

Abstract
Time-resolved wide-angle x-ray scattering (TR-WAXS) is an emerging biophysical method which probes protein conformational changes with time. Here we present a comparative TR-WAXS study of native green-absorbing proteorhodopsin (pR) from SAR86 and a halogenated derivative for which the retinal chromophore has been replaced with 13-desmethyl-13-iodoretinal (13-I-pR). Transient absorption spectroscopy differences show that the 13-I-pR photocycle is both accelerated and displays more complex kinetics than native pR. TR-WAXS difference data also reveal that protein structural changes rise and decay an order-of-magnitude more rapidly for 13-I-pR than native pR. Despite these differences, the amplitude and nature of the observed helical motions are not significantly affected by the substitution of the retinal's C-20 methyl group with an iodine atom. Molecular dynamics simulations indicate that a significant increase in free energy is associated with the 13-cis conformation of 13-I-pR, consistent with our observation that the transient 13-I-pR conformational state is reached more rapidly. We conclude that although the conformational trajectory is accelerated, the major transient conformation of pR is unaffected by the substitution of an iodinated retinal chromophore.
Introduction
Since their discovery during gene sequencing of marine bacteria recovered from ocean seawater (1,2), proteorhodopsins (pR) have been recognized as light-driven energy-transducing integral membrane proteins closely related to the archeal bacteriorhodopsin (bR). Genes containing the pR sequence have been recognized throughout the world's oceans (1–3) and cell culture studies have demonstrated that pR plays an important physiological role in light-enhanced growth and survival during starvation (4,5). In combination with the wide prevalence of pR homologs in diverse marine bacteria and some marine eukaryotes, these findings suggest an important, yet until recently unsuspected role of pR in powering the ocean's ecology (3,6).
Proteorhodopsin cloned and overproduced from the Sar86 pR gene is the best characterized of this family of seven-transmembrane α-helical bacterial membrane proteins (7). A retinal molecule is covalently bound via a protonated Schiff base to Lys231 of helix G. After the absorption of a single photon the all-trans retinal rapidly isomerizes to adopt a 13-cis conformation, which induces a sequence of specific structural changes in the vicinity of the retinal that propagate with time throughout the protein. Several spectroscopic studies show that green-absorbing pR displays pH-dependent color changes and has a photocycle similar to that of bR, with comparable intermediates states (named PR, K, L, M, N, and O) but with a slightly slower turnover (8–13). As with bR, the primary proton transfer event occurs on a timescale of microseconds and proceeds from the protonated Schiff base to Asp97 (8–10), which is located immediately on the extracellular side of the retinal. Unlike bR, however, deprotonated retinal is rapidly reprotonated from Glu108 on the cytoplasmic side and a significant population of the M-intermediate (10), in which the retinal is deprotonated, only builds up at high pH (≥9). This difference has been suggested to be related to the absence of a proton-release complex comparable to Glu194/Glu204 found in bR, for which the equivalent residues are replaced with the hydrophobic Leu209/Leu219 (8).
Time-resolved wide-angle x-ray scattering (TR-WAXS), which was originally applied to study small molecules in solution (14,15), has recently been developed as a novel structural tool for probing protein structural dynamics in real time (16–18). This emerging biophysical method provides information about the number of distinct structural conformations and the rates at which these conformations form and decay within the sample. Moreover, unlike crystallographic methods (19), the nature of the protein conformational changes are not restricted by crystal lattice contacts. Because the extracted difference WAXS spectra correspond to the global protein conformational changes recorded from an ensemble of randomly oriented protein molecules, the information content recorded is lower than that accessed using crystallographic methods. Despite this limitation, significant progress has been made in developing analytical tools for structural refinement of tertiary and secondary structural movements against TR-WAXS difference spectra (17,20). In comparison with advanced spectroscopic methods such as time-resolved Fourier-transform infrared (12) or electron paramagnetic resonance (21) spectroscopy, TR-WAXS has the key advantages that it directly probes protein conformational changes over timescales from picoseconds (18) to seconds without the need to introduce labels that potentially affect the protein's conformations (21). On the other hand, TR-WAXS cannot yet provide local structural information concerning specific amino-acid residues, which (under favorable circumstances) can be achieved using Fourier-transform-infrared spectroscopy (22). It has been proposed to use site-specific heavy-atom labeling to simultaneously record both local and global conformational changes in proteins using TR-WAXS (23).
bR and pR were the first integral membrane proteins to be characterized using TR-WAXS (20). Significant structural rearrangements of α-helices were observed in bR before the primary proton transfer step from the Schiff base to Asp85, and the transition from an intermediate to the late conformational state was regarded as a smooth structural evolution driven by the deprotonation of the retinal Schiff base (20). In contrast, the structural evolution of pR could be described by only one major structural component over the time-window from 20 μs to 100 ms, with additional structural states indicated for shorter timescales (20).
In this study, we chemically modify the retinal chromophore in pR and use TR-WAXS to characterize the resulting perturbations to the evolution of structural changes over time. Specifically, retinal in native pR is replaced with a halogenated derivative whereby the retinal's C-20 methyl group (which is attached to C-13 of the retinal) is substituted with an iodine atom to create 13-desmethyl-13-iodoretinal (13-I-pR). This choice of substitution derives from its relative ease of synthesis and the widely held view that steric interactions of the retinal's C-20 methyl group with the protein play a crucial role in the coupling of retinal isomerization to protein conformational changes (24–26).
Our findings show that the pR photocycle is more dramatically perturbed by this substitution than is the case for bR, with the photocycle kinetics accelerated by an order of magnitude relative to native pR, as compared to only a twofold acceleration for the same substitution in bR (27). TR-WAXS also reveals that the protein's conformational changes are accelerated by an order of magnitude for 13-I-pR relative to native pR yet, perhaps surprisingly, the transient conformational state of 13-I-pR adopts the same structure as the transient conformation of native pR. These accelerated conformational changes were interpreted in light of free energy differences calculated for pR with the two retinal forms. We conclude that although iodination of the retinal chromophore leads to significant changes in the chemical, absorption, and kinetic properties of pR, the nature of the primary conformational changes undertaken by the protein is preserved.
