

Abstract
Fibroblasts are electrophysiologically quiescent in the healthy heart. Evidence suggests that remodeling following myocardial infarction may include coupling of myofibroblasts (Mfbs) among themselves and with myocytes via gap junctions. We use a magnetic resonance imaging-based, three-dimensional computational model of the chronically infarcted rabbit ventricles to characterize the arrhythmogenic substrate resulting from Mfb infiltration as a function of Mfb density. Mfbs forming gap junctions were incorporated into both infarct regions, the periinfarct zone (PZ) and the scar; six scenarios were modeled: 0%, 10%, and 30% Mfbs in the PZ, with either 80% or 0% Mfbs in the scar. Ionic current remodeling in PZ was also included. All preparations exhibited elevated resting membrane potential within and near the PZ and action potential duration shortening throughout the ventricles. The unique combination of PZ ionic current remodeling and different degrees of Mfb infiltration in the infarcted ventricles determines susceptibility to arrhythmia. At low densities, Mfbs do not alter arrhythmia propensity; the latter arises predominantly from ionic current remodeling in PZ. At intermediate densities, Mfbs cause additional action potential shortening and exacerbate arrhythmia propensity. At high densities, Mfbs protect against arrhythmia by causing resting depolarization and blocking propagation, thus overcoming the arrhythmogenic effects of PZ ionic current remodeling.
Introduction
Myocytes, nonmyocytes, and the extracellular matrix are key components of the myocardium. Myocytes are central to the electromechanical function of the heart, but represent only one-third to one-half of the cell population of healthy adult myocardium. The remaining cells are nonmyocytes, the majority of which are fibroblasts. The latter are electrophysiologically quiescent and do not contribute to cardiac electrical behavior under normal physiological conditions. However, pathological conditions such as myocardial infarction (MI) trigger electrical and fibrotic remodeling of the myocardium, resulting in a substrate susceptible to arrhythmias. Although electrical remodeling in surviving myocytes includes primarily ion channel and gap junction alterations (1,2), fibrotic remodeling is manifested as fibroblast proliferation and phenotypic switching, as well as excess collagen deposition (3,4). Specifically, differentiating fibroblasts, herein called myofibroblasts (Mfbs), acquire a migratory phenotype by developing contractile bundles, express α-smooth muscle actin, and exhibit altered connexin distribution (5).
MI results in a scar (electrically inactive) and an adjacent remodeled periinfarct zone (PZ) (2); the latter has been shown to be critical in arrhythmogenesis (6–8). A variety of mechanisms resulting from MI electrophysiological remodeling (e.g., slowed conduction due to ion channel and gap junction modifications (9)) and the development of fibrosis (e.g., functional discontinuities in the excitable substrate due to collagen accumulation (10–12)) have been explored as underlying these arrhythmias. However, recent evidence suggests an additional mechanism: remodeled Mfbs may electrically couple both with myocytes and among themselves in the infarct via gap junctions and dynamically contribute to arrhythmia susceptibility (3,13,14).
The functional coupling of Mfbs with themselves and with myocytes is well established in cell culture (15–18). However, it is difficult to electrically differentiate between a cardiomyocyte and a well-coupled Mfb in situ due to the Mfb's very high membrane resistance, which causes it to mirror the electrical activity of well-coupled myocytes (19,20). Therefore, direct evidence of electrical myocyte-Mfb and inter-Mfb coupling in the intact myocardium remains difficult to obtain experimentally. Alternative in situ experimental methods, including immunohistochemistry and dye transfer, have confirmed that a network of coupled fibroblasts, possibly linked to adjacent myocytes, exists in the rabbit sinoatrial node (20) and have established that Cx43-expressing fibroblasts infiltrate damaged tissue during progressive infarction, supporting the possibility that Mfbs couple with themselves and with myocytes in both the scar and PZ (21).
Due to the limitations associated with obtaining Mfb electrophysiological measurements in the intact myocardium, computational modeling has emerged as an important tool capable of determining the arrhythmogenic role that inter-Mfb and Mfb-myocyte coupling would play if these electrical connections do, in fact, form in the myocardium under disease conditions. Previous computational studies using cellular-level and two-dimensional tissue models showed that Mfbs coupled to myocytes have the capability of modulating the myocyte's action potential (AP) morphology as well as the conduction and upstroke velocities in surrounding tissue (22–27), thus suggesting a possible role of Mfb-myocyte coupling in perturbing the electrical activity in diseased hearts.
The goal of this study is to explore the possible role of inter-Mfb and Mfb-myocyte electrical coupling in the scar and PZ in the mechanisms of arrhythmia generation under the conditions of MI. To achieve this goal, a novel, to our knowledge, magnetic resonance imaging (MRI)-based biophysically detailed three-dimensional (3D) computational model of the infarcted rabbit ventricles is developed. Simulations are conducted to characterize the arrhythmogenic substrate resulting from inter-Mfb and Mfb-myocyte coupling for varying degrees of Mfb infiltration in the PZ and scar. Simulations are then performed to assess the various substrates' propensity to arrhythmia.
Methods
Model generation
We have recently developed a high-throughput pipeline for the generation of 3D models of hearts from ex vivo structural magnetic resonance (MR) and diffusion tensor (DT) MR images (28–30). This pipeline was used in the creation of the infarcted rabbit heart model used in this study.
A rabbit heart at 7.5 weeks postinfarction was scanned (MR and DTMR) at a resolution of 61 × 61 × 60 μm3, as described in (29); a representative long-axis slice of the MR image stack is shown in Fig. 1. The scar and PZ were differentiated from the healthy myocardium and segmented out as described in (29) (Fig. 1). The segmented images were used to generate the finite element mesh of the ventricles using the meshing package Tarantula (CAE-Software Solutions, Eggenburg, Austria); the methodology is described in (31). Each element of the 4.1 million node mesh (average edge lengths: 60 μm in the PZ/scar, 171 μm in healthy tissue) was assigned a fiber orientation based on the calculation of the primary eigenvector of the DT obtained from the DTMR data, as described in (29).
