

Abstract
We present what we believe to be a new mathematical model of Ca2+ leak from the sarcoplasmic reticulum (SR) in the heart. To our knowledge, it is the first to incorporate a realistic number of Ca2+-release units, each containing a cluster of stochastically gating Ca2+ channels (RyRs), whose biophysical properties (e.g., Ca2+ sensitivity and allosteric interactions) are informed by the latest molecular investigations. This realistic model allows for the detailed characterization of RyR Ca2+-release properties, and shows how this balances reuptake by the SR Ca2+ pump. Simulations reveal that SR Ca2+ leak consists of brief but frequent single RyR openings (∼3000 cell−1 s−1) that are likely to be experimentally undetectable, and are, therefore, “invisible”. We also observe that these single RyR openings can recruit additional RyRs to open, due to elevated local (Ca2+), and occasionally lead to the generation of Ca2+ sparks (∼130 cell−1 s−1). Furthermore, this physiological formulation of “invisible” leak allows for the removal of the ad hoc, non-RyR mediated Ca2+ leak terms present in prior models. Finally, our model shows how Ca2+ sparks can be robustly triggered and terminated under both normal and pathological conditions. Together, these discoveries profoundly influence how we interpret and understand diverse experimental and clinical results from both normal and diseased hearts.
Introduction
Ca2+-induced release (CICR) forms the basis for the translation of electrical signals to physical contraction in cardiac myocytes during the process known as excitation-contraction coupling. In heart, L-type Ca2+ channel current is amplified by triggering Ca2+ release from the intracellular Ca2+ store (i.e., sarcoplasmic reticulum, SR) primarily via the ryanodine receptor, type 2 (RyR) Ca2+ channel. These Ca2+-activated RyRs are located on the SR membrane and largely arranged in paracrystalline arrays (10–300 RyRs) that are separated from the sarcolemmal membrane (SL) by the small (15-nm) dyadic subspace (1,2).
During systole, RyRs are activated by Ca2+ influx via adjacent voltage-sensitive L-type Ca2+ channels (LCCs). Together, the LCC and RyR clusters form a functional unit of Ca2+ release known as the Ca2+-release unit (CRU), which is essential to the local control of Ca2+ release during excitation-contraction coupling (3). The synchronized opening of clustered RyRs results in elevations of local (i.e., subspace) [Ca2+] known as “Ca2+ sparks”. During diastole, in the absence of LCC Ca2+ influx, spontaneous Ca2+ sparks are rare but still easy to observe using a Ca2+ indicator where the Ca2+-spark rate reflects the finite opening rate of the RyR channel (4).
Another form of Ca2+ release, “invisible” by standard confocal imaging methods, is the “nonspark” event, which involves the opening of a single RyR (e.g., “Ca2+ quark”) or a few RyRs that fail to trigger a full Ca2+ spark (5).
A third RyR-based Ca2+ release pathway is attributed to a small population of diffusely distributed RyRs termed “rogue” RyRs, which are located away from the junctional cleft (2,6,7). Here, we present a mathematical model that identifies and characterizes these three forms of visible and invisible diastolic RyR Ca2+ release, in a coherent manner. This SR Ca2+ leak or loss of Ca2+ from the SR is experimentally observed (8,9) but flawed in earlier mathematical models. In our fully stochastic model, the simulation now matches the biology and provides what we believe to be new insight into the mechanisms by which SR Ca2+ leak operates in intact cells. This model is fully informed by the latest molecular investigations of heart cells, heart nanoanatomy, and recent characterizations of channels, transporters, and buffers.
SR leak is attributed to RyRs, Ca2+permeant channels whose open probability is controlled by [Ca2+]i, [Ca2+]sr, phosphorylation state, and other factors. In this manner, SR Ca2+ content is regulated by Ca2+ leak and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) Ca2+ pump. [Ca2+]sr is observed to change in response to diverse diseases (e.g., heart failure and arrhythmia) (10,11) and phosphorylation by kinases such as protein kinase A or Ca2+-calmodulin-dependent kinase II (12,13). Additionally, RyR mutations such as those related to catecholaminergic polymorphic ventricular tachycardia can also underlie changes in RyR behavior and thus change SR Ca2+ content (14). These conditions are frequently found to be arrhythmogenic and contribute to Ca2+ waves, Ca2+ alternans, and other forms of cellular instability (11). The dynamics of SR Ca2+ leak are thus critical to our understanding of heart function during both physiological and pathophysiological conditions. Computational models offer an explicit means to investigate nanoscale events related Ca2+ leak not easily measured under experimental settings.
The physiological mathematical model of Ca2+ leak presented here provides fundamentally new, to our knowledge, and important findings that change our understanding of Ca2+ signaling at the nanoscopic and cell-wide level. The five major new findings of this model include the following:
-
1.
We observe the presence of an “invisible” Ca2+ leak that is quantitatively consistent with earlier unexplained experimental findings (15).
-
2.
We find that the fully stochastic activation and termination of RyR-based Ca2+ release within the ventricular myocyte allows us to properly account for SR Ca2+ leak, obviating the inclusion of an ad hoc, non-RyR-mediated Ca2+ leak flux.
-
3.
[Ca2+]sr levels are demonstrated to depend critically on RyR open probability (Po) and vice versa. This reconciles modeling concepts and findings with new experimental results relating to pump/leak balance.
-
4.
Single RyR openings, although brief, are frequent (∼3000 cell−1 s−1) and often fail to trigger a full Ca2+ spark (∼130 cell−1 s−1), suggesting that Ca2+ spark fidelity in response to both single LCC and RyR openings is actually quite low.
-
5.
Simulations predict that increased RyR activity can, somewhat paradoxically, lead to increased SR Ca2+ leak even in the presence of decreased [Ca2+]sr.
Taken together, this model accounts for how the molecular-level characteristics of individual RyRs influence the emergent property of cellular SR Ca2+ leak, and lays the foundation for fully spatially resolved modeling of Ca2+ signaling in single cells.
