

Abstract
Prion diseases are infectious fatal neurodegenerative diseases including Creutzfeldt-Jakob disease in humans and bovine spongiform encephalopathy in cattle. The misfolding and conversion of cellular PrP in such mammals into pathogenic PrP is believed to be the key procedure. Rabbits are among the few mammalian species that exhibit resistance to prion diseases, but little is known about the molecular mechanism underlying such resistance. Here, we report that the crowding agents Ficoll 70 and dextran 70 have different effects on fibrillization of the recombinant full-length PrPs from different species: although these agents dramatically promote fibril formation of the proteins from human and cow, they significantly inhibit fibrillization of the rabbit protein by stabilizing its native state. We also find that fibrils formed by the rabbit protein contain less β-sheet structure and more α-helix structure than those formed by the proteins from human and cow. In addition, amyloid fibrils formed by the rabbit protein do not generate a proteinase K-resistant fragment of 15–16-kDa, but those formed by the proteins from human and cow generate such proteinase K-resistant fragments. Together, these results suggest that the strong inhibition of fibrillization of the rabbit PrP by the crowded physiological environment and the absence of such a protease-resistant fragment for the rabbit protein could be two of the reasons why rabbits are resistant to prion diseases.
Abbreviations used: CJD, Creutzfeldt-Jakob disease; CD, circular dichroism; ESI, electrospray ionization; GPI, glycosylphosphatidylinositol; FTIR, Fourier transform infrared; MS, mass spectrometry; PK, proteinase K; PrP, prion protein; PrPC, the normal cellular PrP molecule; PrPSc, an abnormal disease-causing isoform of prion protein; SDS, sodium dodecyl sulfate; TEM, transmission electron microscopy; ThT, thioflavin T
Introduction
Prions are the heretical protein-only infectious agents responsible for prion diseases including CJD and kuru in humans, bovine spongiform encephalopathy in cattle, scrapie in sheep, and chronic wasting disease in elk and mule deer (1–6). The misfolding and conversion of the normal protease-sensitive cellular prion protein (PrPC) in such mammals into the aberrant protease-resistant pathogenic prion protein (PrPSc) is believed to be the key procedure (1–6), which may require other cofactors (6). Rabbits are among the few animal species that exhibit resistance to prion diseases (7), and they survive challenge with the human kuru and CJD agents as well as with scrapie agent isolated from sheep or mice (7,8). It has been suggested that the resistance of rabbits to prion diseases is due to multiple rabbit PrP-specific amino acid residues (9,10) that may result in a PrP structure that is unable to misfold to the abnormal isoform PrPSc (10). The loop connecting the second β-strand with the second α-helix is well-defined in rabbit, elk, and mule deer PrPC, whereas it exhibits pronounced structural disorder in PrPC of mice, humans, cattle, and sheep (11–13). The x-ray crystallographic structure of rabbit PrPC has identified a key helix-capping motif implicated in the resistance of rabbits to prion diseases (14). Recently, it has been suggested that the rabbit-specific residues surrounding the C-terminal GPI anchor may interfere with a PrPSc interaction site (15). Furthermore, rabbit PrP does not induce spongiform degeneration and does not convert into scrapie-like conformers in transgenic flies (16). However, the molecular mechanism underlying the resistance of rabbits to prion diseases is not well understood.
The misfolding of proteins associated with neurodegenerative diseases such as prion diseases has been traditionally and widely studied in dilute solutions (3,17–21). However, these amyloidogenic proteins form fibrils in a physiological crowded environment (22–29). Macromolecular crowding takes place not only inside the cell (23–25), but also in the extracellular matrix of tissues (23) and at membrane surfaces (30). It has been demonstrated that macromolecular crowding significantly accelerates the misfolding of several amyloidogenic proteins, such as human α-synuclein (31,32), the amyloid β peptide (30), human apolipoprotein C-II (33), human Tau protein (28), and human PrP (28). PrPC is a highly conserved GPI-anchored cell surface glycoprotein and the conformational conversion of PrPC into its pathogenic isoform PrPSc is believed to occur on the cell surface or in the crowded extracellular matrix (34,35). Thus, clarifying how macromolecular crowding on the cell surface or in the extracellular matrix influences the conformational transition and amyloid formation of PrP in vivo is of importance to elucidate the mechanism for the resistance of rabbits to prion diseases.
To understand the molecular mechanism underlying the resistance of rabbits to prion diseases, we have investigated and compared the effects of macromolecular crowding on amyloid formation of the recombinant rabbit/human/bovine full-length PrPs by using several biophysical methods, such as ThT binding, TEM, atomic force microscopy, FTIR spectroscopy, far-ultraviolet CD, and MS. Our results indicated that a crowded, cell-like environment strongly inhibited rabbit PrP misfolding but remarkably accelerated human/bovine PrP misfolding, by fitting the data to a sigmoidal equation (20,28,36). Furthermore, amyloid fibrils formed by the rabbit PrP had remarkably different structure and a PK-resistant feature from those formed by the proteins from human and cow.
Experimental Procedures
Details regarding the experimental procedures are in the Supporting Material.
Materials
The crowding agents, Ficoll 70 and dextran 70, were purchased from Sigma-Aldrich (St. Louis, MO). ThT was also obtained from Sigma-Aldrich. Guanidine hydrochloride was obtained from Promega (Madison, WI). Proteinase K, Triton X-100, and urea were purchased from Ameresco (Solon, OH). All other chemicals used were made in China and were of analytical grade.
Expression and purification of prion proteins
Recombinant full-length rabbit/human/bovine PrPs were expressed in Escherichia coli, isolated on a Ni-NTA agarose column, and further purified by high-performance liquid chromatography on a C4 reversed-phase column (Shimadzu, Kyoto, Japan) as described by Bocharova and co-workers (17). Purified rabbit/human/bovine PrPs were confirmed by SDS-polyacrylamide gel electrophoresis (PAGE) and MS to be single species with an intact disulfide bond. The concentrations of the rabbit PrP, human PrP, and bovine PrP were determined by their absorbance at 280 nm using the molar extinction coefficient values of 57,995, 57,995, and 63,495 M−1 cm−1, respectively, deduced from the composition of the proteins online.
Seeding experiments
Mature fibrils formed by the rabbit/human/bovine PrP were sonicated on ice using a probe sonicator for 30 s (interval of 5 s, 200 W) to produce the seeds used in the seeding experiments. The kinetics of fibril formation of samples with 0.5% (v/v) preformed seed fibrils at the initial time were monitored by ThT binding assays.