Materials and Methods
Protein expression, reconstitution, and purification
Proteo-opsin from the SAR86 clade, containing a C-terminal His6 tag for purification purposes, was heterologously expressed in Escherichia coli UT5600 strain using the pBAD-TOPO (Invitrogen, Carlsbad, CA) plasmid (7). Chromophore reconstitution was performed at the stage of cell lysis by addition of either all-trans retinal (yielding native pR) or 13-desmethyl-13-iodoretinal (yielding 13-I-pR) to the apo-protein membrane. Native pR and 13-I-pR were subsequently solubilized and purified in n-Octyl-β-D-Glucopyranoside (β-OG) using immobilized metal ion chromatography and ion exchange chromatography. Absorption spectra were recorded for solubilized native pR or 13-I-pR (25 mM KPi, 1% w/v β-OG) using a model No. ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) at pH 7.0 and pH 9.0. See the Supporting Material for details on the synthesis of 13-desmethyl-13-iodoretinal and sample preparation.
Transient absorption spectroscopy
Flash-induced absorptions transients of 25 μM native pR or 13-I-pR (25 mM KPi, pH = 9.0, 1% w/v β-OG) were acquired at room temperature (23°C) in the microsecond-to-millisecond time domain on a TDS 3032 digital oscilloscope (Tektronix, Portland, OR) using a previously described flash photolysis system and a 532 nm Nd:YAG laser (6 ns, 5 mJ/pulse, 1 Hz repetition rate; Spectra-Physics, Houston, TX) with 32 transients averaged at 500 and 610 nm and 128 transients averaged at 410 nm (28). The single-wavelength transients were modeled as single exponentials using the software Origin (OriginLab, Northampton, MA).
WAXS data collection and processing
Time-resolved WAXS data were recorded for 0.05 Å−1 < q < 2.2 Å−1 at room temperature (23°C) using a pump-probe data collection scheme at the dedicated time-resolved beamline ID09B at the European Synchrotron Radiation Facility as previously described (20,29). The concentrated (15 mg/mL protein, 25 mM KPi, pH = 9.0, 1% w/v β-OG) native pR or 13-I-pR samples were mounted in 0.8 mm diameter quartz capillaries on a motorized translational stage and were photoactivated using a q-switched 527 nm ND:YLF laser (125 μJ/pulse, DM50; Photonics Industries, Bohemia, NY) focused to a spot of 0.26 × 0.27 mm2 (full width at half-maximum). The polychromatic x-rays (ΔE/E = 3%, Ef = 17.5 keV, spotsize of 60 × 120 μm2), directed perpendicular to the laser, were scattered onto a Mar133 detector (Marresearch, Norderstedt, Germany) located 250 mm from the sample position. X-ray pulses of 2 μs (Δt = 2 μs and 20 μs) and 20 μs (Δt = 20 μs, 65 μs, 200 μs, 650 μs, 2 ms, 20 ms, and 100 ms) duration were isolated from the 7/8 bunch mode using a high-speed rotating chopper (30).
For each image the detector was exposed for either 300 (2-μs pulse duration) or 30 (20-μs pulse duration) x-ray pulses with the sample position translated between each pump-probe cycle. Additional dark images were collected as a reference at the start and the beginning of each time series. The WAXS images were integrated in rings using the data analysis program Fit2D (31) to yield the radial intensity curves, S(q,Δt), expressed as a function of the magnitude of momentum change of the deflected beam (q = 4πsinθ/λ, where 2θ is the angle of the deflected beam and λ is the x-ray wavelength) and the time-delay (Δt) after photoactivation. Each resulting curve was normalized in the region of 1.5–1.7 Å−1 (16,20) before subtracting the normalized dark curve, which was the average of the two images flanking the time-series, yielding the WAXS difference intensity curves ΔS(q,Δt) (laser-on minus laser-off; and see Fig. S2 in the Supporting Material). Each time point was typically averaged for 12–130 images in order to improve the signal/noise ratio.
Photoactivation unavoidably deposits heat into the sample causing a mixing of the structural signal with that of a thermal signal (16,18,20). The thermal signal for pR has previously been characterized through measurements at the Swiss Light Source (located at the Paul Scherrer Institute, Villigen, Switzerland) using a rapid readout x-ray camera (32). This signal was convoluted with the undulator spectrum used in the TR-WAXS experiments and then used to subtract the heating contribution from each individual time point to recover the pure difference scattering intensities. For consistency, the same thermal signal was used to remove the heating contribution for both native pR and 13-I-pR (see Fig. S3).
Singular value decomposition of the time-resolved difference WAXS data
Each dataset was arranged in a m × n matrix consisting of difference-scattering intensities recorded at m number of q-values (m = 674) and n time points (n = 8) and decomposed as
| (1) |
where U(q) is a m × n matrix constituting the orthonormal basis spectra (eigenvectors) of ΔS, Sv is an n × n diagonal matrix indicating the contribution of the basis spectra (singular values) to ΔS, and V(Δt) is an n × n matrix describing the time-dependent variation of the basis spectra (amplitudes of the eigenvectors) (33). The number of signal-containing basis spectra in each of the datasets was determined based on inspection of the eigenvectors, the magnitude of their singular values, and calculation of autocorrelation values of each basis spectra (see Fig. S4 and Fig. S5). The time-dependent amplitudes of each basis spectra were modeled using a single exponential rise followed by a single exponential decay for native pR, and a single exponential decay with an offset for 13-I-pR using the software Origin (OriginLab). The offset for 13-I-pR was required due to the fact that the higher level of noise associated with the last two time delays (Δt = 20 and 100 ms) was fitted as a small-amplitude, yet nonzero, SVD component.