Figure 1.
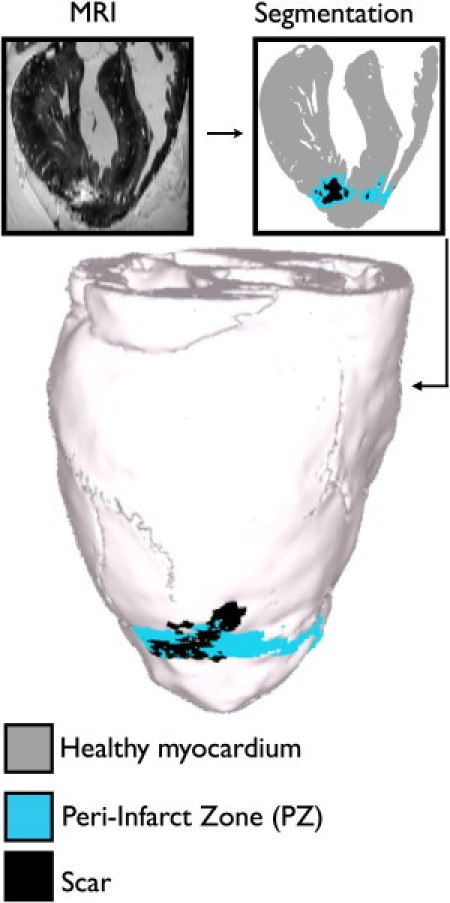
Model generation. Representative MRI slice of the rabbit heart, segmentation of the slice, and whole heart mode.
Representing infarction-related structural remodeling
The PZ was modeled as containing 0% (PZ0), 10% (PZ10), or 30% (PZ30) Mfbs by volume, thus including the range of densities previously quantified in sheep heart PZ tissue over a 30-day postinfarction period (21). This was achieved by assigning Mfb properties, at random distribution, to a percentage of PZ elements in the model. Because the average PZ element edge length is 60 μm, each Mfb-assigned element actually represents a small cluster of interconnected Mfbs due to their small size (620 μm2 average area corresponding to an average cell length ∼25 μm) (32).
Although the scar is known to be densely fibrotic, the question of whether Mfbs in the scar form an interconnected network via electrical coupling or simply act as passive barriers to conduction remains unanswered (21). Therefore, the scar was modeled in two ways: with either 80% Mfbs by volume (Scar80), with the remainder as insulator, or as 100% insulator (Scar0). Thus, a total of six versions of the ventricular model were created when combining the three cases of Mfb density in the PZ with the two cases of Mfb density in the scar: Scar0PZ0, Scar80PZ0, Scar0PZ10, Scar80PZ10, Scar0PZ30, and Scar80PZ30. The case for which the PZ contained no Mfbs and the scar was purely insulator (Scar0PZ0) was considered to be the control.
Representing electrophysiological properties in the model
Mathematical description of current flow in cardiac tissue in the rabbit ventricles was based on the monodomain representation. Myocyte membrane kinetics was represented by Mahajan et al.'s model of the rabbit ventricular AP (33). In the PZ, the myocyte's membrane kinetics was modified to match data reported in the literature. Previous investigations of PZ remodeling in infarcted hearts found reductions in peak currents as follows: Na+ (INa) to 38% of the normal value (9); L-type Ca2+ (ICa,L) to 31% of normal (34); and rapid (IKr) and slow (IKs) components of the delayed rectifier to 30% and 20% of maximum (35), respectively. For the purpose of differentiating between the electrophysiological consequences of Mfb presence in the PZ versus ionic current remodeling in the PZ, additional ventricular models were created with Mfb presence but without ionic current remodeling in the PZ (Scar% PZ%no-ionic). In all models, Mfb membrane kinetics in PZ and scar were represented by the MacCannell model (22), which includes inwardly rectifying (Kir) and time- and voltage-gated (Kv) K+ currents, and a resting membrane potential (VREST) of −49.6 mV.
The three regions of the model (healthy tissue, PZ, and scar) were assigned different conductivities based on recent experimental data. Specifically, healthy myocardium was assigned conductivities such that conduction velocity (CV) in the model matched that recorded in healthy rabbit myocardium (36–38); conductivities of 0.163 S/m in the primary myofiber direction and 0.025 S/m in the cross-fiber direction resulted in a mean CV of 47.9 cm/s in healthy tissue, matching the recorded value of 49.0 cm/s reported in normal rabbit myocardium (38). PZ myocytes were assigned conductivity values that were 90% (longitudinal) and 8.5% (transverse) of that of healthy tissue to match measured changes in gap junctional conductance (39). PZ Mfbs were modeled as isotropic with reduced conductivity (75% of that of healthy myocardium's primary fiber direction), representing the reduction of single gap junction conductance measured in Mfb-myocyte pairs as compared to intermyocyte junctions (15). Similarly, Mfbs in the scar were assigned a conductivity value 50% of that of healthy tissue in the primary fiber direction, thus accounting for the decrease in gap junction conductance measured between two Mfbs as compared to two myocytes (15).
Simulation protocol and data analysis
To achieve steady state in the ventricles, ionic model state variables were first precomputed in a single cell by pacing until a steady state was reached. Next, the ventricular models were paced for 5 beats at 300 ms cycle length from a site on the endocardium far removed from the infarct.
Because Mfbs have a less negative resting potential than myocytes, coupling between the two cell type models elevates the myocyte's resting potential (24). Indeed, experimental results have shown that cardiomyocyte strands coated in Mfbs undergo a Mfb density-dependent gradual depolarization (14). Thus, to characterize the diastolic potential distribution, maps of VREST were constructed for each model.