Sarkosyl-soluble SDS-PAGE
Rabbit/human PrP amyloid formation was carried out as stated above, during the incubation time, 20 μl samples were taken out and added with 2.5 μl of 100 mM Tris-HCl (pH 7.0) and 2.5 μl of 20% Sarkosyl. The mixture was left at room temperature for 30 min, and then mixed with 2X loading buffer (without β-mercaptoethanol and no heating) and separated by 15% SDS-PAGE. Gels were stained by Coomassie Blue.
Results
Effects of macromolecular crowding on kinetics of amyloid formation of the rabbit/human/bovine PrP
The enhanced fluorescence emission of the dye ThT has been frequently used for monitoring the kinetics of amyloid fibril formation, which is a specific marker for the β-sheet conformation of fibril structures (20,28,37). In this study, the effects of two nominally inert polymeric additives, Ficoll 70 and dextran 70, on kinetics of amyloid formation of the recombinant rabbit/human/bovine full-length PrP were examined by ThT binding assays (Fig. 1), as a function of crowder concentration. Ficoll 70 and dextran 70 are widely accepted as reasonable models for the principal crowding components in living cells (25).
Figure 1.
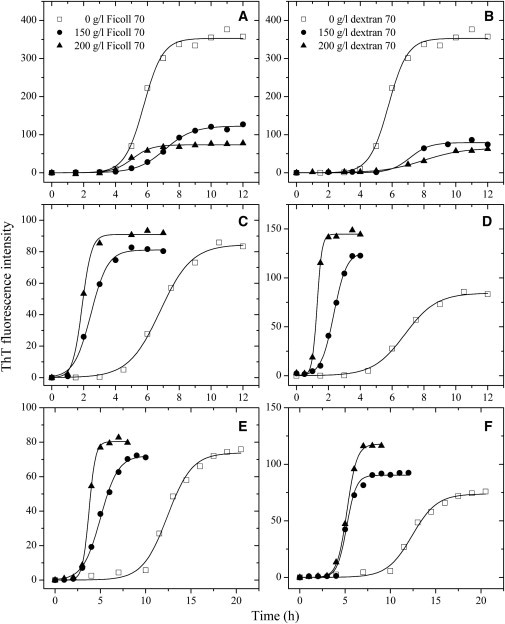
Effects of macromolecular crowding on amyloid formation of rabbit/human/bovine PrPs. Rabbit (A and B), human (C and D), and bovine (E and F) PrPs in the absence and presence of Ficoll 70 (A, C, and E) or dextran 70 (B, D, and F), monitored by ThT fluorescence. All experiments were repeated at least once. The final concentrations of all PrPs were 10 μM. The crowding agent concentrations were 0 (open square), 150 g/l (solid circle), and 200 g/l (solid triangle), respectively. The data were fitted to a sigmoidal equation, and the solid lines represent the best fit. The corresponding kinetic parameters from A–F are summarized in Table 1.
For rabbit PrP, time dependence of ThT fluorescence as a function of crowder concentration is shown in Fig. 1, A and B. As shown in Fig. 1, A and B, the rabbit PrP allowed efficient amyloid formation in the absence of a crowding agent, however, amyloid formation of the rabbit PrP was strongly inhibited by Ficoll 70 and dextran 70 at 150–200 g/l on the investigated timescale, accompanied by a remarkable decline of the maximum ThT intensity.
For comparison, two amyloidogenic proteins, human PrP and bovine PrP, were explored (Fig. 1, C–F). As shown in Fig. 1, C and E, the presence of Ficoll 70 at concentrations of 150 and 200 g/l in the reaction systems both significantly accelerated amyloid formation of the human PrP and bovine PrP on the investigated timescale. Similarly, the presence of dextran 70 at 150–200 g/l in the reaction systems also significantly accelerated fibril formation of the human PrP and bovine PrP on the investigated timescale (Fig. 1, D and F).
To elucidate the mechanisms of the inhibitory effect of macromolecular crowding on amyloid formation of the rabbit PrP and the enhancing effect of macromolecular crowding on fibril formation of the human PrP and bovine PrP, a sigmoidal equation (20,28,36) was used to fit the kinetic data, yielding three kinetic parameters A, the lag time, and k, which are summarized in Table 1. As shown in Table 1 and Fig. 1, in contrast to the proteins from human and cow, fibril formation of the rabbit protein was not accelerated by crowding agents. However, the onset fibril formation in the absence of a crowding agent was very similar. The onset of fibril formation of the rabbit protein appeared even slightly earlier than that of the other two proteins (Fig. 1). The addition of a crowding agent such as dextran 70 at 200 g/l remarkably decreased the rate constant for the growth of the rabbit PrP fibrils and significantly decreased the amount of the rabbit PrP fibrils represented by A (Table 1). On the contrary, the addition of 200 g/l dextran 70 dramatically accelerated both steps of human PrP fibrillization (Table 1), resulting in a lag time of 0.99 h in the presence of dextran 70, which is 4.9-fold decreased compared with that in the absence of a crowding agent (4.82 h), and a rate constant for the growth of fibrils of 6.49 h−1, which is 6.4-fold larger than that in the absence of a crowding agent (1.01 h−1). The addition of 200 g/l dextran 70 also markedly accelerated both steps of bovine PrP fibrillization, albeit to a lower extent than the human PrP (Table 1). These results indicate that the crowding agents Ficoll 70 and dextran 70 have different effects on fibrillization of the recombinant PrPs from different species: although these agents dramatically promote fibril formation of the proteins from human and cow, they appear to significantly inhibit fibrillization of the rabbit protein. Therefore, under crowded physiological conditions, amyloid fibril formation of the rabbit PrP was markedly slower than those of the human/bovine proteins and the amount of the rabbit PrP aggregates was significantly less than that of the human/bovine PrP aggregates (Fig. 1 and Table 1).
Table 1.