Structural refinement
In the absence of a crystallographic structure of pR, a homology model of pR based on the bR structure with an homology score of 0.27 (based on the BLOSUM40 scoring matrix) was used for structural refinement (34). The resting state homology model and the best-fit transient conformation model, identified using rigid body modeling (20), were inserted into an equilibrated preformed micelle consisting of 125 β-OG detergent molecules. Each protein-micelle system was solvated using 25,000 tip3p water molecules and two sodium counterions were used to make the system electrically neutral before energy minimization (400 steps, conjugate gradient method). Three-nanosecond molecular dynamics simulations were performed, using harmonic restraints on the backbone atoms (fc = 1000 kJ mol−1 nm−2). For each resting state and transient conformational state trajectory, 10-ps-spaced snapshots of the protein were extracted and the theoretical scattering for these structures was analyzed pairwise.
Before calculating the theoretical scattering, each pair of structures was placed in the same representative micelle to prevent artificial effects from dominating the difference scattering due to transient differences in the micelle shape. The absolute theoretical scattering was calculated for each resting state (Sig) and transient conformational state (Sit) structure using the program CRYSOL (35). In order to account for the use of a polychromatic beam in the experiment, predicted WAXS spectra were convoluted with the measured undulator spectrum (ΔE/E = 3%) (36). The theoretical difference scattering (ΔSi) was in turn calculated as
| (2) |
where i = 1–300 corresponds to the number of structure pairs and the exponential term, analogous to the Debye-Waller factor used in crystallography, was introduced to account for the highly disordered nature of the micelle surrounding the protein. The value of the B-factor (200 Å2) was extracted from the trajectories based on the root-mean square fluctuation (RMSF) of the detergent molecules.
To evaluate the experimental differences against those from the molecular dynamics trajectories, the following error weighted χ2-value (37) was used as
| (3) |
where ΔSi(q) is the theoretical scattering for structure pair i, ΔSdata(q) is the experimental basis spectrum from singular value decomposition (SVD), σq is the standard deviation (noise) in each q-point of the data as compared against the same basis spectrum after filtering, N is the number of data points included in the fitting range, and M is the number of parameters used for the fitting. The data were modeled over the q-range 0.2–1.0 Å−1 (N = 353) while allowing for an overall scaling factor and a limited offset between the data and theory (M = 2). For the top solutions, based on minimizing χi2, for native pR and 13-I-pR the displacement of the Cα-atoms between the resting state and the transient conformational state was calculated, after superposition, using the program LSQMAN (38). See the Supporting Material for rigid body definitions, force-field parameters, simulation details, and free energy calculations.
Results and Discussion
Spectral characterization and kinetics
Retinal and halogenated retinal were successfully incorporated into E. coli overexpressed His-tagged green-absorbing proteo-opsin (apoprotein lacking the chromophore) and reconstituted and purified in β-octylglucoside to form the solubilized native pR and 13-iodoretinal pR (13-I-pR). The replacement of a methyl group with an iodine atom at the C-13 retinal position resulted in a red spectral shift of 23–26 nm (Fig. 1 A). Moreover, both pR and 13-I-pR underwent a ∼20 nm spectral blue shift when the pH was increased from 7.0 to pH 9.0 (9,10,39). These findings are consistent with earlier studies on bR (27) and indicate that the protonation state of the retinal and all key groups in the local vicinity of the Schiff base are not significantly affected by this substitution at the C-13 position. A potential complication is that the pH dependence, photocycle kinetics, and sequence of proton transfers may be affected by the presence of a His6 tag (12). We therefore chose to perform all time-resolved spectroscopy and TR-WAXS measurements at pH 9.0, which mimics the alkaline pH in oceanic seawater and for which it has been shown that Asp97 is deprotonated in the resting conformation of His-tagged pR at this pH (40–42).
Figure 1.
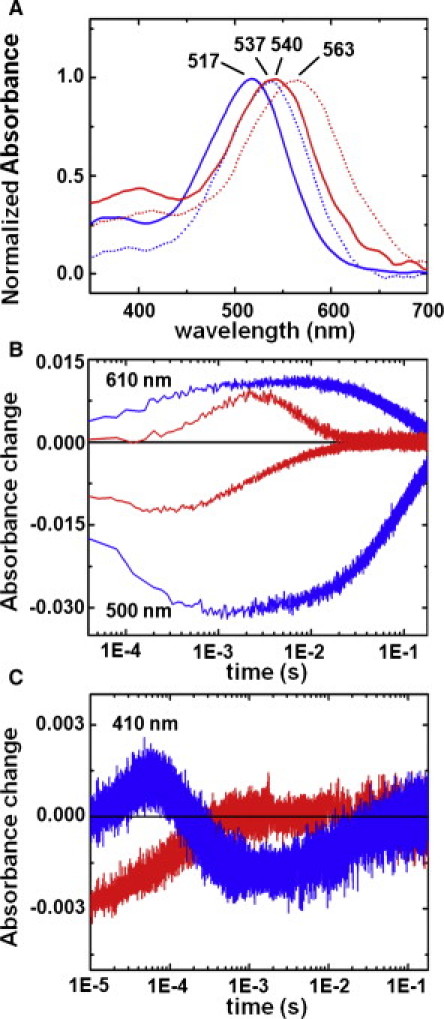
(A) Optical absorption spectra of native pR and 13-I-pR. The absorption spectra of pR (blue) and 13-I-pR (red) at pH 7.0 (dotted lines) and pH 9.0 (solid lines) are shown with the corresponding absorption maxima indicated. (B and C) Single wavelength transient absorption changes. Transient absorption changes at pH 9.0 (23°C) for native pR (blue) and 13-I-pR (red) collected at (B) 500 and 610 nm and (C) 410 nm after photoactivation by a 532 nm laser (6 ns, 5 mJ/pulse).
