar]
- 7.De Clercq E., Naesens L., Neyts J. Antiviral agents active against human herpes viruses HHV-6, HHV-7 and HHV-8. Rev. Med. Virol. 2001;11:381–395. doi: 10.1002/rmv.336. [DOI] [PubMed] [Google Scholar]
- 8.Olsen O., Bredt D.S. Functional analysis of the nucleotide binding domain of membrane-associated guanylate kinases. J. Biol. Chem. 2003;278:6873–6878. doi: 10.1074/jbc.M210165200. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Gutierrez G., Miranda-Laferte E., Hidalgo P. The guanylate kinase domain of the β-subunit of voltage-gated calcium channels suffices to modulate gating. Proc. Natl. Acad. Sci. USA. 2008;105:14198–14203. doi: 10.1073/pnas.0806558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGee A.W., Dakoji S.R., Prehoda K.E. Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol. Cell. 2001;8:1291–1301. doi: 10.1016/s1097-2765(01)00411-7. [DOI] [PubMed] [Google Scholar]
- 11.Tavares G.A., Panepucci E.H., Brunger A.T. Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol. Cell. 2001;8:1313–1325. doi: 10.1016/s1097-2765(01)00416-6. [DOI] [PubMed] [Google Scholar]
- 12.Opatowsky Y., Chen C.-C., Hirsch J.A. Structural analysis of the voltage-dependent calcium channel β-subunit functional core and its complex with the α1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 13.Marcette J., Hood I.V., Prehoda K.E. Allosteric control of regulated scaffolding in membrane-associated guanylate kinases. Biochemistry. 2009;48:10014–10019. doi: 10.1021/bi901160f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan H., Tsai M.-D. Nucleoside monophosphate kinases: structure, mechanism, and substrate specificity. Adv. Enzymol. Relat. Areas Mol. Biol. 1999;73:103–134. doi: 10.1002/9780470123195.ch4. x. [DOI] [PubMed] [Google Scholar]
- 15.Hible G., Renault L., Cherfils J. Calorimetric and crystallographic analysis of the oligomeric structure of Escherichia coli GMP kinase. J. Mol. Biol. 2005;352:1044–1059. doi: 10.1016/j.jmb.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 16.Hible G., Christova P., Cherfils J. Unique GMP-binding site in Mycobacterium tuberculosis guanosine monophosphate kinase. Proteins. 2006;62:489–500. doi: 10.1002/prot.20662. [DOI] [PubMed] [Google Scholar]
- 17.Blaszczyk J., Li Y., Ji X. Crystal structure of unligated guanylate kinase from yeast reveals GMP-induced conformational changes. J. Mol. Biol. 2001;307:247–257. doi: 10.1006/jmbi.2000.4427. [DOI] [PubMed] [Google Scholar]
- 18.Choi B., Zocchi G. Guanylate kinase, induced fit, and the allosteric spring probe. Biophys. J. 2007;92:1651–1658. doi: 10.1529/biophysj.106.092866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tseng C.Y., Wang A., Zocchi G. Mechano-chemistry of the enzyme Guanylate Kinase. Eur. Phys. Lett. 2010;91:18005. [Google Scholar]
- 20.Sacquin-Mora S., Delalande O., Baaden M. Functional modes and residue flexibility control the anisotropic response of guanylate kinase to mechanical stress. Biophys. J. 2010;99:3412–3419. doi: 10.1016/j.bpj.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vonrhein C., Schlauderer G.J., Schulz G.E. Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases. Structure. 1995;3:483–490. doi: 10.1016/s0969-2126(01)00181-2. [DOI] [PubMed] [Google Scholar]
- 22.Miyashita O., Onuchic J.N., Wolynes P.G. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc. Natl. Acad. Sci. USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maragakis P., Karplus M. Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J. Mol. Biol. 2005;352:807–822. doi: 10.1016/j.jmb.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 24.Lou H.F., Cukier R.I. Molecular dynamics of apo-adenylate kinase: a distance replica exchange method for the free energy of conformational fluctuations. J. Phys. Chem. B. 2006;110:24121–24137. doi: 10.1021/jp064303c. [DOI] [PubMed] [Google Scholar]
- 25.Henzler-Wildman K.A., Lei M., Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 26.Henzler-Wildman K.A., Thai V., Kern D. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 27.Chu J.-W., Voth G.A. Coarse-grained free energy functions for studying protein conformational changes: a double-well network model. Biophys. J. 2007;93:3860–3871. doi: 10.1529/biophysj.107.112060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seeliger D., Haas J., de Groot B.L. Geometry-based sampling of conformational transitions in proteins. Structure. 2007;15:1482–1492. doi: 10.1016/j.str.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 29.Chennubhotla C., Bahar I. Signal propagation in proteins and relation to equilibrium fluctuations. PLOS Comput. Biol. 2007;3:1716–1726. doi: 10.1371/journal.pcbi.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arora K., Brooks C.L.I., 3rd Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cukier R.I. Apo adenylate kinase encodes its holo form: a principal component and varimax analysis. J. Phys. Chem. B. 2009;113:1662–1672. doi: 10.1021/jp8053795. [DOI] [PubMed] [Google Scholar]