Kinetic parameters of amyloid formation of rabbit/human/bovine PrPs in the absence and presence of Ficoll 70 or dextran 70 as determined by ThT binding assays at 37°C
| PrP | Crowding agent | A | Lag time (h) | k (h−1) |
|---|---|---|---|---|
| Rabbit PrP | 0 | 353 ± 6 | 4.57 ± 0.14 | 1.67 ± 0.18 |
| 150 g/l Ficoll 70 | 123 ± 2 | 5.35 ± 0.14 | 1.14 ± 0.09 | |
| 200 g/l Ficoll 70 | 73.5 ± 1.7 | 3.85 ± 0.20 | 1.66 ± 0.26 | |
| 150 g/l dextran 70 | 79.1 ± 2.7 | 5.90 ± 0.76 | 1.64 ± 0.63 | |
| 200 g/l dextran 70 | 65.0 ± 2.1 | 5.47 ± 0.19 | 0.80 ± 0.08 | |
| Human PrP | 0 | 84.6 ± 1.9 | 4.82 ± 0.17 | 1.01 ± 0.09 |
| 150 g/l Ficoll 70 | 81.1 ± 1.4 | 1.40 ± 0.11 | 1.89 ± 0.19 | |
| 200 g/l Ficoll 70 | 91.0 ± 1.2 | 1.32 ± 0.11 | 3.40 ± 0.56 | |
| 150 g/l dextran 70 | 126 ± 2 | 1.54 ± 0.05 | 2.50 ± 0.16 | |
| 200 g/l dextran 70 | 145 ± 1 | 0.99 ± 0.03 | 6.49 ± 0.40 | |
| Bovine PrP | 0 | 73.9 ± 2.0 | 9.65 ± 0.34 | 0.73 ± 0.09 |
| 150 g/l Ficoll 70 | 72.2 ± 1.2 | 3.08 ± 0.12 | 1.03 ± 0.07 | |
| 200 g/l Ficoll 70 | 80.4 ± 0.7 | 3.02 ± 0.05 | 2.78 ± 0.17 | |
| 150 g/l dextran 70 | 90.3 ± 1.6 | 4.13 ± 0.15 | 1.91 ± 0.25 | |
| 200 g/l dextran 70 | 118 ± 1 | 4.11 ± 0.04 | 1.84 ± 0.07 |
Best-fit values of these kinetic parameters were derived from nonlinear least squares modeling of a sigmoidal equation as described in the Materials and Methods (in the Supporting Material) to the data plotted in Fig. 1. Errors shown are ±SE.
For a thorough analysis and control of the effects of crowding agents, complementary approaches were used such as testing the effects of additives in seeding experiments (Fig. 2) or measurements of the decrease of monomeric protein (Figs. S1 and S2 in the Supporting Material). As shown in Fig. 2, in the absence of a crowding agent, the addition of seeds significantly reduced the lag time of fibrillization of the recombinant PrPs from rabbit, human, and cow. The addition of 200 g/l Ficoll 70 not only decelerated both steps of seed-induced rabbit PrP fibrillization, resulting in a lag time of 1.62 h in the presence of Ficoll 70, which is 1.6-fold increased compared with that in the absence of a crowding agent (1.02 h), and a rate constant for the growth of fibrils of 2.51 h−1, which is 1.7-fold decreased compared with that in the absence of a crowding agent (4.24 h−1), but also significantly decreased the amount of rabbit PrP fibrils (Fig. 2). By contrast, the addition of 200 g/l Ficoll 70 remarkably accelerated both steps of seed-induced human/bovine PrP fibrillization (Fig. 2). To semiquantify the decrease of monomeric protein in the presence of crowding agents, we carried out Sarkosyl-soluble SDS-PAGE experiments. As shown in Fig. S1, a clear band corresponding to Sarkosyl-soluble human PrP monomers was observed when the human protein was incubated in the absence of a crowding agent for 8 h, whereas the human PrP monomer band was observed when the human protein was incubated with 200 g/l Ficoll 70 for a much shorter time (1–2 h). As shown in Fig. S2, a clear band corresponding to Sarkosyl-soluble rabbit PrP monomers was observed when the rabbit protein was incubated with 150 g/l Ficoll 70 for 8 h, whereas the rabbit PrP monomer band was observed when the rabbit protein was incubated in the absence of a crowding agent for a shorter time (4–6 h). The above results indicate that although the crowding agents dramatically promote fibril formation of the proteins from human and cow, they significantly inhibit fibrillization of the rabbit protein by stabilizing its native state.
Figure 2.
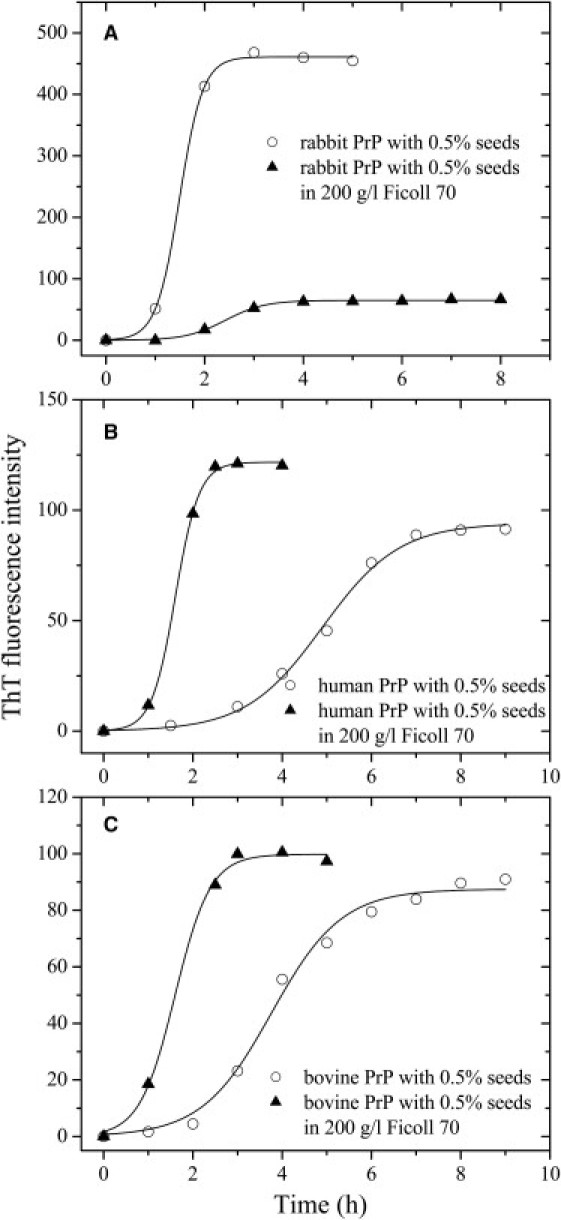
Effects of macromolecular crowding on seed-induced amyloid formation of rabbit/human/bovine PrPs. Rabbit (A), human (B), and bovine (C) PrPs in the absence (open circle) and presence of 200 g/l Ficoll 70 (solid triangle) with 0.5 (v/v) seeds, monitored by ThT fluorescence. All experiments were repeated at least once. The final concentrations of all PrPs were 10 μM. The data were fitted to a sigmoidal equation, and the solid lines represent the best fit.