AP duration (APD) dispersion increases susceptibility to arrhythmia (40,41). Although ionic current remodeling of the PZ has been demonstrated to cause APD dispersion (6,34,42), Mfbs have also been found to lead to APD shortening in adjacent myocytes (9,18,22,24,25,34,35). To characterize APD dispersion in each ventricular model, APD maps were generated by calculating the duration from peak AP amplitude to 60% repolarization (APD60) at each node in the ventricular mesh, a level that allows for comparison of APD under conditions of elevated VREST. Additionally, to assess the sole effect of Mfb coupling on APD, APD distribution was examined in preparations without ionic current remodeling in the PZ.
To assess the arrhythmogenic propensity of the different ventricular models, a programmed electrical stimulation (PES) protocol was used to induce arrhythmias (43,44). The protocol consisted of pacing for 5 beats at 300 ms cycle length, followed by two premature stimuli (S2 and S3) at various coupling intervals (165–180 ms for S2, 115–180 ms for S3) after the last pacing stimulus (S1). The pacing electrode was located at the endocardial apex, a common site for PES. In all cases activity was simulated for 2.5 s following S3. Arrhythmia induction was defined as the formation of a reentrant circuit, which completed a minimum of one cycle.
Results
Characterization of the electrophysiological properties in the ventricles arising from inter-Mfb coupling in the scar and Mfb-myocyte coupling in the PZ
Increasing Mfb density results in a progressive elevation of PZ resting potential
Fig. 2 A displays maps of VREST distribution for each of the ventricular models. Incorporation of Mfbs of increasing densities in both scar and PZ results in the progressive elevation of PZ VREST from −87.8 mV (Scar0PZ0) to −75.1 mV (Scar80PZ30). The boxplots in Fig. 2 B depict the distribution of VREST in each ventricle's PZ; the middle 50% of the PZ's VREST values (represented by each box) cover a wider range and exhibit less negative values as Mfbs are either added to the scar or PZ Mfb densities are increased. Likewise, the largest degree of resting tissue depolarization (indicated by the rightmost whisker of each plot) occurs in ventricular models that incorporate inter-Mfb coupling in the scar, with further increase in maximum resting depolarization as Mfb densities are increased in the PZ. The model with the largest number of Mfbs (Scar80PZ30) has the greatest degree of resting depolarization, reaching up to −61 mV as compared to the control's VREST of −87.8 mV. The VREST map of this densely fibrotic preparation (Fig. 2 A, lower right) exhibits maximum resting depolarization at the core of the interconnected Mfb network (arrow), and thus at the point farthest away from any myocyte connections.
Figure 2.
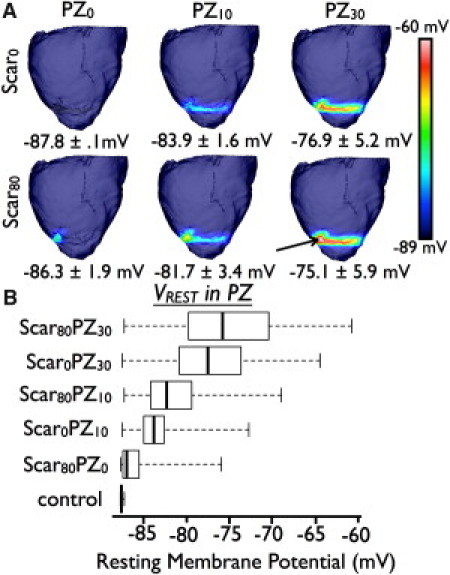
(A) VREST maps in the models. 0% (top row) or 80% (bottom row) Mfbs in the scar region. 0% (left column), 10% (middle column), or 30% (right column) Mfbs in the PZ. Average PZ VREST values and standard deviations are listed below the respective preparations (B). Boxplots of VREST in PZ of each substrate. Whiskers represent minimum and maximum of all values in a given preparation.
Inter-Mfb coupling in the scar and Mfb-myocyte coupling in the PZ lead to heterogeneous APD shortening in the ventricles
APD distribution maps for the six ventricular models are displayed in Fig. 3 A. In the control preparation (Scar0PZ0), APD shortening of ∼25 ms occurs in the PZ as a result of ionic current remodeling, consistent with rabbit experimental findings (45). Therefore, to isolate the Mfb's potential shortening effect on APD, APD distributions for each of the six preparations were additionally calculated without ionic current remodeling in the PZ (Fig. 3 B). Fig. 3 B presents the gradual decrease in average APD throughout the tissue (value listed below each map) as Mfb density is increased in either PZ or scar, ranging from 130.9 ms (Scar0PZ0no-ionic) to 81.9 ms (Scar30PZ80no-ionic). Mfb-induced APD shortening occurs in a spatially heterogeneous fashion—the locations with the shortest APD (green) are nearest to the scar and PZ (refer to Fig. 4 for PZ and scar locations). Largest APD values occur in healthy regions farthest from the infarct and are consistent in all models, including Scar0PZ0no-ionic. Comparing Fig. 3 B (APD distribution due solely to Mfbs) to Fig. 3 A (APD distribution due to Mfbs and ionic current remodeling), one finds that Mfb infiltration, with or without ionic current remodeling in the PZ, results in the same trend of APD shortening. Although average APD in the Scar0PZ0 model (112.8 ms) is shorter than that in Scar0PZ0no-ionic (130.9 ms), a progressive decrease in average APD is still observed as Mfb density is increased in either the PZ or scar, reaching the shortest average duration of 81.9 ms in both the Scar80PZ30 and Scar80PZ30no-ionic models.
Figure 3.

APD maps in the models (A) and those when ionic current remodeling in the PZ is removed (B). 0% (top row) or 80% (bottom row) Mfbs in the scar region. 0% (left column), 10% (middle column), or 30% (right column) Mfbs in the PZ. The average APD and its standard deviation are listed below each preparation. Boxplots represent distribution of APD in the PZ (C) and healthy tissue (D) for both sets of models.
Figure 4.

VREST and APD as a function of distance along the vertical line between points A (in the outer edge of the PZ) and B (10 mm removed from the outer edge of the PZ) in the Scar80PZ30 model.