Time-resolved spectroscopic studies of the photocycle turnover of the native and 13-iodoretinal variants showed that the rate of recovery of the resting state (pR, measured at 500 nm) and the decay of the final O-intermediate (measured at 610 nm) is approximately an order-of-magnitude faster in 13-I-pR as compared to pR (Fig. 1 B, and see Table S1 in the Supporting Material). This perturbation is significantly greater than that observed when bR retinal is substituted with 13-iodoretinal (27) for which the timescales were accelerated only twofold. More strikingly, however, the growth, plateau, and decay of differences at 500 nm and 610 nm follow almost exactly the same temporal dependence in native pR (98% negative correlation), suggesting that the changes in the transient populations of the PR and O states are almost perfectly complementary over this time window.
In contrast, these timescales separate in 13-I-pR (77% negative correlation), with the absorption minima at 500 nm being reached after ∼200 μs and the absorption maxima at 610 nm occurring an order-of-magnitude later. Transient absorption spectra recorded at 410 nm (Fig. 1 C) indicate that a small population of the M-intermediate of native pR (for which the retinal is deprotonated) reaches a maximum after ∼50 μs and rapidly decays. In contrast, no appreciable population of the M-intermediate accumulates for 13-I-pR, as is the case for native pR at lower pH (9,43). From these time-dependent absorption spectroscopy observations it is apparent that a more complex photochemistry arises for 13-I-pR than native pR.
Time-resolved WAXS difference data
Representative difference WAXS spectra ΔS(q,Δt) (after the removal of the thermal signal) are shown in Fig. 2 as a function of the time-delay Δt between photoexcitation and the arrival of the x-ray probe. Without any further analysis it is apparent that the amplitude of the difference signal ΔS(q,Δt) is stronger at Δt = 2 μs for the modified retinal case (Fig. 2 B) when compared to the corresponding signal from the native protein (Fig. 2 A). Similarly, for the two later time points (Δt = 20 ms, 100 ms) the difference signal due to protein conformational changes is not visible above noise for 13-I-pR, whereas a significant structural component was clearly visible at Δt = 20 ms and even persisted until Δt = 100 ms for native pR.
Figure 2.
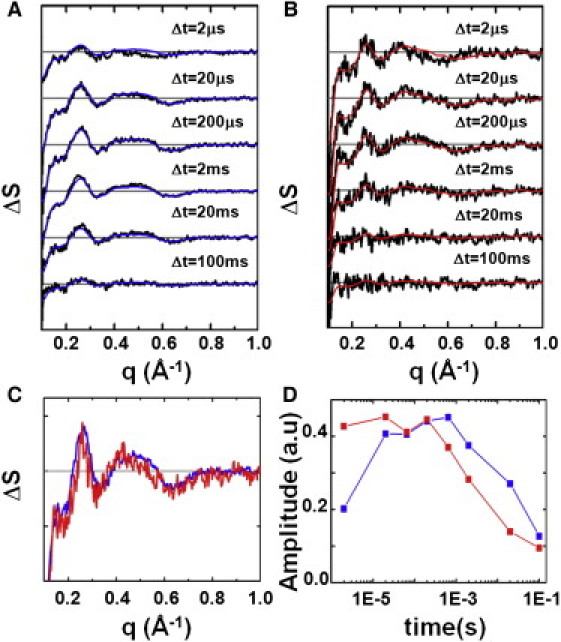
Time-resolved difference WAXS data and spectral decomposition. Time-resolved difference WAXS data collected at pH 9.0 (23°C) for (A) native pR and (B) 13-I-pR for the representative time delays after laser excitation. All curves have had the solvent response due to sample heating removed. (Blue and red solid lines) Each time-delay recalculated using only the major component from SVD analysis (see Materials and Methods). (Gray line) ΔS(q,Δt) = 0. Basis spectra (C) and amplitudes (D) of the major signal containing component from SVD for native pR (blue) and 13-I-pR (red).
Singular value decomposition of these data quantified the experimental differences in the structural response of the protein with time (33). For each of the pR and 13-I-pR datasets one signal-containing component (Fig. 2 C) was identified based on singular vectors, singular values, and autocorrelations (see Fig. S4 and Fig. S5). The fact that a single component is sufficient to describe these data from 2 μs < Δt < 100 ms is also seen from the agreement between the individual time points and those recalculated using only these basis spectra, singular values, and amplitudes of the major component (Fig. 2, A and B, solid blue and red lines).
The temporal dependence of the light-induced conformational changes in pR can be seen in Fig. 2 D, which overlays the amplitudes of the major components extracted from TR-WAXS difference data for native pR and 13-I-pR with time. From these results it is observed that, over the temporal window sampled in this study, the pR photocycle contains one transient conformational state that builds up and decays with timescales of 2 μs and 49 ms, respectively (see Table S1). When the C-20 retinal methyl group is chemically substituted by an iodine atom, these rates are significantly accelerated such that, by Δt = 2 μs, 13-I-pR had reached a maximum population of its transient conformational state, an order-of-magnitude faster than that observed for the native pR, and that this conformation decays on a timescale of 2 ms (see Table S1). That the structural changes within pR are accelerated when the retinal is iodinated is consistent with conclusions drawn from spectroscopic analysis (Fig. 1 B, and see Table S1).
Structural refinement
In contrast with the observation that TR-WAXS could observe very clear differences in the rates of the rise and decay of the major structural components it is striking that the basis spectra derived from single value decomposition are almost identical for both samples of pR within the observed signal/noise (Fig. 2 C). This apparent similarity is quantified by a specific correlation of 91% between the two basis spectra (see the Supporting Material), indicating that the nature of the structural changes occurring within pR are not significantly affected by the substitution of the C-20 methyl group with iodine.