Effects of macromolecular crowding on morphology of the rabbit/human/bovine PrP samples
TEM was employed to study the morphology of rabbit/human/bovine PrP samples incubated under different conditions. Fig. 3, A–C, shows TEM images of rabbit PrP samples incubated in dilute solutions or in the presence of Ficoll 70 at concentrations of 150 and 200 g/l. The amount of fibrils formed by the rabbit PrP in the presence of 150–200 g/l Ficoll 70 (Fig. 3, B and C), appears to be markedly less than that in the absence of a crowding agent (Fig. 3 A) on the same timescale. The results from TEM are in accordance with those from ThT binding assays, further supporting the conclusion that macromolecular crowding significantly inhibits fibril formation of rabbit PrP. Atomic force microscopy was used to probe the structural order in the rabbit PrP aggregates (Fig. S3). In the absence of a crowding agent, the fibrils formed by the rabbit PrP appear as a long and straight structure after incubation for 8 h (Fig. S3 A). In the presence of 150 g/l Ficoll 70, however, sporadic short fibrils and granular aggregates were observed when rabbit PrP samples were incubated for 8 h (Fig. S3 B). Fig. 3, D–F, shows TEM images of bovine PrP samples incubated in diluted buffer or in the presence of 150–200 g/l Ficoll 70. In the absence of a crowding agent, the fibrils formed by the bovine PrP appear as a long, twisted, and branched structure after incubation for 16 h (Fig. 3 D), which is similar to those produced from the human PrP (28), but different from those produced from the rabbit PrP (Fig. 3 A) in the absence of a crowding agent. In the presence of 150 or 200 g/l Ficoll 70, however, abundant short fibrillar fragments and spherical or ellipsoidal particles were observed when bovine PrP samples were incubated for 10 h (Fig. 3, E and F), which is similar to those produced from the human PrP in the presence of 150 g/l Ficoll 70 (28). The above results indicated that macromolecular crowding also caused the bovine PrP to form short fibrils and nonfibrillar particles. Furthermore, the morphology of bovine PrP fibrils formed in crowded solutions is similar to that of PrPSc purified from scrapie-infected hamster brain (38) or bovine spongiform encephalopathy-affected cow brain (39). Together, we showed that a crowded physiological environment could play an important role in the pathogenesis of prion diseases by accelerating human/bovine PrP misfolding and inducing human/bovine PrP fibril fragmentation.
Figure 3.
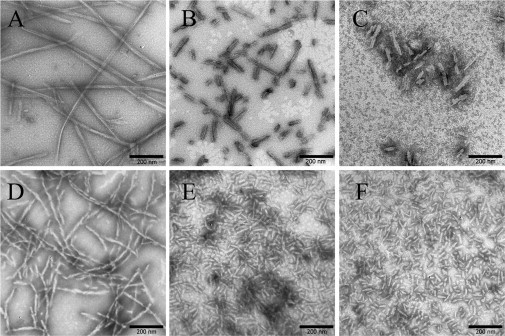
Transmission electron micrographs of rabbit/bovine PrP samples at physiological pH after incubation under different conditions. Rabbit (A–C) and bovine (D–F) PrP samples were incubated for 8 h (A–C), 16 h (D), or 10 h (E and F) in the absence of a crowding agent (A and D) and in the presence of 150 g/l Ficoll 70 (B and E) or 200 g/l Ficoll 70 (C and F), respectively. A 2% (w/v) uranyl acetate solution was used to negatively stain the fibrils. The scale bars represent 200 nm.
Comparison of the secondary structures of the rabbit/human/bovine PrP fibrils
Prion propagation involves the conversion of PrPC into PrPSc, shifting from a predominantly α-helical to β-sheet structure (40–42). Herein, FTIR was used to compare the secondary structures of rabbit/human/bovine PrP fibrils. The shape and position of amide I' (1600–1700 cm−1) of FTIR bands provide detailed information on the secondary structure of proteins (40,42), and amide I' peaks in the range of 1611–1630 cm−1 are characteristic for β-sheet formed by amyloid fibrils (43). The infrared spectra of rabbit/human/bovine PrP fibrils in the absence of a crowding agent were fitted using a combination of Gaussian peaks with maxima taken from second derivatives of FTIR spectra (Fig. S4, A–C), and the secondary structure contents were estimated by area of different peaks, which are summarized in Table 2. As shown in Table 2, the percentage of β-sheet in rabbit PrP fibrils was 37.5%, whereas those in human PrP fibrils and bovine PrP fibrils were 43.9% and 41.0%, respectively. The percentage of α-helix in rabbit PrP fibrils was 18.7%, whereas those in human PrP fibrils and bovine PrP fibrils were 12.5% and 15.1%, respectively. Clearly, amyloid fibrils formed by the rabbit protein contained more α-helix structure and less β-sheet structure than the human/bovine proteins.
Table 2.
Secondary structure composition of amyloid fibrils formed by rabbit/human/bovine PrPs as determined from deconvolution and curve fitting of the FTIR amide I' band
| Wavenumber(cm−1) | Secondary structure (%) |
|||
|---|---|---|---|---|
| Rabbit PrP fibrils | Human PrP fibrils | Bovine PrP fibrils | ||
| α-Helix | ∼1655 | 18.7 | 12.5 | 15.1 |
| β-Sheet | ∼1614–1630 | 37.5 | 43.9 | 41.0 |
| Turn | ∼1663–1689 | 17.1 | 23.4 | 25.5 |
| Random coil | ∼1645 | 26.7 | 20.2 | 18.4 |
CD spectroscopy was used to confirm the significant differences in secondary structures between rabbit PrP fibrils and human/bovine PrP fibrils. Fig. 4 shows the CD spectra of native rabbit/human/bovine PrP and rabbit/human/bovine PrP fibrils formed in the absence of a crowding agent. As shown in Fig. 4, A–C, in the absence of a crowding agent and before incubation, the CD spectra measured for rabbit/human/bovine PrP sample had double minima at 208 and 222 nm, indicative of predominant α-helical structure. After incubation for 12 and 16 h, respectively, a single minimum around 218 nm was observed for human PrP and bovine PrP samples (Fig. 4, B and C), which is typical of predominant β-sheet structure and a characteristic for amyloid formation. After incubation for 12 h, however, a single minimum around 210 nm (but not 218 nm) was observed for rabbit PrP fibril samples (Fig. 4 A), further supporting the conclusion reached by FTIR spectroscopy that rabbit PrP fibrils contained more α-helix structure and less β-sheet structure than human/bovine PrP fibrils. In other words, amyloid fibrils formed by the rabbit PrP had a remarkably different structure feature from those formed by the proteins from human and cow.
Figure 4.