Methods
Model formulation
Here we present a whole-cell model for CICR in cardiac myocytes, which builds and expands upon the local SR Ca2+ release model previously introduced by Sobie et al. (16). Both the RyR cytosolic [Ca2+]i sensitivity and luminal SR [Ca2+] ([Ca2+])sr dependency have been updated to reproduce recently reported single channel gating behavior (17). The previous ad hoc formulation of cooperative gating has been replaced with an energetic coupling formulation (18) derived from allosteric behavior observed in biological systems (19,20). Our model (see Fig. 1) consists of 20,000 independent Ca2+ release units (CRUs), each containing a cluster of stochastically gating RyRs that are instantaneously coupled via local subspace [Ca2+] ([Ca2+]ds). These CRUs are coupled via a bulk myoplasmic [Ca2+]i, which also includes a small fraction (5%) of diffusely distributed, nonjunctional or “rogue” RyRs. SERCA pumps Ca2+ from the myoplasm back into the SR (21). The SR itself is composed of junctional SR (JSR) and network SR (NSR) components, each with appropriate volumes and Ca2+ buffers. The sarcolemma (SL) has a background Ca2+ leak and two transsarcolemmal calcium transporters (the Na+-Ca2+ exchanger, and the plasmalemmal Ca2+-ATPase) to extrude Ca2+ from the cell.
Figure 1.
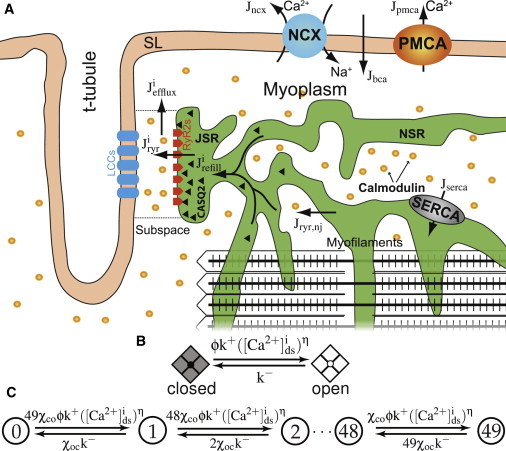
Diagram of SR Ca2+ leak model and release site schematic. (A) Model compartments and Ca2+ fluxes (solid arrows). (B) Transition-state diagram for the two-state Markov chain describing a single RyR. (C) Transition-state diagram for the Markov chain representing the RyR cluster where each state indicates the number of open RyRs (No) in the CRU (e.g., 0, 1, 2, …48, 49).
Although we also envision a cluster of L-type Ca2+ channels (LCCs) apposing the RyR cluster that trigger the CRUs during electrical activity, the LCCs have been disabled for this simulation that focuses on diastolic Ca2+ signaling when LCCs are quiescent. We are assuming minimal or no mitochondrial Ca2+ buffering (22), therefore potential contributions of this organelle to Ca2+ handling are omitted. The mathematical description of key elements is presented below. The seeming novelty of this model lies in the details of its formulation and the inclusion of the known properties and functions of each model component that had been overlooked, ignored, or strategically excluded in prior models. We have consequently identified several major findings—believed by us to be entirely new—as described above and presented below.
RyR model
In this study, the RyR signaling that underlies cardiac EC coupling is studied by observations of Ca2+ sparks in resting cells in which LCCs are quiescent. The dyad, or dyadic subspace, is a restricted space located between the transverse tubule and JSR membranes that contains a cluster of RyRs that gate stochastically, display coupled gating (23), and have a Po that depends both cytosolic and luminal [Ca2+] (17). Here, each Ca2+-activated RyR is represented by a two-state Markov chain containing one closed (C) and one open (O) state. Transitions from C to O are dependent on both [Ca2+]ds and junctional SR [Ca2+]([Ca2+])jsr (see Fig. 1 B), with k+ as the association rate constant for [Ca2+]ds binding with units of μM−η s−1, and k− as the corresponding dissociation rate constant in s−1. A luminal regulation function (ϕ = ϕm[Ca2+]sr + ϕb) serves to modify the channel opening rate where [Ca2+]sr is the luminal [Ca2+] associated with each RyR (which is [Ca2+]jsr [Ca2+]sr and [Ca2+]nsr for junctional and nonjunctional RyRs, respectively).
Combining 49 of these identical two-state RyRs into a cluster and assuming they are instantaneously coupled via [Ca2+]ds yields a M = 50 state release site where each state indicates the number of open RyRs (No) for the CRU (0 ≤ No ≤ 49), as shown in Fig. 1 C where terms on the arrows are transition rates. In these rate terms, χoc and χco represent mean-field allosteric coupling factors (18) given by
| (1) |
| (2) |
where a∗ represents the average allosteric connectivity (based on a 7 × 7 grid with nearest-neighbor coupling), and εcc is a dimensionless free energy of interaction (units of kBT) that specifies the change in free energy experienced by a channel in state C when allosterically coupled to another channel in state C, and similarly for εoo. The coefficients Nc (number of closed RyRs) and No serve to partition allosteric coupling between the forward and reverse transitions. In addition to dyadic RyRs, a small fraction (5%) of RyRs are assumed to be found away from the junction as diffusely distributed, single channels.
SERCA formulation
SERCA consumes ATP to pump Ca2+ into the SR from the myoplasm. Here, we have implemented a SERCA formulation recently developed by Tran et al. (21). The SERCA flux takes the form, Jserca = 2vcycleAp, where vcycle is the cycling rate per pump molecule (see the Supporting Material) and Ap is the concentration of SERCA molecules per liter cytosol. The Tran-Crampin SERCA formulation was selected because it provides a realistic representation of SERCA-dependent Ca2+ flux that displays physiological behavior at low [Ca2+]i (see Fig. S6 A in the Supporting Material) and is sensitive to changes in [Ca2+]sr (see Fig. S6 B), which is essential when allowing RyR-based leak to balance SERCA. Whole-cell Ca2+ dynamics obtained using this SERCA formulation is provided in the Supporting Material.
Concentration balance equations
The Markov chain Monte Carlo model used here consists of 2N + 2 (N = 20,000) ordinary differential equations representing the time-evolution of (Ca2+) in the bulk myoplasm ([Ca2+]i), the NSR ([Ca2+]nsr), the JSR ([Ca2+]jsr), and the dyadic subspace ([Ca2+]ds) compartments, and N Markov chains representing the stochastic RyR clusters (see Fig. 1 A). The concentration balance equations and formulation for all fluxes can be found in the Supporting Material. The flux through the RyR cluster associated with the ith CRU is given by
| (3) |
where 1 ≤ i ≤ N, vryr is the RyR Ca2+ release rate in s−1, and Nio is the number of open RyR channels at the ith release site. Similar to previous work (24,25), model parameters lead to rapid equilibrium of [Ca2+]ds with the [Ca2+]i and [Ca2+]jsr allowing [Ca2+]ds to be approximated using an algebraic expression of [Ca2+]i, [Ca2+]jsr, and No (see Eq. S23 in the Supporting Material).