APD distribution is quantified in PZ and healthy tissue in Fig. 3, C and D, respectively, with data compared, in each case, to that without ionic current changes in the PZ. At lower Mfb densities, the Mfb's APD shortening effect is augmented by the APD shortening due to ionic current remodeling in the PZ, as evidenced by a left shift of the Scar80PZ0 data's white box (representing data from PZ tissue with ionic current remodeling) compared to its gray box (representing data from PZ tissue without ionic current changes) in Fig. 3 C. As Mfb densities are increased in the models, an increasing portion of PZ tissue does not activate (compare black regions in Fig. 3 A). Because inactivated tissue is not included in the boxplot calculations, the PZ APD for these models (Scar80PZ10, Scar0PZ30, and Scar80PZ30) is shortened to a lesser degree as Mfb densities increase (Fig. 3 C, slight right shift of boxes for the top three models). As Mfbs are added to the PZ or scar in these three cases, PZ myocytes, which previously exhibited a very short APD, become inactivated and therefore, no longer contribute to the shortening trend in the APD data for that model. As more PZ tissue becomes inactivated, less excitable tissue remains in close proximity to Mfbs, resulting in less tissue experiencing the highest degrees of APD shortening.
VREST and APD were calculated as functions of distance from the PZ for the Scar80PZ30 model at the locations shown in Fig. 4. VREST exhibits a sharp drop-off with distance away from the PZ, reaching 90% of the average control value over a short distance of 2.2 mm. Thus, the partial INa inactivation that results from membrane depolarization (and may contribute to conduction block) is confined to the infarct regions and tissue within close proximity to the PZ. APD, on the other hand, attenuates with distance from PZ at a slower rate; APD restoration to 90% of the average control value occurs over a distance of 5.3 mm. That said, tissue in regions farthest from the PZ and scar remains unaffected by the Mfbs; maximum APD in healthy tissue (as indicated by the rightmost whiskers in Fig. 3 D) is 144 ms in all preparations, including control.
Mechanisms of arrhythmia propensity resulting from inter-Mfb and Mfb-myocyte coupling
Low Mfb densities do not alter the propensity to arrhythmias
PES was used to assess each substrate's susceptibility to reentry. Because ionic current changes in the PZ and Mfbs in the infarct both create potentially arrhythmogenic APD dispersion (Fig. 3, A and B), reentry generation in preparations both with and without PZ ionic current remodeling was examined. Outcomes of PES are summarized in Tables 1 and 2, respectively. All induced arrhythmias were unsustained, consistent with experimental evidence; pharmacological intervention is necessary to induce sustained arrhythmias in the chronic MI rabbit ventricles (38). In the Scar0PZ0no-ionic model (Table 2, at top left), where no electrophysiological remodeling (ionic current changes in PZ or Mfb infiltration) takes place, S3 encounters tissue of uniform recovery and results in either complete conduction block or undisturbed propagation, depending on the S3 coupling interval. In the Scar0PZ0 preparation (Table 1, at top left), arrhythmia induction following PES is due exclusively to ionic current remodeling in the PZ, as no Mfbs are present in the preparation. As shown in Fig. 3 A (at top left), PZ ionic current remodeling alone creates APD dispersion and results in arrhythmia, as demonstrated by a body of experimental research (40,41). Adding 80% Mfbs to the scar results in the same differences in the outcome of PES (compare Tables 1, lower left, with Table 2, lower left), indicating that low levels of Mfb infiltration are not a major player in post-MI ventricular arrhythmogenesis.
Table 1.
Outcomes of PES on ventricles with ionic current remodeling in the PZ
| PZ0 | PZ10 | PZ30 | |
|---|---|---|---|
| Scar0 | Arrhythmia | Arrhythmia | No Arrhythmia |
| Scar80 | Arrhythmia | Arrhythmia | No Arrhythmia |
Table 2.
Outcomes of PES on ventricles without ionic current remodeling in the PZ
| PZ0no-ionic | PZ10no-ionic | PZ30no-ionic | |
|---|---|---|---|
| Scar0 | No Arrhythmia | Arrhythmia | No Arrhythmia |
| Scar80 | No Arrhythmia | Arrhythmia | No Arrhythmia |
Intermediate levels of Mfb infiltration increase arrhythmia susceptibility via APD dispersion
In all substrates with 10% Mfbs by volume in the PZ (with and without PZ ionic current remodeling), PES results in arrhythmia induction, which is caused by APD dispersion, as characterized in Fig. 3. In those substrates without PZ ionic current remodeling (middle column of Table 2), Mfbs themselves generate sufficient APD dispersion to support arrhythmogenesis. In the presence of PZ ionic current remodeling (middle column of Table 1), Mfbs contribute synergistically to the induction of arrhythmia. This is illustrated in the top row of Fig. 5, which displays snapshots of transmembrane potential maps in a cross-sectional view of the Scar80PZ10 ventricles following PES (see Movie S1 in the Supporting Material). S3 results in unidirectional block (black line) and transmural reentry (manifested as breakthrough activity on the endocardial surface) due to the APD dispersion concurrently generated by Mfbs and PZ ionic current remodeling.
Figure 5.

Transmembrane potential maps of arrhythmia (top row) and termination of conduction (bottom row) at four time instants in two preparations. Cross-sectional views show endocardial surfaces for the right and left ventricles (RV and LV, respectively, marked at bottom right). Red arrows indicate the location of S3 on the endocardial apex. Line of unidirectional block is marked in black. Conduction direction is indicated by the black arrows.