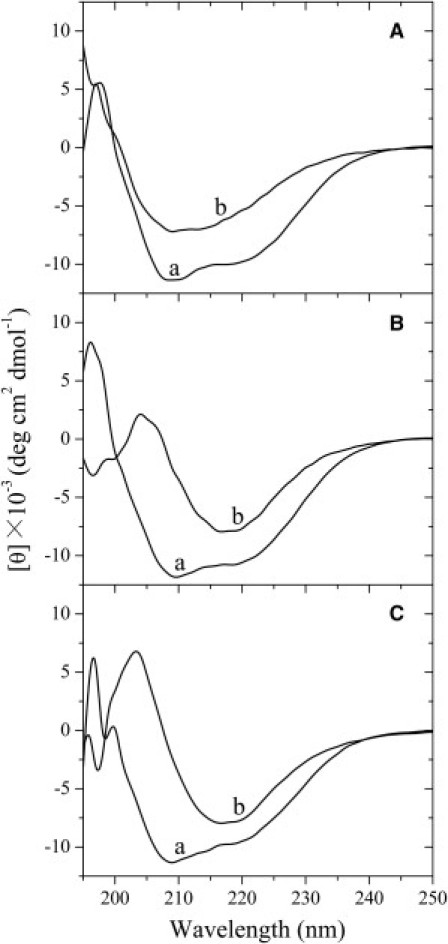
Secondary structural changes of rabbit (A), human (B), and bovine (C) PrP isoforms monitored by far-ultraviolet CD. Curve a: native rabbit/human/bovine PrPs in the absence of a crowding agent. Curve b: amyloid fibrils produced from rabbit, human, and bovine PrPs incubated for 12, 12, 16 h, respectively, in the absence of a crowding agent.
Comparison of proteinase K-digestion products of the rabbit/human/bovine PrP fibrils
PK resistance activity has been widely used to distinguish PrPC from PrPSc since the pioneering studies of Prusiner and co-workers (44,45). Limited exposure of PrPSc to PK digestion generates a fragment of 19–21-kDa encompassing residues ∼90–231, named PrP-27–30 (17,38,44–46). As shown in Fig. 5, B and C, and Fig. S5, B and C, amyloid fibrils produced from the human/bovine prion proteins generated PK-resistant fragments of 15–16-kDa after PK digestion for 1 h, which share the primary N-terminal sequence with PrP-27–30 type 2 but lacks the GPI anchor (46,47). As PK concentration or digestion time increased, the bands corresponding to such fragments became fainter. Human/bovine PrP fibrils also generated three short fragments (12-, 10-, and 8-kDa bands), which are similar to those of mouse PrP reported previously (18). By contrast, amyloid fibrils produced from the rabbit PrP did not generate a PK-resistant fragment of 15–16-kDa at all PK concentrations (or digestion time) examined, but generated two short fragments (11- and 9-kDa bands) (see Fig. 5 A and Fig. S5 A).
Figure 5.
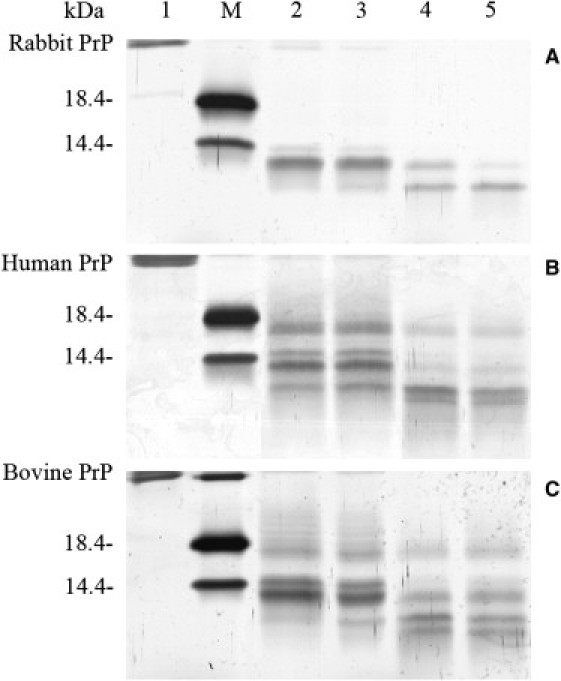
Concentration-dependent proteinase K-digestion assays of rabbit (A), human (B), and bovine (C) PrP fibrils. Samples were treated with PK for 1 h at 37°C at PK/PrP molar ratios as follows: 1:1000 (lane 2), 1:500 (lane 3), 1:100 (lane 4), and 1:50 (lane 5). PK concentration: 0.4 μg/ml (lane 2), 0.8 μg/ml (lane 3), 4 μg/ml (lane 4), and 8 μg/ml (lane 5). The controls with zero protease in the absence of a crowding agent were loaded in lane 1. Protein molecular mass markers were loaded on lane M: restriction endonuclease Bsp98 I (25.0 kDa), β-lactoglobulin (18.4 kDa), and lysozyme (14.4 kDa). Amyloid fibrils were produced from rabbit, human, and bovine PrPs incubated for 12, 12, and 16 h, respectively, in the absence of a crowding agent. Protein fragments were separated by SDS-PAGE and detected by silver staining.
Matrix-assisted laser desorption/ionization time-of-flight MS was used to examine PK-digestion products of rabbit/human/bovine PrP fibrils in the absence of a crowding agent. As shown in Fig. 6 A, amyloid fibrils formed by the rabbit PrP did not generate a PK-resistant fragment of 15–16-kDa, but generated several short fragments (11.43-, 10.98-, 10.58-, 9.02-, and 5.93-kDa bands). On the contrary, amyloid fibrils formed by the human and bovine proteins did generate such PK-resistant fragments of 15.42- and 15.71-kDa, respectively (Fig. 6, B and C).
Figure 6.