Results
Ca2+ signaling in heart cells involves interactions among many proteins in a spatially and temporally complex environment as suggested in Fig. 1 A. Teasing apart the causal links in this cellular environment is aided enormously by mathematical models informed by experimental results. Here, we present results from a whole-cell model of SR Ca2+ signaling that seeks to investigate local Ca2+ dynamics using fresh modeling elements constrained and informed by the recent experimental findings described below.
RyR Ca2+ sensitivity and allosteric coupling
SR Ca2+ release in cardiac myocytes is a multiscale process ranging from small invisible Ca2+ leak events due to brief openings of a single RyR, to microscopic Ca2+ sparks involving numerous RyRs at single a CRU, to cell-wide [Ca2+]i transients involving release from numerous CRUs within the cell. This process depends critically on the sensitivity of the RyRs to local cytosolic and luminal [Ca2+] as well as interactions between neighboring channels (i.e., allosteric coupling). It has been shown that Ca2+ sparks result from synchronized openings of clustered RyRs (4), mediated by [Ca2+]ds with a strong dependence on [Ca2+]sr (26). Clustered RyRs undergo probabilistic interactions here referred to as “allosteric coupling” but first described more rigidly as “coupled gating” (23). In our sticky-cluster model (16), we attempted to capture this known property of RyR gating by including an ad hoc formulation for cooperativity among the RyRs and demonstrated that this feature enabled robust Ca2+ spark termination. Here, we have implemented an energetic coupling formulation (18) based on models of protein-protein interactions (19).
Fig. 2 shows how the sensitivity of single RyR channels to be triggered by Ca2+ depends on the local [Ca2+]i (Fig. 2 A), local [Ca2+]sr (Fig. 2 B), and also on allosteric coupling between the individual RyRs within the CRUs (Fig. 2, C and D). When plotted as a function of [Ca2+]i (Fig. 2 A), the steady-state single RyR Po is an increasing function of [Ca2+]i with a half-maximal point (Km) of ∼12 μM when [Ca2+]sr is held constant at 1 mM (solid line). This result corresponds well with results from lipid bilayer measurements (solid circles) that display a Km of 12.2 μM and a Hill coefficient of 2.6 (27). Fig. 2 B shows how the RyR Po increases as a function of increasing [Ca2+]sr when [Ca2+]i is held constant at 8 μM, which also agrees well with a corresponding experimental observation (solid squares) from Qin et al. (17).
Figure 2.
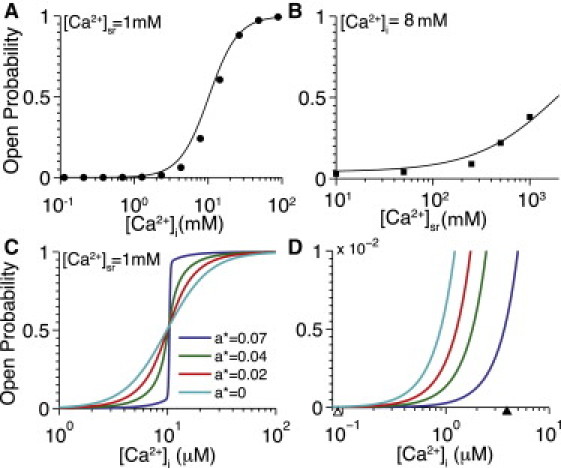
Single channel RyR Ca2+ sensitivity and influence of allosteric coupling on RyR steady-state open probability (Po). (A) Single channel RyR Po as a function of cytosolic [Ca2+] ([Ca2+]jsr = 1 mM). (Solid line) Model result in which data points (solid circles) indicate best fit; normalized Po (Kd = 12.2 μM, η = 2.6) from Fig. 1B in Qin et al. (27). (B) Single RyR Po as a function of luminal [Ca2+] ([Ca2+]ds = 8 μM). (Solid line) Model result in which data points (solid squares) indicate experimental results from Fig. 3B in Qin et al. (27). (C) Po for a cluster of 49 RyRs as a function of [Ca2+]i with varying levels of average RyR allosteric connectivity (a∗). As a∗ increases, Po exhibits steeper dependence [Ca2+]i. (D) Zoom of Fig. 2C showing lower [Ca2+]i levels. For reference, markers on x axis indicate the diastolic [Ca2+]i and peak [Ca2+]ds resulting from a single open RyR (blue triangle and red triangle, respectively).
Fig. 2 C displays the steady-state Po for a cluster of RyRs versus [Ca2+]i with varying levels of average RyR allosteric connectivity (a∗) and Fig. 2 D shows the same behavior for low [Ca2+]i levels. Note that decreasing a∗ is equivalent to removing allosteric interactions between some RyRs. In the absence of coupling (i.e., a∗ = 0), RyRs are coupled only by [Ca2+]ds and exhibit higher Po at low local [Ca2+]i, similar to isolated RyR channels. However, with the allosteric coupling levels used in this study (i.e., a∗ = 0.07; blue line), clustered RyRs display decreased Po at low cytosolic [Ca2+]i due to stabilization of closed channels by the allosteric interactions. Because closed channels have a stabilizing influence on their neighbors, the RyR cluster is more likely to remain closed at low [Ca2+]i and transition rapidly from all-closed to all-open at elevated [Ca2+]i. Note, we also observed that NRyR has a modest impact on CRU Po (see Fig. S2).
Local Ca2+ dynamics
In our previous model (16), we showed that SR Ca2+ release events via a cluster of cooperatively gating RyRs with both cytosolic and luminal Ca2+ sensitivity provided robust Ca2+ sparks with characteristics comparable to those observed experimentally including frequency, duration, termination, and restitution (4). However, this single CRU model did not attempt to characterize the leak of Ca2+ out of the SR nor was the dynamic Ca2+ sensitivity examined. The formulation of RyR gating presented here as new and the inclusion of a thermodynamically constrained SERCA formulation allows significant insights into the dynamics of SR Ca2+ leak shown below.