High levels of Mfb infiltration protect against reentrant activity via VREST elevation
As apparent from the right columns in Tables 1 and 2, increasing PZ Mfb density to 30% by volume protects the preparation from arrhythmia induction, regardless of ionic current remodeling in the PZ (Table 1). Thus, high levels of Mfb infiltration have an opposite effect on arrhythmogenesis than intermediate levels. The bottom row of Fig. 5 illustrates the failure of arrhythmia induction in the Scar80PZ30 preparation (see Movie S2), arising from conduction failure due to the higher VREST elevation as compared to the Scar80PZ10 case. VREST elevation, caused by PZ Mfb-myocyte coupling (Fig. 2) causes partial INa inactivation and contributes to conduction failure (14,46). Overall, preparations with the highest VREST elevation (Scar0PZ30 and Scar80PZ30, as seen in Fig. 2 B) are also those that exhibit conduction failure of S3 and thus do not result in arrhythmia (right column, Table 1). Therefore, the antiarrhythmic effect of VREST elevation counteracts the proarrhythmic APD dispersion created by both the Mfbs and the PZ ionic current remodeling.
Discussion
The goal of this study was to examine the role of inter-Mfb and Mfb-myocyte electrical coupling in the scar and PZ in establishing the arrhythmogenic substrate in MI. Propensity to arrhythmia following PES was evaluated as a function of Mfb density in the infarcted ventricles. It was found that the Mfbs' contribution to arrhythmia propensity is a function of Mfb density. At low densities, Mfbs do not alter arrhythmia susceptibility, which is determined exclusively by PZ ionic current remodeling. At intermediate densities, Mfbs act synergistically with PZ ionic current remodeling to support arrhythmogenesis by creating additional APD dispersion. At high densities, Mfbs cause resting depolarization sufficient to suppress the propensity to reentry, despite other proarrhythmogenic effects.
To the best of our knowledge, this study is the first to use a high-resolution MRI-based 3D model of the infarcted ventricles in a study of electrophysiological mechanisms associated with the Mfb infiltration levels in the infarct. The model structure is highly detailed, including papillary muscles and trabeculations. Realistic scar and PZ morphology as well as fiber architecture are implemented from MRI and DTMRI scans. These model features, combined with the small spatial discretization aimed at resolving the presence of Mfbs (or small Mfb clusters), and with a complex ionic model featuring Markovian calcium handling, represents possibly the most sophisticated 3D model of the infarcted ventricles to date.
The Mfb's role as a current sink and source
Numerous studies, both experimental and computational, have aimed to determine the electrophysiological effects of myocyte-Mfb coupling on cardiomyocyte electrophysiological properties. None, however, have investigated the ensuing arrhythmogenic effects in the realistic 3D substrate resulting from MI. As this study confirmed, the roles of the Mfb as a current sink and current source act synergistically to promote or protect against arrhythmia susceptibility:
Current sink. During cardiomyocyte AP upstroke and plateau, coupled Mfbs act as passive load, as a result of their time and voltage-dependent outward K+ currents (47,48), sinking current from neighboring excited myocytes. Previous modeling studies (22,24,25) have shown that coupling of Mfbs to ventricular myocytes shortens the myocyte APD when the Mfb's VREST is sufficiently low (∼49 mV, as presented here). In this study, the role of the Mfb as an electrophysiological current sink underlies the APD shortening experienced by the myocardium.
Current source. Mfbs have a relatively depolarized VREST compared to that of cardiomyocytes. In vitro studies have found that Mfbs act as a current source, causing diastolic depolarization of the connected myocytes (14), which may give rise to ectopic excitation (49–51). In this study, the Mfb's role as a current source results in VREST elevation in PZ. The resulting partial sodium channel inactivation contributes to conduction failure when PZ Mfb densities are relatively high.
Mfb densities modulate arrhythmogenesis
The Mfb's dual role as a current source and sink, as described previously, results in APD dispersion and VREST elevation in substrates with electrical inter-Mfb and Mfb-myocyte connections in the scar and PZ. The unique combination of these consequences resulting from the specific Mfb density, combined with the particular structure of the infarct, contributes to or protects against the substrate's susceptibility to arrhythmia. At relatively low densities (Scar0PZ0 and Scar80PZ0), Mfbs do not alter arrhythmia propensity; the latter results from APD dispersion stemming from PZ ionic current remodeling. When Mfbs are present at intermediate densities (Scar0PZ10 or Scar80PZ10), however, the additional APD dispersion caused by the Mfbs' role as current sinks exacerbates arrhythmia propensity. At higher Mfb density the scale is tipped to an antiarrhythmic behavior as the Mfbs act as current sources causing resting depolarization; it overcomes the PZ ionic current remodeling arrhythmogenic effect and prevents the establishment of reentry.
Experimental evidence indicates that remodeling in the PZ alone underlies the mechanisms for arrhythmogenesis in the post-MI heart (7,44,52). Likewise, several experimental studies suggest that Mfb-myocyte coupling can contribute to arrhythmia propensity (3,14,17,27). In our study, both classes of mechanisms are manifested; we dissect their interplay as a function of Mfb density. Research has reported that mouse survival rates during the first month after MI is highest during the first 6 days (53), corresponding to the time that Mfbs show maximal accumulation in mouse infarct tissue (54); this evidence supports our finding that Mfbs at high densities have an antiarrhythmic effect. As mouse infarct Mfb densities begin to decrease significantly at day 7 (54), survival rates for postinfection mice plummet (53); this corroborates our finding that intermediate levels of Mfbs exacerbate arrhythmogenesis. Finally, mouse survival rates have been documented to proceed at a steady rate after day 15 (53), a time frame during which Mfb densities are very low (54), suggesting that arrhythmogenesis is not due to Mfb infiltration.
In certain conditions, such as those associated with aging or genetic disease, myocardial fibrotic remodeling (and proliferation of Mfbs) might occur without concomitant ionic current remodeling (3). Because aging is positively correlated with increased incidence of ventricular arrhythmia, even when excluding populations with coronary artery disease (55), it is feasible that Mfbs could underlie arrhythmogenicity in aged hearts. We speculate that such conditions would correspond to the effect of intermediate Mfb densities because the distribution of Mfbs is diffuse in fibrosis (56).