Secondary conformational changes were modeled against the basis spectra extracted by SVD analysis using rigid movements of a limited number of possible conformational changes (cytoplasmic regions of helices E, F, and G (44) and the cytoplasmic region of helix C (45)) extracted from crystallographic studies of bR, as previously described in Andersson et al. (20). These structural assumptions are supported by solid-state NMR studies of green-absorbing pR that show an increased mobility in helices C, F, G, and the EF-loop (46). Moreover, studies of conformational changes in visual rhodopsin have also identified movements in these helices (47–49). In this work we extend this protocol by introducing molecular-dynamics simulations (17) to sample conformational space about the best-fit model identified using rigid body motions (20). We also extended the q-domain over which we previously performed the structural minimization and performed the minimization against an error-weighted difference (37) (Eq. 3) rather than an R-factor (17).
Somewhat counterintuitively, the introduction of a strongly scattering iodine atom onto the retinal only affects the difference x-ray scattering to a minor extent (23). This is illustrated in Fig. S6, which overlays the difference WAXS curve calculated between the resting and transient conformational states assuming identical protein structures but with one structure pair having an iodine atom at the retinal's C-13 position. This may be understood because, unlike the case of two- (27) or three-dimensional diffraction, there is no regular lattice which amplifies the contribution of the single heavy atom when using WAXS.
Fig. 3 A overlays the predicted WAXS differences resulting from the best-fit structural model for native pR overlaid upon the experimental difference WAXS basis spectra. A similar representation of the results of structural refinement is given in Fig. 3 B for 13-I-pR. Best-fit models are compared in Fig. 3 C, where the displacement of the protein backbone Cα-atoms between the resting state and the transient conformational state are overlaid, and the final α-helix movements are represented as Fig. 3 D. As was indicated above from the overlay of the difference basis spectra extracted from SVD analysis (Fig. 2 C), the secondary structural changes in pR are almost indistinguishable for the two models. Thus we conclude that the introduction of an iodine atom on the retinal C-20 position does not significantly perturb the global conformational changes that the protein undergoes during its photocycle.
Figure 3.
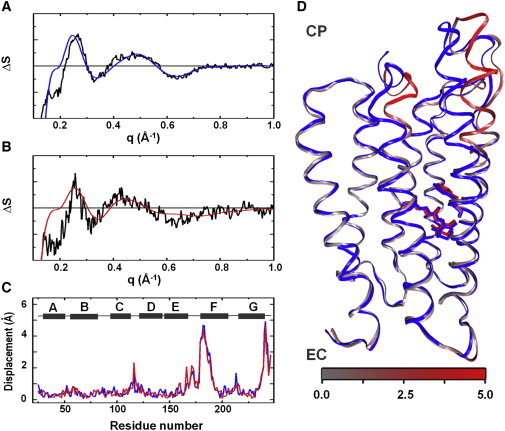
Structural modeling of WAXS data. Overlay between the basis spectrum from SVD (black) and the optimal theoretical difference WAXS curve (blue, red) from structural refinement for (A) native pR and (B) 13-I-pR. (C) Displacement plot for Cα-atoms between the resting state and the transient conformation as a function residue number for the optimal models of native pR (blue) and 13-I-pR (red). Helix locations (gray) are represented as a function of residue number (above). (D) Overlay of the resting state homology model (blue) and the refined transient conformation model of native pR. The transient conformation is colored gray to red depending upon the amplitude of the movement on Cα atoms. The all-trans and 13-cis retinal conformations and the Trp197 are represented as sticks. EC, Extracellular side; CP, cytoplasmic side.
Free energy estimates and the accelerated conformational transition of 13-I-pR
Free energy differences (ΔG) associated with the substitution of 13-iodo-retinal into pR were estimated using molecular-dynamics simulations, with the retinal in both the all-trans and 13-cis configuration. These were computed using an alchemical transformation of the methyl group at C-20 into an iodine atom, employing the technique of thermodynamic integration (see the Supporting Material). For each of the retinal states this transformation was conducted with the retinal inside pR (yielding ΔGpR) and with the retinal in vacuum (yielding ΔGvac). ΔΔG, defined as the difference between ΔGpR and ΔGvac, estimates the impact of the alchemical transformation on the surrounding protein matrix, where a large positive (negative) ΔΔG indicates that the transformation destabilizes (stabilizes) the protein conformation. To avoid conformational instabilities during long-trajectories, position restraints were applied on the protein backbone during these simulations. Because the calculated ΔΔG values depend upon the force constant fc of the backbone position restraints, ΔΔG values should be interpreted only qualitatively. To test for bias in the specific choice of fc, we have computed ΔΔG using a wide range of fc values between 200 and 1000 kJ/mol.
Our calculations suggest that the 13-iodoretinal substantially destabilizes the 13-cis state of pR when compared to wild-type retinal by ∼10–20 kJ/mol (see Fig. S7) and these qualitative findings are independent of the choice of harmonic restraints (fc). Assuming that the transition state along the path toward the transient conformational state is less affected by the introduction of 13-iodoretinal, then these findings imply that structural transitions after the trans-cis isomerization would be accelerated by an iodinated retinal, in qualitative agreement with the experimental findings (Fig. 2 D).
In contrast, the stability of the all-trans state is hardly affected by the presence of the iodine. These conclusions may be understood by reference to Fig. 4, which illustrates that the iodine atom is bulkier than the methyl group. In the all-trans state (Fig. 4, A and C), relaxation of the retinal helps to avoid a steric clash of the iodine atom with the adjacent side chains of residues Trp197 and Leu105. In 13-cis (Fig. 4, B and D), however, a similar structural relaxation is impeded by the kink in the retinal, leading to an unfavorable steric interaction between the iodine and the Trp197 and Leu105 residues and consequently an increase in the free energy. It is generally believed that steric interactions of the retinal's C-20 methyl group with the neighboring aromatic residues (e.g., Trp197 in pR; Trp186 in bR) is crucial for coupling the retinal isomerization to protein conformational changes (24–26). Our findings, where this methyl group has been substituted with a bulkier iodine atom causing accelerated light-induced structural changes in pR, support this viewpoint.
Figure 4.