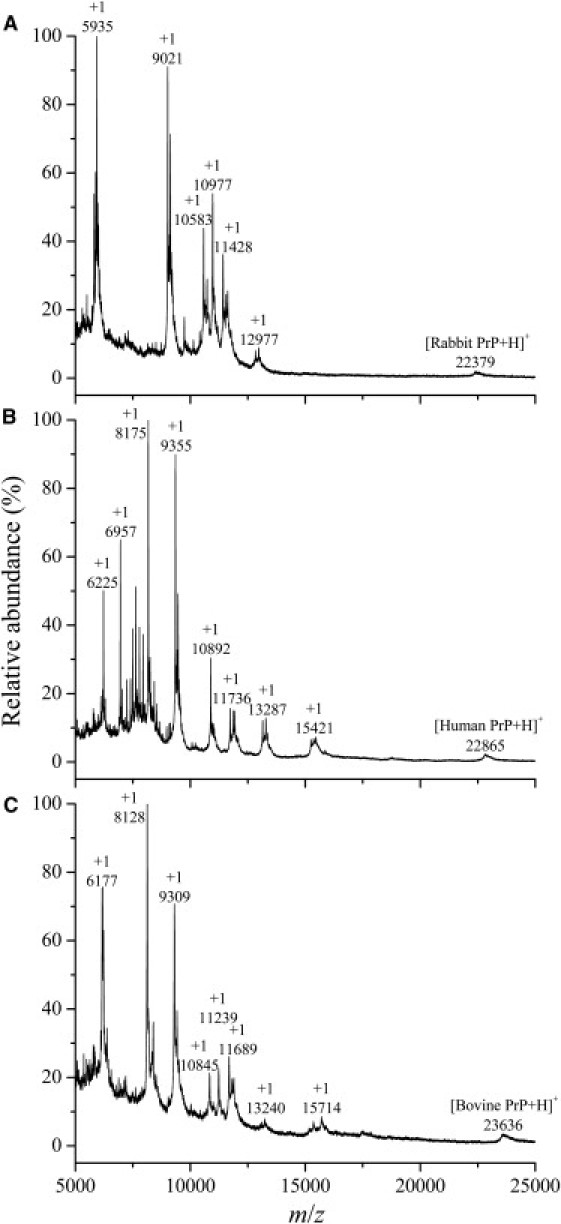
Matrix-assisted laser desorption/ionization time-of-flight mass spectra of proteinase K-digestion products of rabbit (A), human (B), and bovine (C) PrP fibrils. Amyloid fibrils, produced from rabbit, human, and bovine PrPs incubated for 12, 12, and 16 h, respectively, in the absence of a crowding agent, were treated with 4 μg/ml PK for 40 min at 37°C. Amyloid fibrils formed by rabbit PrP did not generate a PK-resistant core fragment of 15–16-kDa (A), but those formed by human and bovine PrPs generated such PK-resistant core fragments of 15.42-kDa (B) and 15.71-kDa (C), respectively.
Nano-liquid chromatography LTQ Orbitrap MS (ThermoFisher, Waltham, MA) was used to identify PK-digestion products of rabbit/human/bovine PrP fibrils in the absence of a crowding agent. By carefully analyzing the total ion chromatogram of PK-digestion products of rabbit PrP fibrils (Fig. S6 A), we once again showed that amyloid fibrils produced from the rabbit PrP did not generate a PK-resistant fragment of 15–16-kDa, but generated several short fragments, such as fragments encompassing residues 134–228 (Fragment 1, measured mass 11488.29 Da, theoretical mass 11489.68 Da) and 138–228 (Fragment 2, measured mass 11034.46 Da, theoretical mass 11036.14) (Fig. S6 B and Table S1). By contrast, three PK-resistant fragments encompassing residues 97–231 (Fragment 1, measured mass 15554.06 Da, theoretical mass 15554.40 Da), residues 99–231 (Fragment 2, measured mass 15337.85 Da, theoretical mass 15339.19 Da), and residues 92–231 (Fragment 3, measured mass 15961.41 Da, theoretical mass 15963.80 Da) for human PrP fibrils were identified by comparison of measured and theoretical masses (Fig. S7, A and B, and Table S1). Two PK-resistant fragments encompassing residues 108–241 (Fragment 1, measured mass 15355.91 Da, theoretical mass 15356.19 Da) and 102–240 (Fragment 2, measured mass 15821.91 Da, theoretical mass 15822.64 Da) for bovine PrP fibrils were identified (Table S1). In summary, amyloid fibrils formed by the rabbit PrP had remarkably different PK-resistant features from those formed by the proteins from human and cow.
Discussion
The inability of a protein to adopt its native and soluble conformation (protein misfolding) in its crowded physiological environment is the origin of an increasing number of human diseases such as CJD (22). The direct consequence of macromolecular crowding is that little space is left for additional macromolecules, which reduce their configurational entropy and therefore increase their free energy (24). As a consequence, protein misfolding is theoretically favored by macromolecular crowding (24). Experimentally, macromolecular crowding has been shown to accelerate amyloid formation of a wide range of proteins, such as human α-synuclein (31,32), human Tau protein (28), and human PrP (28). Two recent studies have suggested that macromolecular crowding can significantly accelerate protein aggregation and may play a significant role in aggregation-related diseases (48,49). This study demonstrated that macromolecular crowding significantly inhibited fibrillization of the rabbit PrP by stabilizing its native state, but dramatically accelerated fibril formation by the human/bovine PrP, indicating yet another biologically relevant system that is strongly influenced and distinguished by the crowding of inert macromolecules. Our additional experiments indicated that the two crowding agents had a similar impact on the structure of rabbit PrP monomers and aggregates (Fig. S8). Clearly, it is the crowding effect but not the direct PrP-crowder interactions that blocked fibril formation of the rabbit PrP.
For the resistance to prion diseases, some widely accepted generalizations are as follows.1), Laboratory animals such as mice, hamsters, and rats, are susceptible to a variety of prions, including those from sheep, humans, and cattle (8). 2), Rabbits are among the few animal species that exhibit resistance to prion diseases from different species (8,9). 3), Critical amino acid residues inhibiting PrPSc formation are located throughout the rabbit PrP sequence and the resistance of rabbits to prion diseases is due to multiple rabbit PrP-specific amino acid residues (9,10). The overall sequence homology between the rabbit PrP and the human/bovine PrP is 88.6%/87.5%, respectively, and only 22/28 amino acids are different in the mature PrP molecules between rabbit and human/cattle (9,10). Our data showed that in contrast to the proteins from human and cow, fibril formation of the rabbit protein was not accelerated by crowding agents. However, the onset fibril formation in the absence of a crowding agent was very similar. Therefore, the strong inhibition of fibril formation of the rabbit protein from the crowded physiological environment could be one of the reasons why rabbits are resistant to prion diseases. We then designed a novel (to our knowledge) triple mutant G99N/L137I/S173N of the rabbit PrP, in which three rabbit-specific amino acid residues (99G, 137L, and 173S) (10) were replaced by the corresponding amino acids of the human PrP. As shown in Fig. S9, the fibrillization kinetics of G99N/L137I/S173N differed greatly from that of wild-type rabbit PrP (Fig. 1 A) and macromolecular crowding did not inhibit but dramatically promoted fibril formation of such a mutant of the rabbit PrP. The above results demonstrated that the sequence did affect both the kinetics and the inhibitory effect of crowding agents on fibrillization of the rabbit PrP.