Studies on cultured strands of cells have found that Mfbs at high densities cause ectopic excitation as the myocyte's VREST elevates (due to the Mfb's inherently high VREST) and surpasses the myocyte's activation threshold (51). In the current study, however, maximal VREST elevation reaches −60.7 mV, well below the used myocyte model's fast sodium channel activation threshold of −40 mV (33); no automaticity was observed. It is possible that increasing Mfb levels beyond the high densities used here could result in ectopic activity, thus adding another layer of complexity to the Mfb's influence presented herein. In our study, however, Mfb densities were assigned in the model as low, intermediate, and high based on in vitro experimental data (21), thus we did not represent high Mfb densities as could be found in cultured strand studies.
In vitro studies have found that Mfbs alter their electrophysiological contribution as a function of density, consistent with the findings presented here. Specifically, Morita et al. (57) used histological analysis to measure the percentage of area occupied by fibrosis (compared to total area) at sites of early afterdepolarization (EAD) origination in aged hearts; it was found that sites of frequent EAD origination had an intermediate percent fibrosis (mean of 28%) compared to other regions of the heart. Likewise, the two-dimensional computational model of fibrosis (including Mfb-myocyte coupling) used to further elucidate their findings found that an intermediate range of Mfb/myocyte cell number ratios supported EADs and repetitive triggered activity, whereas high and low did not (57). Similarly, Zlochiver et al. (27) used Cx43 silencing and overexpression techniques to modify the degree of Mfb-myocyte coupling in heterocellular cocultures; CV was found to have a biphasic dependency on Mfb Cx43 expression levels, and thus on electrical coupling, whereby CV initially decreased and then increased as heterocellular coupling levels increased. Increasing heterocellular coupling is analogous to increasing Mfb density, as both result in APD shortening and VREST elevation. Miragoli et al. (14) found that an increase in Mfb/myocyte ratios up to 7:100 caused an increase in both CV and maximal upstroke velocity, followed by a progressive decline at higher ratios.
The antiarrhythmic behavior of Mfbs at high densities opens a new door for therapeutic targets. A pharmacological agent that increases gap junction conductance between myocytes and Mfbs would enhance the Mfb's ability to act as a current source at rest, which could prevent reentrant activity. Research is under way to design peptides that can prevent closure of Cx43 gap junctions between myocytes (58,59) because reduced Cx43 expression among myocytes in the PZ is thought to underlie disruption of conduction (51,52). This novel treatment of cardiac arrhythmias could be extended based on our findings to involve targeting of Mfb-myocyte gap junctions.
Study limitations
Although it is agreed upon that Mfbs have a relatively elevated VREST compared to that of cardiomyocytes, the actual value is debated (15,22,60). If the Mfb's VREST is different than the one used here (−49.6 mV), it may affect APD distribution and thus conduction block and reentry initiation; however, the mechanisms uncovered here will remain valid. Furthermore, our continuum finite element model does not resolve individual cells, and the need for computational tractability of this large model prevents the decrease in the spatial discretization of the PZ and scar beyond element edge lengths of 60 μm (larger than a Mfb). Thus, we are unable to model individual Mfbs scattered among larger myocytes; instead, our model represents clusters of a few interconnected Mfbs scattered among larger myocytes. However, because it has previously been shown in vitro that interconnected Mfbs can serve as a bridge for electrical connection between myocyte groups over distances of up to 300 μm (5× as long as the average edge length in the PZ and scar) (17), it is unlikely that this modeling methodology would produce different results regarding the contribution of Mfbs to arrhythmogenesis in MI.
Our model does not account for transmural heterogeneity of ion channel expression, which would cause APD dispersion across the transmural wall. However, because the Mfb's influence on APD reported herein is much greater than the difference in APD experienced across the transmural wall in the adult rabbit (∼21 ms (61)), it is unlikely that such inclusion will alter the uncovered mechanisms. Likewise, the His bundle and Purkinje network are not represented in the model to limit model complexity. This is a reasonable simplification because we assess arrhythmogenesis following PES; experimental studies have shown that the Purkinje network does not play a role under such circumstances (62,63). Furthermore, this study was performed using one MI rabbit heart model. Although the specific pattern of the reentrant arrhythmia is likely to be different in different infarcted hearts, we expect that the basic mechanisms of arrhythmogenesis associated with Mfb-myocyte coupling uncovered here will remain valid regardless of the specific infarct morphology.
Finally, the distinction between an activated fibroblast and a myofibroblast is debatable. Some argue that the development of contractile bundles does not necessarily transform the fibroblast into a new cell type, and therefore do not acknowledge the myofibroblast as a distinct cell type (64). Thus, what we refer to herein as a myofibroblast (Mfb) may represent what others see as a specific fibroblast phenotype. Because we are unaware of a mathematical model that fully represents the myofibroblast, we have chosen to represent this cell type with MacCannell et al.'s ionic model of an active fibroblast phenotype, which includes time- and voltage-gated K+ currents (22). The model choice seems suitable for an electrophysiological study because inwardly rectifying K+ (Kir) current densities were found to be the same between myofibroblasts and fibroblasts with measurable Kir currents (48). However, further studies are needed to fully elucidate the electrophysiological differences between activated fibroblasts and myofibroblasts.
Acknowledgments
The authors gratefully acknowledge support of this work by the National Science Foundation Graduate Research Fellowship to K.M. and the National Institutes of Health (grants HL082729 and HL067322) to N.T.