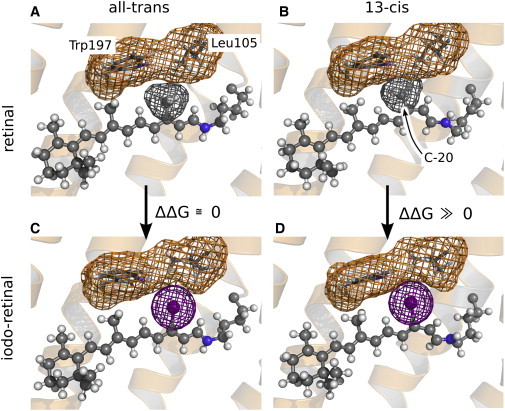
Native retinal (A and B) and iodoretinal (C and D), either in the all-trans (A and C) or in the 13-cis state (B and D). The retinal is shown in ball-and-sticks representation, and the side chains of residues Leu105 and Trp197 as sticks. The van der Waals surface of the C-20 methyl group, the iodine, and of Leu105/Trp197 is indicated (gray, purple, or orange mesh, respectively). (B and D) In 13-cis, replacing C-20 by iodine induces a steric clash with Leu105 and Trp197, leading to an unfavorable increased free energy of the protein conformation. (A and C) In all-trans, that replacement has little effect on the free-energy of the conformation. For ease of understanding, we refer to the 13-I-pR retinal conformation as all-trans and 13-cis in analogy to that used for describing native pR, although (strictly speaking) the opposite nomenclature should be applied for the 13-I-pR retinal.
Steric effects and the accelerated conformational relaxation of 13-I-pR
TR-WAXS (Fig. 2) and time-resolved spectroscopy (Fig. 1) both demonstrate that 13-I-pR completes its photocycle an order-of-magnitude more rapidly than native pR. This may be understood by reflecting that for bR the replacement of Leu93 (corresponding to Leu105 of pR) with an alanine residue greatly slows the recovery of the resting state conformation (50). In that work it was argued that steric interactions with Leu93 in bR restrict the flexibility of the retinal and this accelerates the retinal reisomerization back to the all-trans conformation. Similarly, it can be argued that the rate of relaxation back to the resting state (Fig. 1 B and Fig. 2 D) is faster in 13-I-pR than native pR because the van der Waals contacts of the iodine atom with Leu105 are significantly stronger for the isomerized 13-iodoretinal (Fig. 4 D).
Structural implications
Structural analysis of the difference WAXS data show that both the number of transient conformational states (Fig. 2) and their structures (Fig. 3) are not affected by the substitution of 13-iodoretinal into pR. Thus the replacement of the C-20 retinal methyl group by a bulkier iodine atom does not drive the protein into a new conformational state. Instead, we observe that the underlying structural mechanism of the pR photocycle tolerates the increased volume of the retinal without any significant structural consequences. We are thus led to the conclusion that it is subtly balanced interactions within the protein rather than the bulk of the retinal molecule which determines the end point of pR's major transient conformational state.
Because one major transient conformational state is sufficient to describe both the pR and 13-I-pR photocycles (Fig. 2), then differences in their transient absorption properties do not arise from large-scale conformational differences between the native and 13-I-pR forms. Instead, perturbations in the rates at which transient spectral features rise and decay must be due to local differences in the vicinity of the modified retinal and specific chemical differences between the two retinal forms. Thus we are led to an understanding whereby proteorhodopsin rapidly adopts one predominant transient conformation, and it is upon the scaffold defined by this state that the proton exchange steps occur. This picture is consistent with the picture which emerged from TR-WAXS studies of bR, where TR-WAXS data showed that approximately two-thirds of the protein's global conformational change occurs before the primary proton transfer event (20) and only the amplitude of the helix movements increase during the latter part of the bR photocycle.
Conclusions
In this work a comparative TR-WAXS study was performed which analyzed the transient conformational dynamics of native proteorhodopsin and iodoretinal substituted proteorhodopsin. The 13-iodoretinal substitution caused the pR absorption maxima to be red-shifted by ∼20 nm and accelerated the photocycle absorption kinetics by an order of magnitude. TR-WAXS revealed that the protein conformational changes were also accelerated by an order of magnitude upon the modified retinal substitution. Despite this significant change in conformational dynamics, TR-WAXS data demonstrated that the dominant transient conformational state of 13-I-pR had the same structure as that of the corresponding conformation of native pR. Thus, although changes to the retinal chromophore caused significant changes in the chemical, absorption, and kinetic properties of pR, the nature of the primary conformational changes undertaken by the protein was preserved.
These findings further develop the emerging biophysical method of TR-WAXS by establishing that the technique is powerful enough to accurately characterize similarities and differences for very closely related protein samples. Moreover, the successful labeling using 13-iodoretinal is an important step in realizing the proposal (23) that TR-WAXS, when used in combination with site-specific heavy atom labels, could simultaneously probe both local and global conformational changes. This bodes well for future comparative studies of protein structural dynamics using TR-WAXS.
Acknowledgments
We thank R. Birge for providing coordinates for a model of pR developed from homology with bR and S. Hayashi for providing us with the AMBER topology files for the retinal. Time resolved WAXS experiments were performed on the ID09B beamline at the European Synchrotron Radiation Facility (ESRF), Grenoble, France, and heating characterization on the cSAXS beamline at the Swiss Light Source (SLS), Villigen, Switzerland.
E.M. and Z.O. were supported by the Human Frontier Science Program and J.S.H. and S.W. were supported through a Marie Curie Intra-European fellowship within the 7th European Community framework program. Financial support from the Swedish Research Council and the Harald and Greta Jeanssons Foundation is also gratefully acknowledged.
Footnotes
Ziad Omran's present address is School of Pharmacy, University of Nottingham, University Park, Nottingham, UK.
Magnus Anderson's present address is Department of Physiology and Biophysics, University of California at Irvine, Irvine, CA.