Besides the opposite influences of macromolecular crowding on fibril formation of the rabbit PrP and the human/bovine PrP, rabbit PrP aggregates and human/bovine PrP aggregates also show some differences in their secondary structures. As judged from FTIR, the percentages of β-sheet in human PrP fibrils and bovine PrP fibrils were 43.9% and 41.0%, respectively, which are similar to those of PrPSc purified from scrapie-infected hamster brain (43%) (42) and fibrils formed by recombinant full-length hamster PrP (40–44%) (50). However, the percentage of β-sheet in rabbit PrP fibrils was 37.5%. Clearly, amyloid fibrils formed by the rabbit PrP contained less β-sheet structure than the human/bovine/hamster PrPs. It should be mentioned that an in-register parallel β-sheet structure formed by the C-terminal end may be a general feature of PrP fibrils prepared in vitro (51–53). Most recently, Fernandez-Funez and co-workers (16) have found that the 15B3 conformational antibody does not recognize rabbit PrP aggregates in elder flies but hamster and mouse PrP accumulate 15B3-positive (PrPSc-like) conformers in elder flies. Taken together, these results suggest that compared with other mammalian PrP aggregates, rabbit PrP aggregates possess unique structural characteristics.
In this study, we demonstrate that under crowded physiological conditions, the amount of rabbit PrP aggregates is significantly less than that of human/bovine PrP aggregates. These results are consistent with recent data concerning rabbit/hamster/mouse PrP misfolding in vivo (16). The recent data have shown that the amounts of PrP aggregates in the elder flies expressing hamster PrP and mouse PrP are large and moderate, respectively, whereas the elder flies expressing rabbit PrP reveal only traces of PrP aggregates (16). Rabbit PrP does not induce spongiform degeneration and does not convert into scrapie-like conformers in transgenic flies (16), supporting the hypothesis that the rabbit PrP cannot be converted into the pathogenic PrPSc-like conformers spontaneously (10). It is known that if the process of PrPSc multiplication is slower than the degradation, PrPSc will be cleared throughout an animal's lifetime (54,55). In a crowded physiological environment, only traces of rabbit PrP aggregates are observed (16), likely due to the strong inhibition of rabbit PrP misfolding by the crowded environment (this work). Such a crowded environment, however, remarkably accelerates human/bovine PrP misfolding. Furthermore, we find that human/bovine PrP aggregates generate PK-resistant fragments of 15–16-kDa but rabbit PrP aggregates do not generate such a fragment at all PK concentrations or digestion time examined. We show that crowding alters reaction rates in different protein forms and that there are some structural differences in the fibrils. They are both contributors to the different kinetics of fibrillization. Together, our results suggest that the strong inhibition of fibrillization of the rabbit PrP by the crowded physiological environment and the difference in structure and PK-resistant feature between amyloid fibrils formed by the rabbit PrP and those formed by the proteins from human and cow could be two of the reasons why rabbits are resistant to prion diseases, and the different kinetics of fibrillization is the important effect to the biological phenomenon. It should be pointed out that PrPSc is a complicated assembly that contains the PrP core and other cofactors (6,56). These cofactors, which we shall study in detail in the future, could play an important role in the pathogenic function of human/bovine PrPSc and in the resistance of rabbits to prion diseases. Future issues also include: what is the reason for the poor infectivity of amyloid fibrils derived from recombinant PrPC (56)?
Acknowledgments
We sincerely thank Prof. Robert L. Baldwin (Stanford University School of Medicine) and Prof. Chih-chen Wang (Institute of Biophysics, Chinese Academy of Sciences) for their continuous encouragement to our work during the past several years. We thank Prof. Song-Ping Liang and Dr. Ji-Xian Xiong (College of Life Sciences, Hunan Normal University) and Dr. Li Li in this college for their technical assistances on MALDI-TOF-TOF MS and TEM, respectively.
The research of Y.L., Z.Z., X.Y., K.P., and J.C. is supported by National Key Basic Research Foundation of China (grants 2012CB911003 and 2006CB910301 to Y.L.), National Natural Science Foundation of China (grants 30970599 and 31170744 to Y.L.), and Fundamental Research Funds for the Central Universities of China (grant 1104006 to Y.L.).
Supporting Material
References
- 1.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguzzi A., Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- 3.Stöhr J., Weinmann N., Riesner D. Mechanisms of prion protein assembly into amyloid. Proc. Natl. Acad. Sci. USA. 2008;105:2409–2414. doi: 10.1073/pnas.0712036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguzzi A., Sigurdson C., Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu. Rev. Pathol. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- 5.Sandberg M.K., Al-Doujaily H., Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- 6.Soto C. Prion hypothesis: the end of the controversy? Trends Biochem. Sci. 2011;36:151–158. doi: 10.1016/j.tibs.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlow R.M., Rennie J.C. The fate of ME7 scrapie infection in rats, guinea-pigs and rabbits. Res. Vet. Sci. 1976;21:110–111. [PubMed] [Google Scholar]
- 8.Gibbs C.J., Jr., Gajdusek D.C. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science. 1973;182:67–68. doi: 10.1126/science.182.4107.67. [DOI] [PubMed] [Google Scholar]
- 9.Loftus B., Rogers M. Characterization of a prion protein (PrP) gene from rabbit; a species with apparent resistance to infection by prions. Gene. 1997;184:215–219. doi: 10.1016/s0378-1119(96)00598-7. [DOI] [PubMed] [Google Scholar]
- 10.Vorberg I., Groschup M.H., Priola S.A. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J. Virol. 2003;77:2003–2009. doi: 10.1128/JVI.77.3.2003-2009.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soto C. Constraining the loop, releasing prion infectivity. Proc. Natl. Acad. Sci. USA. 2009;106:10–11. doi: 10.1073/pnas.0811625106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sigurdson C.J., Nilsson K.P., Aguzzi A. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc. Natl. Acad. Sci. USA. 2009;106:304–309. doi: 10.1073/pnas.0810680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen Y., Li J., Lin D. Unique structural characteristics of the rabbit prion protein. J. Biol. Chem. 2010;285:31682–31693. doi: 10.1074/jbc.M110.118844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan M.Q., Sweeting B., Chakrabartty A. Prion disease susceptibility is affected by β-structure folding propensity and local side-chain interactions in PrP. Proc. Natl. Acad. Sci. USA. 2010;107:19808–19813. doi: 10.1073/pnas.1005267107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nisbet R.M., Harrison C.F., Hill A.F. Residues surrounding the glycosylphosphatidylinositol anchor attachment site of PrP modulate prion infection: insight from the resistance of rabbits to prion disease. J. Virol. 2010;84:6678–6686. doi: 10.1128/JVI.02709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez-Funez P., Zhang Y., Rincon-Limas D.E. Sequence-dependent prion protein misfolding and neurotoxicity. J. Biol. Chem. 2010;285:36897–36908. doi: 10.1074/jbc.M110.174391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bocharova O.V., Breydo L., Baskakov I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrPSc. J. Mol. Biol. 2005;346:645–659. doi: 10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 18.Bocharova O.V., Makarava N., Baskakov I.V. Annealing prion protein amyloid fibrils at high temperature results in extension of a proteinase K-resistant core. J. Biol. Chem. 2006;281:2373–2379. doi: 10.1074/jbc.M510840200. [DOI] [PubMed] [Google Scholar]