Fig. 3 and Fig. S3 show diastolic Ca2+ sparks and nonspark Ca2+-release events as measured by [Ca2+]ds (Fig. 3 A and Fig. S3 A), stochastic RyR gating as measured by No (Fig. 3 B and Fig. S3 B), and Ca2+ blinks as measured by [Ca2+]jsr (Fig. 3 C and Fig. S3 C). In Fig. 3, all 20,000 CRUs were simulated and the traces shown depict release activity recorded in a subpopulation of randomly selected CRUs (i.e., 10% of total) over a period of 200 ms. In this subpopulation, out of the 58 CRUs that exhibited release activity, only five produced Ca2+ sparks during that period of time. This is representative of the whole-cell behavior, which resulted in 132 Ca2+ sparks cell−1 s−1. The five CRUs displaying Ca2+ sparks did not interact directly and were coupled only via [Ca2+]i. Fig. 3 shows not only the [Ca2+]ds changes due to Ca2+ sparks (colored lines) but also the changes in [Ca2+]ds due to the opening of individual RyRs within CRUs that did not lead to Ca2+ sparks (gray lines). In the latter case, CRUs that do not display Ca2+ sparks are still capable of SR Ca2+ leak through uncoordinated openings of single or small groups of RyRs that would likely be invisible experimentally. We also tested a wide range of CRUs sizes (16 ≤ NRyR ≤ 100) and found robust Ca2+-spark triggering and termination (see Fig. S3 and Fig. S4).
Figure 3.
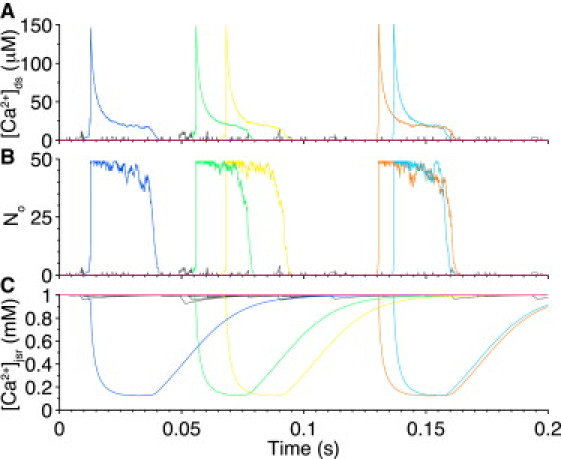
Local Ca2+ dynamics. (A) Subspace [Ca2+] ([Ca2+]ds) associated with each RyR cluster during spontaneous, diastolic Ca2+ release events. (B) No at each CRU. (C) Junctional SR [Ca2+] ([Ca2+]jsr) present on the luminal side of RyR cluster. (Each colored line represents a different CRU where a Ca2+ spark is observed and nonspark RyR activity is shown in shading.) Note: For visual clarity, only 10% of total CRU activity is displayed here.
Dynamics of Ca2+-spark triggering and termination
The model demonstrates that diastolic Ca2+ sparks arise from elevations in [Ca2+]ds caused by brief but frequent openings of single RyRs. The stochastic nature of RyR gating also allows for prolonged openings of single RyRs that increase the probability of a Ca2+ spark due to sustained periods of elevated [Ca2+]ds. Fig. 4 A shows the details of the RyR currents (IRyR) that underlie the Ca2+ spark dynamics in Fig. 3, and Fig. 4 B provides a closer view of these IRyR events. The current associated with a Ca2+ spark (Ispark) peaks at ∼10 pA (see Fig. 4 A) whereas the current associated with an individual RyR (Iquark) peaks at 0.2 pA (see Fig. 4 B). These values are consistent with experimental results (28). Examination of the upstroke of Ispark reveals a stepwise increase in IRyR that precedes the regenerative CICR rise to the peak of the Ispark. Ca2+ sparks arise from frequent Iquark events within each CRU. However, there are many Iquark records that do not initiate an Ispark and some that even trigger one or more RyR to open without triggering a Ca2+ spark. This recruitment pattern is clearly consistent with the stochastic nature of local CICR and with the inference that RyR clusters are relatively difficult to trigger (29,30).
Figure 4.
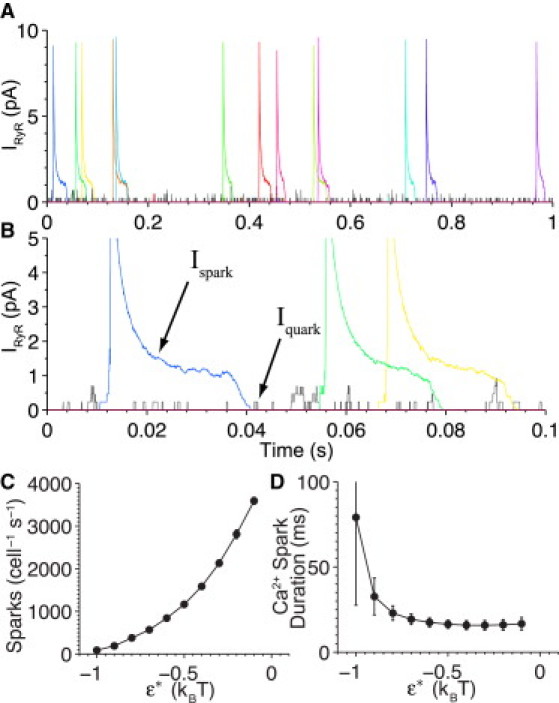
Dynamics of diastolic Ca2+ spark triggering and termination. (A) Spontaneous RyR Ca2+ current in pA during a 1-s simulation. (B) Zoom of RyR current showing spark initiation and termination profiles. (Colored lines) Different CRUs, respectively, where a Ca2+ spark is observed. (Shading) Nonspark RyR activity for the remaining CRUs. Note: For visual clarity, only 10% of the whole cell activity is displayed here. (C) Number of Ca2+ sparks as function of the strength of allosteric coupling (coupling strength is assumed to be symmetrical, i.e., ε∗ = εcc = εoo). (D) Average Ca2+ spark duration versus ε∗.
During Ispark, [Ca2+]jsr declines because Ca2+ moves from the JSR to the dyadic subspace via the open RyRs, causing IRyR to decrease. This can be readily appreciated by examining the fluctuations in IRyR produced during the initial phase of a Ca2+ spark as compared to fluctuations visible near the end of each Ca2+ spark (see Fig. 4 B). Consequently, fluctuations in the release current due to the gating of individual RyRs becomes increasingly small during the final phase of Ispark. Eventually [Ca2+]jsr (see Fig. 3 C) is depleted to a point where the Ca2+ spark terminates stochastically—facilitated by allosteric coupling and reduced RyR reopenings due to lower [Ca2+]ds and [Ca2+]jsr.
Although this result is consistent with the termination mechanism proposed in Sobie et al. (16), many of the key elements appear novel. For example, the clear character of Iquark and how it initiates Ispark reveals that triggering of a Ca2+ spark is not an all-or-none process as we had previously hypothesized (16). Significantly, Fig. 4, A and B, shows that the overwhelming majority of single RyR gating activity does not lead to Ca2+ sparks. The duration of these nonspark, single-channel events are consistent with the biophysical properties of the RyR (e.g., its inherent closing rate) that display mean open times in the range of 1–4 ms in lipid bilayer experiments (31). Ca2+ released via this mechanism is likely to go unaccounted for experimentally and lends itself to the concept of invisible leak. A closer examination of this nonspark leak mechanism is presented below.