- 32.Beckstein O., Denning E.J., Woolf T.B. Zipping and unzipping of adenylate kinase: atomistic insights into the ensemble of open↔closed transitions. J. Mol. Biol. 2009;394:160–176. doi: 10.1016/j.jmb.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brokaw J.B., Chu J.W. On the roles of substrate binding and hinge unfolding in conformational changes of adenylate kinase. Biophys. J. 2010;99:3420–3429. doi: 10.1016/j.bpj.2010.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armenta-Medina D., Pérez-Rueda E., Segovia L. Identification of functional motions in the adenylate kinase (ADK) protein family by computational hybrid approaches. Proteins. 2011;79:1662–1671. doi: 10.1002/prot.22995. [DOI] [PubMed] [Google Scholar]
- 35.Sekulic N., Shuvalova L., Lavie A. Structural characterization of the closed conformation of mouse guanylate kinase. J. Biol. Chem. 2002;277:30236–30243. doi: 10.1074/jbc.M204668200. [DOI] [PubMed] [Google Scholar]
- 36.Stehle T., Schulz G.E. Refined structure of the complex between guanylate kinase and its substrate GMP at 2.0 Å resolution. J. Mol. Biol. 1992;224:1127–1141. doi: 10.1016/0022-2836(92)90474-x. [DOI] [PubMed] [Google Scholar]
- 37.El Omari K., Dhaliwal B., Stammers D.K. Structure of Staphylococcus aureus guanylate monophosphate kinase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006;62:949–953. doi: 10.1107/S174430910603613X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambert C., Léonard N., Depiereux E. ESyPred3D: prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 39.Berman H.M., Battistuz T., Zardecki C. The Protein DataBank. Acta Crystallogr. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 40.Delalande O., Férey N., Baaden M. Multi-resolution approach for interactively locating functionally linked ion binding sites by steering small molecules into electrostatic potential maps using a haptic device. Pac. Symp. Biocomput. 2010;2010:205–215. doi: 10.1142/9789814295291_0023. [DOI] [PubMed] [Google Scholar]
- 41.Sacquin-Mora S., Lavery R. Investigating the local flexibility of functional residues in hemoproteins. Biophys. J. 2006;90:2706–2717. doi: 10.1529/biophysj.105.074997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee R.A., Razaz M., Hayward S. The DynDom database of protein domain motions. Bioinformatics. 2003;19:1290–1291. doi: 10.1093/bioinformatics/btg137. [DOI] [PubMed] [Google Scholar]
- 43.Law R.J., Capener C., Sansom M.S. Membrane protein structure quality in molecular dynamics simulation. J. Mol. Graph. Model. 2005;24:157–165. doi: 10.1016/j.jmgm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 44.Chavent M., Vanel A., Baaden M. GPU-accelerated atom and dynamic bond visualization using HyperBalls: a unified algorithm for balls, sticks, and hyperboloids. J. Comput. Chem. 2011;32:2924–2935. doi: 10.1002/jcc.21861. [DOI] [PubMed] [Google Scholar]
- 45.Sacquin-Mora S., Laforet E., Lavery R. Locating the active sites of enzymes using mechanical properties. Proteins. 2007;67:350–359. doi: 10.1002/prot.21353. [DOI] [PubMed] [Google Scholar]
- 46.Dietz H., Berkemeier F., Rief M. Anisotropic deformation response of single protein molecules. Proc. Natl. Acad. Sci. USA. 2006;103:12724–12728. doi: 10.1073/pnas.0602995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y., Zhang Y., Yan H. Kinetic and thermodynamic characterizations of yeast guanylate kinase. J. Biol. Chem. 1996;271:28038–28044. doi: 10.1074/jbc.271.45.28038. [DOI] [PubMed] [Google Scholar]
- 48.Kandeel M., Kitade Y. Binding dynamics and energetic insight into the molecular forces driving nucleotide binding by guanylate kinase. J. Mol. Recognit. 2011;24:322–332. doi: 10.1002/jmr.1074. [DOI] [PubMed] [Google Scholar]
- 49.Schrift G.L., Waldron T.T., Murphy K.P. Molecular basis for nucleotide-binding specificity: role of the exocyclic amino group “N2” in recognition by a guanylyl-ribonuclease. J. Mol. Biol. 2006;355:72–84. doi: 10.1016/j.jmb.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 50.Prinz H., Lavie A., Konrad M. Binding of nucleotides to guanylate kinase, p21(ras), and nucleoside-diphosphate kinase studied by nano-electrospray mass spectrometry. J. Biol. Chem. 1999;274:35337–35342. doi: 10.1074/jbc.274.50.35337. [DOI] [PubMed] [Google Scholar]
- 51.Lavery R., Sacquin-Mora S. Protein mechanics: a route from structure to function. J. Biosci. 2007;32:891–898. doi: 10.1007/s12038-007-0089-x. [DOI] [PubMed] [Google Scholar]
- 52.Sparks J.W., Brautigan D.L. Molecular basis for substrate specificity of protein kinases and phosphatases. Int. J. Biochem. 1986;18:497–504. doi: 10.1016/0020-711x(86)90159-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.