- 19.Lührs T., Zahn R., Wüthrich K. Amyloid formation by recombinant full-length prion proteins in phospholipid bicelle solutions. J. Mol. Biol. 2006;357:833–841. doi: 10.1016/j.jmb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Mo Z.Y., Zhu Y.Z., Liang Y. Low micromolar zinc accelerates the fibrillization of human Tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 2009;284:34648–34657. doi: 10.1074/jbc.M109.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu H.L., Fernández C., Liang Y. Quantitative characterization of heparin binding to Tau protein: implication for inducer-mediated Tau filament formation. J. Biol. Chem. 2010;285:3592–3599. doi: 10.1074/jbc.M109.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellotti V., Chiti F. Amyloidogenesis in its biological environment: challenging a fundamental issue in protein misfolding diseases. Curr. Opin. Struct. Biol. 2008;18:771–779. doi: 10.1016/j.sbi.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Ellis R.J. Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 2001;11:114–119. doi: 10.1016/s0959-440x(00)00172-x. [DOI] [PubMed] [Google Scholar]
- 24.Ellis R.J., Minton A.P. Protein aggregation in crowded environments. Biol. Chem. 2006;387:485–497. doi: 10.1515/BC.2006.064. [DOI] [PubMed] [Google Scholar]
- 25.Zhou H.X., Rivas G., Minton A.P. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou B.R., Liang Y., Chen J. Mixed macromolecular crowding accelerates the oxidative refolding of reduced, denatured lysozyme: implications for protein folding in intracellular environments. J. Biol. Chem. 2004;279:55109–55116. doi: 10.1074/jbc.M409086200. [DOI] [PubMed] [Google Scholar]
- 27.Du F., Zhou Z., Liang Y. Mixed macromolecular crowding accelerates the refolding of rabbit muscle creatine kinase: implications for protein folding in physiological environments. J. Mol. Biol. 2006;364:469–482. doi: 10.1016/j.jmb.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z., Fan J.B., Liang Y. Crowded cell-like environment accelerates the nucleation step of amyloidogenic protein misfolding. J. Biol. Chem. 2009;284:30148–30158. doi: 10.1074/jbc.M109.002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiao M., Li H.T., Liang Y. Attractive protein-polymer interactions markedly alter the effect of macromolecular crowding on protein association equilibria. Biophys. J. 2010;99:914–923. doi: 10.1016/j.bpj.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bokvist M., Gröbner G. Misfolding of amyloidogenic proteins at membrane surfaces: the impact of macromolecular crowding. J. Am. Chem. Soc. 2007;129:14848–14849. doi: 10.1021/ja076059o. [DOI] [PubMed] [Google Scholar]
- 31.Shtilerman M.D., Ding T.T., Lansbury P.T., Jr. Molecular crowding accelerates fibrillization of α-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- 32.Uversky V.N., M. Cooper E., Bowers K.S., Li J., Fink A.L. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 2002;515:99–103. doi: 10.1016/s0014-5793(02)02446-8. [DOI] [PubMed] [Google Scholar]
- 33.Hatters D.M., Minton A.P., Howlett G.J. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem. 2002;277:7824–7830. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 34.Baron G.S., Wehrly K., Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrPSc) into contiguous membranes. EMBO J. 2002;21:1031–1040. doi: 10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chesebro B., Race B., Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6:e1000800. doi: 10.1371/journal.ppat.1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chattopadhyay M., Durazo A., Valentine J.S. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. USA. 2008;105:18663–18668. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naiki H., Higuchi K., Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 38.Prusiner S.B., McKinley M.P., Glenner G.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 39.Hope J., Reekie L.J., Wells G.A. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature. 1988;336:390–392. doi: 10.1038/336390a0. [DOI] [PubMed] [Google Scholar]
- 40.Gasset M., Baldwin M.A., Prusiner S.B. Perturbation of the secondary structure of the scrapie prion protein under conditions that alter infectivity. Proc. Natl. Acad. Sci. USA. 1993;90:1–5. doi: 10.1073/pnas.90.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson G.S., Hosszu L.L., Collinge J. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science. 1999;283:1935–1937. doi: 10.1126/science.283.5409.1935. [DOI] [PubMed] [Google Scholar]
- 42.Pan K.M., Baldwin M., Prusiner S.B. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zandomeneghi G., Krebs M.R., Fändrich M. FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 2004;13:3314–3321. doi: 10.1110/ps.041024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolton D.C., McKinley M.P., Prusiner S.B. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 45.McKinley M.P., Bolton D.C., Prusiner S.B. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62. doi: 10.1016/0092-8674(83)90207-6. [DOI] [PubMed] [Google Scholar]
- 46.Notari S., Strammiello R., Parchi P. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J. Biol. Chem. 2008;283:30557–30565. doi: 10.1074/jbc.M801877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parchi P., Zou W., Chen S.G. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. USA. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin S., Zhou H.X. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys. J. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batra J., Xu K., Zhou H.X. Effect of macromolecular crowding on protein binding stability: modest stabilization and significant biological consequences. Biophys. J. 2009;97:906–911. doi: 10.1016/j.bpj.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ostapchenko V.G., Sawaya M.R., Baskakov I.V. Two amyloid States of the prion protein display significantly different folding patterns. J. Mol. Biol. 2010;400:908–921. doi: 10.1016/j.jmb.2010.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cobb N.J., Sönnichsen F.D., Surewicz W.K. Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc. Natl. Acad. Sci. USA. 2007;104:18946–18951. doi: 10.1073/pnas.0706522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu X., Wintrode P.L., Surewicz W.K. β-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc. Natl. Acad. Sci. USA. 2007;104:1510–1515. doi: 10.1073/pnas.0608447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tycko R., Savtchenko R., Baskakov I.V. The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel β-sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry. 2010;49:9488–9497. doi: 10.1021/bi1013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baskakov I.V. Branched chain mechanism of polymerization and ultrastructure of prion protein amyloid fibrils. FEBS J. 2007;274:3756–3765. doi: 10.1111/j.1742-4658.2007.05916.x. [DOI] [PubMed] [Google Scholar]
- 55.Peretz D., Williamson R.A., Prusiner S.B. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 56.Caughey B., Baron G.S., Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.