The effect of allosteric coupling strength (ε∗) on frequency and duration of Ca2+ sparks is examined in Fig. 4, C and D. Note that decreasing ε∗ is analogous to increasing the amount by which RyR interactions stabilize both closed and open channel pairs. Ca2+-spark rate rises exponentially and Ca2+-spark duration decreases as coupling strength is lowered (i.e., increased ε∗). This demonstrates that RyRs exhibit elevated activity in the presence of weak allosteric coupling—caused by the increased opening rate of uncoupled channels (see Fig. 2 C). Although the Ca2+-spark rate is very sensitive to ε∗, the average spark duration is much less sensitive to ε∗, suggesting that RyR luminal [Ca2+] dependency is playing the dominant role for Ca2+-spark duration and termination. Note, simulations indicate that complete removal of allosteric coupling yields Ca2+-spark rates much higher than those observed in FK506-based uncoupling experiments (32). This supports the idea that RyR-RyR coupling may involve factors in addition to FKBP12.6.
Visibility of SR Ca2+ leak
Although SR Ca2+-release events originating from a fully activated cluster of RyRs are easily detectable experimentally (4), Ca2+ release by an individual or even a few RyRs is well below the experimental detection threshold (16). Iquark is comparable to unitary LCC current at −30 mV (i.e., 0.2 pA). This local Ca2+-signaling mass is far less than that of a Ca2+ sparklet (33)—which is detectable because of its long mean open time (≥10 ms due to channel agonists) and high external (Ca2+) (20 mM) or special optical conditions (e.g., the 100-nm-thin optical section of total internal reflection microscopy) or both. In Fig. 5, the visibility of Ca2+ release due to Ispark and Iquark is assessed in the presence of realistic noise (see Sobie et al. (16)). In Fig. 5 A, a simulated linescan image of the SR Ca2+ release activity shown in Fig. 5 B (black line, right axis) is given. Although the signal due to the Ispark event is clearly visible, the signal emitted by the two Iquark events (generated by one and four open RyRs) that precede the Ca2+ spark cannot be discerned. The fluorescent profile (Fig. 5 B (blue line, left axis)) suggests that such SR Ca2+ release events that fail to induce a full Ca2+ spark are likely to go undetected. We also generated a simulated Ca2+ spark/blink pair (see Fig. S5) for comparison with recent experimental measurements (34). Below, we examine the propensity of these nonspark occurrences during diastole.
Figure 5.
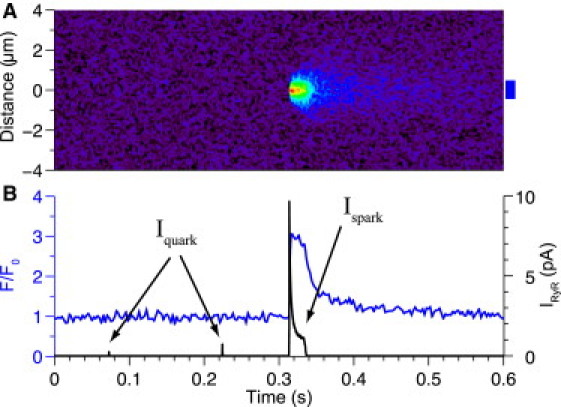
Experimental detection of SR Ca2+ release. (A) Simulated linescan of IRyR activity. (B) Time course of IRyR exhibiting both spark (Ispark) and nonspark (Iquark) events (black line) and the corresponding fluorescence profile (F/F0) of Fluo3 (blue line). The F/F0 profile was obtained by averaging fluorescence from a 1-μm-wide region (blue box). Simulated linescan based on previously published methods (see Sobie et al. (16) and Smith et al. (40)).
Ca2+-spark latency and fidelity
The cardiac SR Ca2+-leak pathway includes both Ca2+ sparks and nonspark SR Ca2+-release events. The nonspark events occur when RyR channels open but do not trigger enough RyRs to produce a Ca2+ spark. Statistical analysis of all spontaneous Ca2+ release events provides information on SR Ca2+-leak dynamics such as Ca2+-spark latency (the time from spark initiation to spark peak) and fidelity (the probability of individual RyR openings triggering a full Ca2+ spark). During diastolic simulations (see Figs. 3 and 4) the time required to reach the peak of a Ca2+ spark varies from less than a millisecond to several milliseconds and decreases as a function of No (Fig. 6 A). The fidelity of Ca2+-spark triggering has been anticipated by the examples of Iquark and Ispark shown in Fig. 4, A and B. Single RyR openings occur at a rate of ∼3000 cell−1 s−1; however, most fail to trigger a Ca2+ spark.
Figure 6.
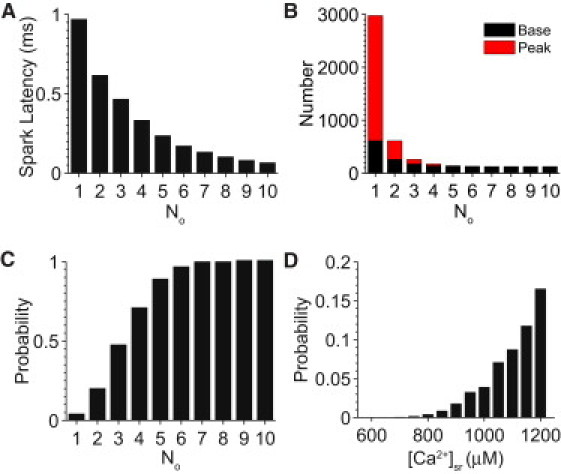
Analysis of Ca2+ spark latency and fidelity. (A) Average mean time to spark peak as a function of the No. (B) Number of Ca2+ release events versus the No where events are segregated between events that lead to additional RyR openings (solid bars) and events that do not (shaded bars). Note that the bar above No = 10 also corresponds to the number of Ca2+ sparks during the simulation. (C) Probability of triggering a spark as a function of peak No. (D) Histogram of the probability of a single RyR opening triggering a spark as a function of [Ca2+]sr.