Marco Cammarata's present address is Linear Coherent Light Source, Stanford Linear Accelator Center, Menlo Park, CA.
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Béjà O., Aravind L., DeLong E.F. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science. 2000;289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- 2.Béjà O., Spudich E.N., DeLong E.F. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411:786–789. doi: 10.1038/35081051. [DOI] [PubMed] [Google Scholar]
- 3.de la Torre J.R., Christianson L.M., DeLong E.F. Proteorhodopsin genes are distributed among divergent marine bacterial taxa. Proc. Natl. Acad. Sci. USA. 2003;100:12830–12835. doi: 10.1073/pnas.2133554100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gómez-Consarnau L., González J.M., Pinhassi J. Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature. 2007;445:210–213. doi: 10.1038/nature05381. [DOI] [PubMed] [Google Scholar]
- 5.Gómez-Consarnau L., Akram N., Pinhassi J. Proteorhodopsin phototrophy promotes survival of marine bacteria during starvation. PLoS Biol. 2010;8:e1000358. doi: 10.1371/journal.pbio.1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keeling P.J., Slamovits C.H., James E.R. A bacterial proteorhodopsin proton pump in marine eukaryotes. Nat. Commun. 2011;2:183. doi: 10.1038/ncomms1188. [DOI] [PubMed] [Google Scholar]
- 7.Gourdon P., Alfredsson A., Neutze R. Optimized in vitro and in vivo expression of proteorhodopsin: a seven-transmembrane proton pump. Protein Expr. Purif. 2008;58:103–113. doi: 10.1016/j.pep.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Krebs R.A., Alexiev U., Braiman M.S. Detection of fast light-activated H+ release and M intermediate formation from proteorhodopsin. BMC Physiol. 2002;2:5. doi: 10.1186/1472-6793-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedrich T., Geibel S., Bamberg E. Proteorhodopsin is a light-driven proton pump with variable vectoriality. J. Mol. Biol. 2002;321:821–838. doi: 10.1016/s0022-2836(02)00696-4. [DOI] [PubMed] [Google Scholar]
- 10.Dioumaev A.K., Brown L.S., Lanyi J.K. Proton transfers in the photochemical reaction cycle of proteorhodopsin. Biochemistry. 2002;41:5348–5358. doi: 10.1021/bi025563x. [DOI] [PubMed] [Google Scholar]
- 11.Dioumaev A.K., Wang J.M., Lanyi J.K. Proton transport by proteorhodopsin requires that the retinal Schiff base counterion Asp-97 be anionic. Biochemistry. 2003;42:6582–6587. doi: 10.1021/bi034253r. [DOI] [PubMed] [Google Scholar]
- 12.Xiao Y., Partha R., Braiman M. Time-resolved FTIR spectroscopy of the photointermediates involved in fast transient H+ release by proteorhodopsin. J. Phys. Chem. B. 2005;109:634–641. doi: 10.1021/jp046314g. [DOI] [PubMed] [Google Scholar]
- 13.Bergo V.B., Sineshchekov O.A., Spudich J.L. His-75 in proteorhodopsin, a novel component in light-driven proton translocation by primary pumps. J. Biol. Chem. 2009;284:2836–2843. doi: 10.1074/jbc.M803792200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neutze R., Wouts R., Wulff M. Visualizing photochemical dynamics in solution through picosecond x-ray scattering. Phys. Rev. Lett. 2001;87:195508. doi: 10.1103/PhysRevLett.87.195508. [DOI] [PubMed] [Google Scholar]
- 15.Plech A., Wulff M., Anfinrud P.A. Visualizing chemical reactions in solution by picosecond x-ray diffraction. Phys. Rev. Lett. 2004;92:125505. doi: 10.1103/PhysRevLett.92.125505. [DOI] [PubMed] [Google Scholar]
- 16.Cammarata M., Levantino M., Ihee H. Tracking the structural dynamics of proteins in solution using time-resolved wide-angle x-ray scattering. Nat. Methods. 2008;5:881–886. doi: 10.1038/nmeth.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahn S., Kim K.H., Ihee H. Protein tertiary structural changes visualized by time-resolved x-ray solution scattering. J. Phys. Chem. B. 2009;113:13131–13133. doi: 10.1021/jp906983v. [DOI] [PubMed] [Google Scholar]
- 18.Cho H.S., Dashdorj N., Anfinrud P. Protein structural dynamics in solution unveiled via 100-ps time-resolved x-ray scattering. Proc. Natl. Acad. Sci. USA. 2010;107:7281–7286. doi: 10.1073/pnas.1002951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westenhoff S., Nazarenko E., Neutze R. Time-resolved structural studies of protein reaction dynamics: a smorgasbord of x-ray approaches. Acta Crystallogr. A. 2010;66:207–219. doi: 10.1107/S0108767309054361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson M., Malmerberg E., Neutze R. Structural dynamics of light-driven proton pumps. Structure. 2009;17:1265–1275. doi: 10.1016/j.str.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Thorgeirsson T.E., Xiao W., Shin Y.K. Transient channel-opening in bacteriorhodopsin: an EPR study. J. Mol. Biol. 1997;273:951–957. doi: 10.1006/jmbi.1997.1362. [DOI] [PubMed] [Google Scholar]
- 22.Yoshitsugu M., Yamada J., Kandori H. Color-changing mutation in the E-F loop of proteorhodopsin. Biochemistry. 2009;48:4324–4330. doi: 10.1021/bi900228a. [DOI] [PubMed] [Google Scholar]