Supporting Material
References
- 1.Peters N.S., Wit A.L. Myocardial architecture and ventricular arrhythmogenesis. Circulation. 1998;97:1746–1754. doi: 10.1161/01.cir.97.17.1746. [DOI] [PubMed] [Google Scholar]
- 2.Pinto J.M., Boyden P.A. Electrical remodeling in ischemia and infarction. Cardiovasc. Res. 1999;42:284–297. doi: 10.1016/s0008-6363(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 3.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6:848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 4.Porter K.E., Turner N.A. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol. Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Hinz B. Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 6.Ursell P.C., Gardner P.I., Wit A.L. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ. Res. 1985;56:436–451. doi: 10.1161/01.res.56.3.436. [DOI] [PubMed] [Google Scholar]
- 7.Lue W.M., Boyden P.A. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992;85:1175–1188. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar]
- 8.Wong S.S., Bassett A.L., Myerburg R.J. Dissimilarities in the electrophysiological abnormalities of lateral border and central infarct zone cells after healing of myocardial infarction in cats. Circ. Res. 1982;51:486–493. doi: 10.1161/01.res.51.4.486. [DOI] [PubMed] [Google Scholar]
- 9.Pu J., Boyden P.A. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ. Res. 1997;81:110–119. doi: 10.1161/01.res.81.1.110. [DOI] [PubMed] [Google Scholar]
- 10.Danik S.B., Liu F.Y., Gutstein D.E. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 2004;95:1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spach M.S., Boineau J.P. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 1997;20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 12.de Bakker J.M., van Capelle F.J.L., Lahpor J.R. Slow conduction in the infarcted human heart. ‘Zigzag’ course of activation. Circulation. 1993;88:915–926. doi: 10.1161/01.cir.88.3.915. [DOI] [PubMed] [Google Scholar]
- 13.McDowell K.S., Trayanova N., Kohl P. Fibroblasts and cardiac electrophysiology. In: Turner N.A., editor. The Cardiac Fibroblast. Research Signpost; Kerala, India: 2011. pp. 9–28. [Google Scholar]
- 14.Miragoli M., Gaudesius G., Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ. Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 15.Rook M.B., van Ginneken A.C.G., Jongsma H.J. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am. J. Physiol. 1992;263:C959–C977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 16.Goldsmith E.C., Hoffman A., Borg T.K. Organization of fibroblasts in the heart. Dev. Dyn. 2004;230:787–794. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 17.Gaudesius G., Miragoli M., Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ. Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 18.Vasquez, C., P. Mohandas, …, G. E. Morley. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 107:1011–1020. [DOI] [PMC free article] [PubMed]
- 19.Camelliti P., Green C.R., Kohl P. Structural and functional coupling of cardiac myocytes and fibroblasts. Adv. Cardiol. 2006;42:132–149. doi: 10.1159/000092566. [DOI] [PubMed] [Google Scholar]
- 20.Camelliti P., Green C.R., Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004;94:828–835. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 21.Camelliti P., Devlin G.P., Green C.R. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc. Res. 2004;62:415–425. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 22.MacCannell K.A., Bazzazi H., Giles W.R. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys. J. 2007;92:4121–4132. doi: 10.1529/biophysj.106.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sachse F.B., Moreno A.P., Abildskov J.A. Electrophysiological modeling of fibroblasts and their interaction with myocytes. Ann. Biomed. Eng. 2008;36:41–56. doi: 10.1007/s10439-007-9405-8. [DOI] [PubMed] [Google Scholar]
- 24.Maleckar M.M., Greenstein J.L., Trayanova N.A. Electrotonic coupling between human atrial myocytes and fibroblasts alters myocyte excitability and repolarization. Biophys. J. 2009;97:2179–2190. doi: 10.1016/j.bpj.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacquemet V., Henriquez C.S. Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2040–H2052. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Y.F., Garfinkel A., Qu Z. Cardiac alternans induced by fibroblast-myocyte coupling: mechanistic insights from computational models. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H775–H784. doi: 10.1152/ajpheart.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zlochiver S., Muñoz V., Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys. J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vadakkumpadan F., Rantner L.J., Trayanova N. Image-based models of cardiac structure with applications in arrhythmia and defibrillation studies. J. Electrocardiol. 2009;42 doi: 10.1016/j.jelectrocard.2008.12.003. 157.e1–157.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vadakkumpadan F., Arevalo H., Trayanova N. Image-based models of cardiac structure in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2:489–506. doi: 10.1002/wsbm.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vigmond E., Vadakkumpadan F., Trayanova N. Towards predictive modelling of the electrophysiology of the heart. Exp. Physiol. 2009;94:563–577. doi: 10.1113/expphysiol.2008.044073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prassl A.J., Kickinger F., Plank G. Automatically generated, anatomically accurate meshes for cardiac electrophysiology problems. IEEE Trans. Biomed. Eng. 2009;56:1318–1330. doi: 10.1109/TBME.2009.2014243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson S.A., Copeland C.R., Tung L. Mechanical coupling between myofibroblasts and cardiomyocytes slows electric conduction in fibrotic cell monolayers. Circulation. 2011;123:2083–2093. doi: 10.1161/CIRCULATIONAHA.110.015057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahajan A., Shiferaw Y., Weiss J.N. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys. J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dun W., Baba S., Boyden P.A. Dynamic remodeling of K+ and Ca2+ currents in cells that survived in the epicardial border zone of canine healed infarcted heart. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1046–H1054. doi: 10.1152/ajpheart.00082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang M., Cabo C., Tseng G. Delayed rectifier K currents have reduced amplitudes and altered kinetics in myocytes from infarcted canine ventricle. Cardiovasc. Res. 2000;48:34–43. doi: 10.1016/s0008-6363(00)00159-0. [DOI] [PubMed] [Google Scholar]