Fig. 6 B shows a histogram of the number of Ca2+-release events versus the No involved in the event. Some events serve as the base for triggering additional RyRs to open (solid bars) whereas other events peak at No and then terminate (shaded bars). Note that most (85%) of the 20,000 CRUs have no RyRs that open at all during the 1-s simulation. There are between 100 and 150 diastolic Ca2+ sparks cell−1 s−1 in rat ventricular myocytes (4) and this is faithfully reproduced by the model. Of the ∼3000 RyR opening events, only ∼130 lead to Ca2+ sparks, indicating that the fidelity of an Iquark triggering an Ispark is low. This is further illustrated in Fig. 6 C, which shows the likelihood of a Ca2+ spark increases with No, but is quite low (∼5%) when No = 1. This indicates the probability of a single or a few open channels to trigger a Ca2+ spark is small under normal conditions but increases rapidly as [Ca2+]sr is increased (see Fig. 6 D). In summary, Fig. 6 shows that Ca2+ sparks are a dynamic balance between the regenerative nature of CICR and the stabilizing influence of allosteric coupling on the RyR channels.
SR Ca2+ leak in permeabilized cells
SR Ca2+ content is known to influence the rate of diastolic loss of Ca2+ from the SR (9). To examine SR Ca2+ leak and its dependence on Ca2+ sparks and IRyR, we mimicked the inhibition of SERCA by thapsigargin (TG) and observed the loss of Ca2+ from the SR under permeabilized conditions (similar to experiments by Zima et al. (8)). Fig. 7 A shows the temporal decline in [Ca2+]sr and Ca2+-spark rate after SERCA inhibition. The high cytosolic [Ca2+]i (150 nM) used in these permeabilized cell experiments results in an elevated Ca2+ spark rate (∼1000 sparks cell−1 s−1)—six times the Ca2+ spark rate seen in rat myocytes during diastole. In Fig. 7 A Ca2+ sparks cease after [Ca2+]sr falls below 700 μM. However, even in the absence of Ca2+ sparks, Ca2+ continues to leak from the SR. The model suggests that this is due to the background IRyR that is experimentally “invisible” (see Fig. 5), and the SR loses half of its Ca2+ within 5 min of SERCA inhibition.
Figure 7.
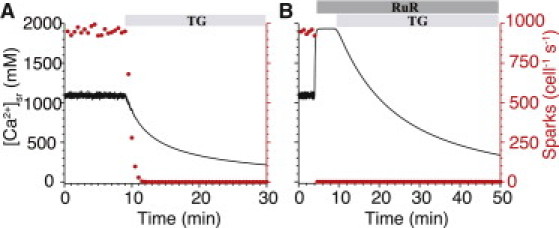
Effect of SERCA inhibition on SR [Ca2+]. (A) Simulated effect of SERCA inhibition by thapsigargin (TG, Ap = 0) on [Ca2+]nsr (black line) and whole-cell Ca2+ spark rate (red circles). (B) Simulated effect of a RyR-specific antagonist, ruthenium red (RuR) on [Ca2+]nsr (black line) and whole-cell Ca2+ spark rate (red circles), before and after SERCA inhibition by TG. In each case, [Ca2+]i was fixed at 150 nM to mimic experimental conditions in permeabilized cells (see Zima et al. (8)). RuR is modeled here by disabling all but 2% of the RyRs in the model cell and giving the functional RyRs the kinetics of nonjunctional, rogue RyRs (vryr = 0, vryr,nj = 1.01 s−1).
To further investigate invisible leak, 98% of the RyRs in the cell were disabled by ruthenium red (RuR)—completely preventing the generation of Ca2+ sparks (Fig. 7 B). During the brief period when most RyRs were blocked but SERCA remained active, [Ca2+]sr increases to nearly twice its initial value due to the shift in pump/leak balance. However, after SERCA inhibition, Ca2+ still leaks out of the SR via the nonspark pathway even as [Ca2+]sr declines below 1 mM, albeit at a much slower rate than in Fig. 7 A. These simulations suggest that the unidentified leak pathway highlighted by Zima et al. (8) may, in fact, be incomplete inhibition of the cell's nearly 1,000,000 RyRs.
Dependence of SR Ca2+ leak on SR Ca2+ content
The opening of RyRs depends on both [Ca2+]i and [Ca2+]sr and makes experimental analysis of SR Ca2+ leak complicated. However, the model allows us to directly examine how SR Ca2+ leak varies based on changes in [Ca2+]sr. Fig. 8 shows the characteristics of SR Ca2+ leak in our whole-cell SR Ca2+ release model and its Ca2+-spark and -nonspark Ca2+ leak-pathways via junctional and nonjunctional rogue RyRs.
Figure 8.
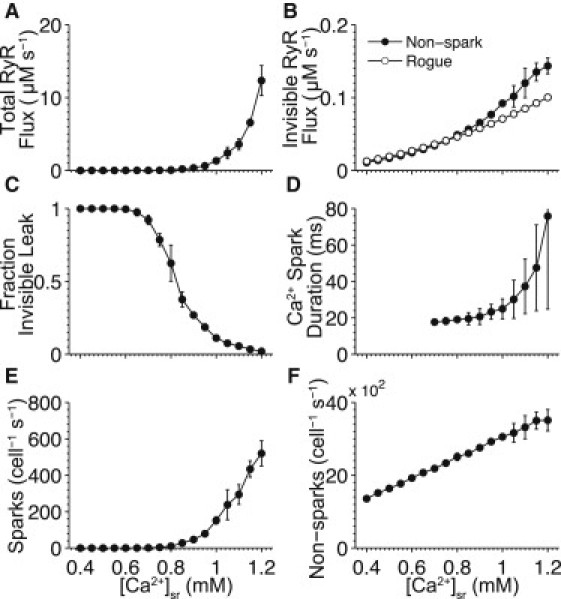
Effect of SR Ca2+ load ([Ca2+]sr) on SR Ca2+ leak. (A) Total integrated RyR flux during a 1-s simulation. (B) Integrated nonspark RyR flux via junctional RyRs (solid line, solid circles) and nonjunctional RyRs (solid line, open circles). (C) Fraction of RyR flux not associated with a Ca2+ spark (e.g., nonspark RyR flux divided by total RyR flux). (D) Average Ca2+ spark duration. (E) Ca2+ spark rate. (F) Number of nonspark events versus [Ca2+]sr. The junctional nonspark flux is defined as RyR activity that does not precede a Ca2+ spark. (Data points) Average over 10 simulations. (Error bars) Standard deviation from the mean. Note that [Ca2+]i was held constant at 90 nM.