- 23.Andersson M., Vincent J., Neutze R. A proposed time-resolved x-ray scattering approach to track local and global conformational changes in membrane transport proteins. Structure. 2008;16:21–28. doi: 10.1016/j.str.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 24.Subramaniam S., Gerstein M., Henderson R. Electron diffraction analysis of structural changes in the photocycle of bacteriorhodopsin. EMBO J. 1993;12:1–8. doi: 10.1002/j.1460-2075.1993.tb05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kralj J.M., Spudich E.N., Rothschild K.J. Raman spectroscopy reveals direct chromophore interactions in the Leu/Gln105 spectral tuning switch of proteorhodopsins. J. Phys. Chem. B. 2008;112:11770–11776. doi: 10.1021/jp802629e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neutze R., Pebay-Peyroula E., Landau E.M. Bacteriorhodopsin: a high-resolution structural view of vectorial proton transport. Biochim. Biophys. Acta. 2002;1565:144–167. doi: 10.1016/s0005-2736(02)00566-7. [DOI] [PubMed] [Google Scholar]
- 27.Hiraki K., Hamanaka T., Kito Y. Bacteriorhodopsin analog regenerated with 13-desmethyl-13-iodoretinal. Biophys. J. 2002;83:3460–3469. doi: 10.1016/S0006-3495(02)75345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oliveberg M., Malmström B.G. Internal electron transfer in cytochrome c oxidase: evidence for a rapid equilibrium between cytochrome a and the bimetallic site. Biochemistry. 1991;30:7053–7057. doi: 10.1021/bi00243a003. [DOI] [PubMed] [Google Scholar]
- 29.Wulff M., Schotte F., Mourou G. Time-resolved structures of macromolecules at the ESRF: single-pulse Laue diffraction, stroboscopic data collection and femtosecond flash photolysis. Nucl. Instrum. Meth. A. 1997;398:69–84. [Google Scholar]
- 30.Cammarata M., Eybert L., Polachowski S. Chopper system for time resolved experiments with synchrotron radiation. Rev. Sci. Instrum. 2009;80:015101. doi: 10.1063/1.3036983. [DOI] [PubMed] [Google Scholar]
- 31.Hammersley, A. P. 1997. FIT2D: An Introduction and Overview. ESRF Internal Report, ESRF97HA02T. European Synchrotron Radiation Facility, Grenoble, France.
- 32.Westenhoff S., Malmerberg E., Neutze R. Rapid readout detector captures protein time-resolved WAXS. Nat. Methods. 2010;7:775–776. doi: 10.1038/nmeth1010-775c. [DOI] [PubMed] [Google Scholar]
- 33.Henry E.R., Hofrichter J. Singular value decomposition: application and analysis of experimental data. Methods Enzymol. 1992;210:129–192. [Google Scholar]
- 34.Rangarajan R., Galan J.F., Birge R.R. Mechanism of spectral tuning in green-absorbing proteorhodopsin. Biochemistry. 2007;46:12679–12686. doi: 10.1021/bi700955x. [DOI] [PubMed] [Google Scholar]
- 35.Svergun D., Barberato C., Koch M.H.J. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 36.Glatter O. A new method for the evaluation of small-angle scattering data. J. Appl. Cryst. 1977;10:415–421. [Google Scholar]
- 37.Haldrup K., Christensen M., Nielsen M.M. Analysis of time-resolved x-ray scattering data from solution-state systems. Acta Crystallogr. A. 2010;66:261–269. doi: 10.1107/S0108767309054233. [DOI] [PubMed] [Google Scholar]
- 38.Kleywegt G.J. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 39.Wang W.W., Sineshchekov O.A., Spudich J.L. Spectroscopic and photochemical characterization of a deep ocean proteorhodopsin. J. Biol. Chem. 2003;278:33985–33991. doi: 10.1074/jbc.M305716200. [DOI] [PubMed] [Google Scholar]
- 40.Shi L., Ahmed M.A., Ladizhansky V. Three-dimensional solid-state NMR study of a seven-helical integral membrane proton pump—structural insights. J. Mol. Biol. 2009;386:1078–1093. doi: 10.1016/j.jmb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 41.Shi L., Lake E.M., Ladizhansky V. Solid-state NMR study of proteorhodopsin in the lipid environment: secondary structure and dynamics. Biochim. Biophys. Acta. 2009;1788:2563–2574. doi: 10.1016/j.bbamem.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Huber R., Köhler T., Wachtveitl J. pH-dependent photoisomerization of retinal in proteorhodopsin. Biochemistry. 2005;44:1800–1806. doi: 10.1021/bi048318h. [DOI] [PubMed] [Google Scholar]
- 43.Lakatos M., Lanyi J.K., Váró G. The photochemical reaction cycle of proteorhodopsin at low pH. Biophys. J. 2003;84:3252–3256. doi: 10.1016/S0006-3495(03)70049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramaniam S., Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 45.Royant A., Edman K., Neutze R. Helix deformation is coupled to vectorial proton transport in the photocycle of bacteriorhodopsin. Nature. 2000;406:645–648. doi: 10.1038/35020599. [DOI] [PubMed] [Google Scholar]
- 46.Yang J., Aslimovska L., Glaubitz C. Molecular dynamics of proteorhodopsin in lipid bilayers by solid-state NMR. J. Am. Chem. Soc. 2011;133:4874–4881. doi: 10.1021/ja109766n. [DOI] [PubMed] [Google Scholar]
- 47.Dunham T.D., Farrens D.L. Conformational changes in rhodopsin. Movement of helix F detected by site-specific chemical labeling and fluorescence spectroscopy. J. Biol. Chem. 1999;274:1683–1690. doi: 10.1074/jbc.274.3.1683. [DOI] [PubMed] [Google Scholar]
- 48.Nakamichi H., Okada T. Local peptide movement in the photoreaction intermediate of rhodopsin. Proc. Natl. Acad. Sci. USA. 2006;103:12729–12734. doi: 10.1073/pnas.0601765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choe H.W., Kim Y.J., Ernst O.P. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 50.Delaney J.K., Subramaniam S. The residues Leu 93 and Asp 96 act independently in the bacteriorhodopsin photocycle: studies with the Leu 93→Ala, Asp 96→Asn double mutant. Biophys. J. 1996;70:2366–2372. doi: 10.1016/S0006-3495(96)79803-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.