- 36.Roth B.J. Electrical conductivity values used with the bidomain model of cardiac tissue. IEEE Trans. Biomed. Eng. 1997;44:326–328. doi: 10.1109/10.563303. [DOI] [PubMed] [Google Scholar]
- 37.Poelzing S., Akar F.G., Rosenbaum D.S. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H2001–H2009. doi: 10.1152/ajpheart.00987.2003. [DOI] [PubMed] [Google Scholar]
- 38.Ripplinger C.M., Lou Q., Efimov I.R. Panoramic imaging reveals basic mechanisms of induction and termination of ventricular tachycardia in rabbit heart with chronic infarction: implications for low-voltage cardioversion. Heart Rhythm. 2009;6:87–97. doi: 10.1016/j.hrthm.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao J.A., Hussain W., Wit A.L. Remodeling of gap junctional channel function in epicardial border zone of healing canine infarcts. Circ. Res. 2003;92:437–443. doi: 10.1161/01.RES.0000059301.81035.06. [DOI] [PubMed] [Google Scholar]
- 40.Kuo C.S., Munakata K., Surawicz B. Characteristics and possible mechanism of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation. 1983;67:1356–1367. doi: 10.1161/01.cir.67.6.1356. [DOI] [PubMed] [Google Scholar]
- 41.Han J., Moe G.K. Nonuniform recovery of excitability in ventricular muscle. Circ. Res. 1964;14:44–60. doi: 10.1161/01.res.14.1.44. [DOI] [PubMed] [Google Scholar]
- 42.Boyden P.A., Gardner P.I., Wit A.L. Action potentials of cardiac muscle in healing infarcts: response to norepinephrine and caffeine. J. Mol. Cell. Cardiol. 1988;20:525–537. doi: 10.1016/s0022-2828(88)80079-8. [DOI] [PubMed] [Google Scholar]
- 43.Ashikaga H., Sasano T., Halperin H.R. Magnetic resonance-based anatomical analysis of scar-related ventricular tachycardia: implications for catheter ablation. Circ. Res. 2007;101:939–947. doi: 10.1161/CIRCRESAHA.107.158980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasano T., McDonald A.D., Donahue J.K. Molecular ablation of ventricular tachycardia after myocardial infarction. Nat. Med. 2006;12:1256–1258. doi: 10.1038/nm1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilensky R.L., Tranum-Jensen J., Janse M.J. The subendocardial border zone during acute ischemia of the rabbit heart: an electrophysiologic, metabolic, and morphologic correlative study. Circulation. 1986;74:1137–1146. doi: 10.1161/01.cir.74.5.1137. [DOI] [PubMed] [Google Scholar]
- 46.Nerbonne J.M., Kass R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 47.Shibukawa Y., Chilton E.L., Giles W.R. K+ currents activated by depolarization in cardiac fibroblasts. Biophys. J. 2005;88:3924–3935. doi: 10.1529/biophysj.104.054429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chilton L., Ohya S., Giles W.R. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H2931–H2939. doi: 10.1152/ajpheart.01220.2004. [DOI] [PubMed] [Google Scholar]
- 49.Kohl P., Noble D. Mechanosensitive connective tissue: potential influence on heart rhythm. Cardiovasc. Res. 1996;32:62–68. [PubMed] [Google Scholar]
- 50.Kohl P., Kamkin A.G., Noble D. Mechanosensitive fibroblasts in the sino-atrial node region of rat heart: interaction with cardiomyocytes and possible role. Exp. Physiol. 1994;79:943–956. doi: 10.1113/expphysiol.1994.sp003819. [DOI] [PubMed] [Google Scholar]
- 51.Miragoli M., Salvarani N., Rohr S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ. Res. 2007;101:755–758. doi: 10.1161/CIRCRESAHA.107.160549. [DOI] [PubMed] [Google Scholar]
- 52.Cabo C., Boyden P.A. Electrical remodeling of the epicardial border zone in the canine infarcted heart: a computational analysis. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H372–H384. doi: 10.1152/ajpheart.00512.2002. [DOI] [PubMed] [Google Scholar]
- 53.Shishido T., Nozaki N., Kubota I. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 54.Dewald O., Ren G., Frangogiannis N.G. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fleg J.L., Kennedy H.L. Cardiac arrhythmias in a healthy elderly population: detection by 24-hour ambulatory electrocardiography. Chest. 1982;81:302–307. doi: 10.1378/chest.81.3.302. [DOI] [PubMed] [Google Scholar]
- 56.Iles L., Pfluger H., Taylor A.J. Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrast-enhanced T1 mapping. J. Am. Coll. Cardiol. 2008;52:1574–1580. doi: 10.1016/j.jacc.2008.06.049. [DOI] [PubMed] [Google Scholar]
- 57.Morita N., Sovari A.A., Karagueuzian H.S. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1594–H1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verma V., Larsen B.D., Delmar M. Design and characterization of the first peptidomimetic molecule that prevents acidification-induced closure of cardiac gap junctions. Heart Rhythm. 2010;7:1491–1498. doi: 10.1016/j.hrthm.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eloff B.C., Gilat E., Rosenbaum D.S. Pharmacological modulation of cardiac gap junctions to enhance cardiac conduction: evidence supporting a novel target for antiarrhythmic therapy. Circulation. 2003;108:3157–3163. doi: 10.1161/01.CIR.0000101926.43759.10. [DOI] [PubMed] [Google Scholar]
- 60.Kamkin A., Kiseleva I., Isenberg G. Activation and inactivation of a non-selective cation conductance by local mechanical deformation of acutely isolated cardiac fibroblasts. Cardiovasc. Res. 2003;57:793–803. doi: 10.1016/s0008-6363(02)00775-7. [DOI] [PubMed] [Google Scholar]
- 61.Idriss S.F., Wolf P.D. Transmural action potential repolarization heterogeneity develops postnatally in the rabbit. J. Cardiovasc. Electrophysiol. 2004;15:795–801. doi: 10.1046/j.1540-8167.2004.03622.x. [DOI] [PubMed] [Google Scholar]
- 62.Wyman B.T., Hunter W.C., McVeigh E.R. Mapping propagation of mechanical activation in the paced heart with MRI tagging. Am. J. Physiol. 1999;276:H881–H891. doi: 10.1152/ajpheart.1999.276.3.H881. [DOI] [PubMed] [Google Scholar]
- 63.Prinzen F.W., Hunter W.C., McVeigh E.R. Mapping of regional myocardial strain and work during ventricular pacing: experimental study using magnetic resonance imaging tagging. J. Am. Coll. Cardiol. 1999;33:1735–1742. doi: 10.1016/s0735-1097(99)00068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Camelliti P., Borg T.K., Kohl P. Structural and functional characterization of cardiac fibroblasts. Cardiovasc. Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.