A pivotal role of [Ca2+]sr is governing the overall RyR-mediated SR Ca2+ efflux as shown in Fig. 8 A. The low overall Ca2+ leak seen at low [Ca2+]sr (≤ 800 μM) is due not only to decreased RyR opening rates but also decreased Ca2+-spark triggering fidelity caused by reduced IRyR. The contribution of nonspark Ca2+ leak to this process is shown in Fig. 8 B. For [Ca2+]sr levels used here (i.e., rodent), Ca2+ sparks dominate SR Ca2+ leak whereas at lower [Ca2+]sr levels (i.e., larger mammals) SR Ca2+ leak is primarily via the invisible, nonspark pathway. Not surprisingly, the frequency of both Ca2+ sparks and nonspark Ca2+-release events are increasing functions of [Ca2+]sr (Fig. 8, E and F), with Ca2+ sparks exhibiting an approximately exponential increase. Note that this rapid rise of Ca2+-spark rate at elevated [Ca2+]sr levels would likely eventually give way to propagating Ca2+ waves in a fully spatial model. This rapid increase in Ca2+-spark rate at elevated [Ca2+]sr levels is due to increased Ca2+-spark fidelity (ability of Iquark to trigger Ispark) and RyR [Ca2+]i sensitivity. In summary, Ca2+ sparks dominate SR Ca2+ leak under normal SR Ca2+ load; however, at lower [Ca2+]sr, the nonspark-based leak contributes a greater fraction of the overall leak. Note that rogue RyRs can be excluded from this model with modest effect on results.
SR Ca2+ leak under pathological conditions
Pathological conditions such as heart failure are often associated with SR Ca2+ mishandling due to downregulation of SERCA and/or excessive phosphorylation of RyR. With this in mind, simulations were conducted with the model to study the effects of these two changes, the goal being to acquire novel insights into the development of the pathology. Fig. 9 demonstrates the effect of phosphorylation on SR Ca2+ leak, simulated via changes in the RyR opening rate (k+) and changes in SERCA's [Ca2+]i sensitivity (Kd,i).
Figure 9.
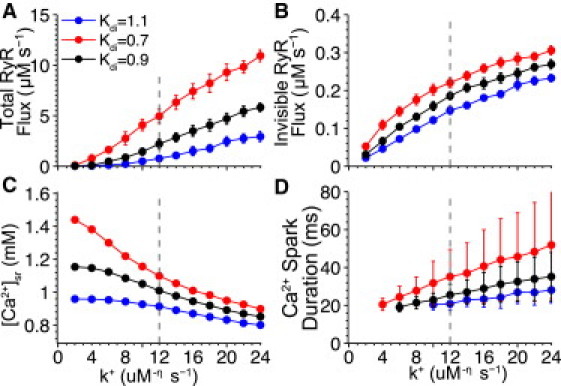
Effect of pathology on SR Ca2+ leak modeled by adjusting RyR opening rate (k+) and SERCA's [Ca2+]i sensitivity (Kd,i). (A) Total integrated RyR flux during a 1-s simulation. (B) Integrated nonspark RyR release flux via both junctional and nonjunctional RyRs. (C) Steady-state [Ca2+]sr. (D) Average Ca2+ spark duration versus k+. In all simulations, [Ca2+]i was fixed at 90 nM and [Ca2+]nsr was allowed to equilibrate to its steady-state value. (Data points) Average of 10 simulations. (Error bars) Standard deviation from the mean. (Vertical dashed lines) Parameter values under normal conditions.
In all simulations, [Ca2+]i was fixed at 90 nM and [Ca2+]sr was allowed to equilibrate to its steady-state value. The whole-cell Ca2+-leak rate is shown to increase with RyR phosphorylation levels (Fig. 9 A), and similarly for nonspark-based leak (Fig. 9 B). This response, along with the decrease in steady-state [Ca2+]sr shown in Fig. 9 C, is expected and is attributed to the phosphorylation-induced increase of the RyR open probability. The model suggests that Ca2+ sparks produced at these diminished SR Ca2+ levels are of longer durations (Fig. 9 D) and exhibit greater variability—which is, to our knowledge, a novel prediction that has yet to be tested experimentally. Also, simulations show that increased RyR activity can, somewhat paradoxically, lead to increased SR Ca2+ leak even in the presence of decreased [Ca2+]sr (see Fig. 9, A and C).
Discussion
We have presented what we believe to be a new mathematical model that successfully and robustly characterizes Ca2+ sparks and Ca2+ leak in the heart during diastole. The model includes a realistic number of CRUs each containing a cluster of stochastically gating RyRs with properties constrained by experimental observations. The CRUs function independently of each other but are sensitive to the [Ca2+] in the cytosol, through which they combine to produce whole-cell phenomena as observed experimentally. The model presented here improves upon our previous Ca2+-spark model (16) that modeled Ca2+ sparks from a single CRU incorporating RyR regulation by subspace and luminal calcium. It incorporates improved RyR Ca2+ sensitivity and allosteric interactions as well as complete descriptions of diastolic Ca2+ cycling. It embodies several well-characterized properties of Ca2+ sparks (duration, termination, activation, and sensitivity to Ca2+ and [Ca2+]sr). In doing this, it describes and characterizes SR Ca2+ balance including both Ca2+ spark- and nonspark-based SR Ca2+ leak. The work provides a conceptual framework and tools for an improved understanding of a number of perplexing issues previously presented by us and others as noted below.
Importantly also, the stochastic model of Ca2+ sparks and Ca2+ leak during diastole, which (to our knowledge) we present here as new, lays the foundation for more complex dynamic models with fully integrated, spatially resolved, time-dependent Ca2+ signaling.
Inactivation of RyR not needed
There is no need for Ca2+-dependent RyR inactivation to terminate Ca2+ sparks as demonstrated here and in our previous model (16). Ca2+ sparks terminate stochastically facilitated by [Ca2+]jsr depletion and RyR allosteric coupling. Although the physical self-association of RyRs in biochemical preparations of RyRs supports the physical interactions among the homotetramers, superresolution imaging and additional experiments are needed to explore this aspect. Finally, to date, no Ca2+-dependent inactivation process involving RyRs has been demonstrated experimentally with the right timing to account for the termination of Ca2+ efflux from a CRU. Moreover, recent experimental studies on Ca2+-spark termination argue against the involvement of Ca2+-dependent inactivation in spark termination (35,36).
Leaky RyR
That RyRs open during diastolic conditions as individual channels to produce Iquark and as ensembles to produce Ispark is consistent with this new Ca2+ spark model. Indeed, it is necessary to include such properties of RyRs in the model to reproduce the recent experimental observation that when SERCA is blocked, [Ca2+]sr decreases due to both spark-mediated and nonspark-mediated mechanisms (8). The need for pump-leak balance in the SR has been nicely demonstrated both experimentally (8,9) and in the model here. The model also suggests how increased phosphorylation of the RyR during heart failure might increase the leakiness of the RyRs. Additionally, despite nonjunctional RyRs having increased Po due to the lack of allosteric coupling, they contribute only a small fraction of the total SR Ca2+ leak in this model.
SERCA
The Tran-Crampin SERCA formulation reproduces the experimental observation that Ca2+ uptake into the SR depends on both [Ca2+]i and [Ca2+]sr (37,38). The model predicts virtually no backflux of Ca2+ under physiological conditions, allowing entirely RyR-based leak to balance SERCA—providing a simple yet physiological system of pump/leak balance to the model. However, we acknowledge that the system may indeed be more complex (i.e., other leak pathways such as IP3Rs may also contribute to SR Ca2+ leak).
Ca2+-induced Ca2+ release
The manner by which Ca2+ sparks are produced suggests that the process is dominated by Ca2+-induced Ca2+ release (CICR). The initial probabilistic opening of a single RyR depends on its sensitivity to both [Ca2+]i and [Ca2+]sr. The opening and closing rates also depend on modulatable properties of RyR and those of interacting proteins (see above). The increase in [Ca2+]ds produced by the initial RyR opening is the primary trigger for diastolic Ca2+ sparks. However, during EC coupling the triggering is due largely to the increase in [Ca2+]ds from the opening LCCs in the sarcolemmal or transtubule membranes opposite the RyR cluster in the CRU. Our simulations suggest that when either a single RyR or LCC opens, the local RyR cluster is unlikely to be triggered. This feature, which is required here to reproduce the phenomenon of invisible RyR-mediated leak (7,8,15), has been suggested previously based on analysis of voltage-clamp experiments (29,30). As the intrinsic sensitivity of RyR increases due to phosphorylation, mutation, or other change, the remarkable stability of the system can change. Ca2+ instability of CICR may arise when [Ca2+]sr becomes elevated or when RyR Ca2+ sensitivity increases. Exploration of the features of instability will be addressed in a spatially resolved Ca2+-spark model.
Ca2+-spark characteristics
An important part of this model is to fit all major features of the experimental findings, in respect to Ca2+ sparks and SR Ca2+ leak, with the simplest model possible. These goals were achieved even as we constrained the behavior of RyRs and SERCA by their measured biophysical properties. The Ca2+-spark rate is similar to that observed in rats during diastole. Ca2+-spark characteristics for other species are largely the same, except for the Ca2+-spark rate, which is lower. Only modest changes to the model are needed to lower the Ca2+-spark rate. For example, slight reductions in diastolic [Ca2+]sr dramatically decrease Ca2+-spark rate.
Trigger characteristics
The likelihood of a Ca2+ spark occurring due to the opening of a single RyR is <10%. During the upstroke of a cardiac action potential, RyRs are triggered by openings of the voltage-gated LCCs. Given that a single LCC opening is known to contribute roughly the same Ca2+ current as an individual RyR and LCC openings are brief (∼0.5 ms) (33), therefore many LCCs must open during systole in order to trigger a spark. However, during systole, LCC openings are well synchronized by depolarization, ensuring adequate Ca2+ influx to trigger Ca2+ sparks. In fact, the presence of seven LCCs per CRU (consistent with the LCC/RyR ratio of 1:7 (39)) allows for robust triggering of the CRUs during a simulated depolarization (see Fig. S1).
In summary, the relatively simple mathematical model of Ca2+ sparks put forward by Sobie et al. (16) has been advanced significantly by the improvements in RyR Ca2+ sensitivity, inclusion of a physiological SERCA formulation, and the incorporation of these Ca2+-handling mechanisms into a detailed whole-cell model of CICR. This self-consistent, and to our knowledge, new model is highly constrained by experimental observations and forms the basis for a new generation of Ca2+ signaling simulations.
Acknowledgments
We acknowledge support from National Science Foundation DMS-0443843; National Institutes of Health 01 HL67849, R01 HL36974, F32 HL108604, and S10 RR023028; the Leducq North American-European Atrial Fibrillation Research Alliance; the European Union Seventh Framework Program (FP7); the Georg-August University “Identification and Therapeutic Targeting of Common Arrhythmia Trigger Mechanisms” program; and the Maryland Stem Cell Research Fund.
Supporting Material
References
- 1.Franzini-Armstrong C., Protasi F., Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann. N. Y. Acad. Sci. 1998;853:20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x. [DOI] [PubMed] [Google Scholar]
- 2.Baddeley D., Jayasinghe I.D., Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. USA. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stern M.D. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng H., Lederer W.J., Cannell M.B. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 5.Brochet D.X., Xie W., Lederer W.J. Quarky calcium release in the heart. Circ. Res. 2011;108:210–218. doi: 10.1161/CIRCRESAHA.110.231258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukyanenko V., Ziman A., Lederer W.J. Functional groups of ryanodine receptors in rat ventricular cells. J. Physiol. 2007;583:251–269. doi: 10.1113/jphysiol.2007.136549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobie E.A., Guatimosim S., Lederer W.J. The Ca2+ leak paradox and rogue ryanodine receptors: SR Ca2+ efflux theory and practice. Prog. Biophys. Mol. Biol. 2006;90:172–185. doi: 10.1016/j.pbiomolbio.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zima A.V., Bovo E., Blatter L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shannon T.R., Ginsburg K.S., Bers D.M. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 10.George C.H. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc. Res. 2008;77:302–314. doi: 10.1093/cvr/cvm006. [DOI] [PubMed] [Google Scholar]
- 11.Wehrens X.H., Lehnart S.E., Marks A.R. Intracellular calcium release and cardiac disease. Annu. Rev. Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 12.Marx S.O., Reiken S., Marks A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 13.Ai X., Curran J.W., Pogwizd S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 14.Wehrens X.H., Lehnart S.E., Marks A.R. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 15.Santiago D.J., Curran J.W., Shannon T.R. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys. J. 2010;98:2111–2120. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobie E.A., Dilly K.W., Jafri M.S. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys. J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin J., Valle G., Fill M. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J. Gen. Physiol. 2008;131:325–334. doi: 10.1085/jgp.200709907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groff J.R., Smith G.D. Ryanodine receptor allosteric coupling and the dynamics of calcium sparks. Biophys. J. 2008;95:135–154. doi: 10.1529/biophysj.107.119982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duke T.A., Bray D. Heightened sensitivity of a lattice of membrane receptors. Proc. Natl. Acad. Sci. USA. 1999;96:10104–10108. doi: 10.1073/pnas.96.18.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stern M.D., Song L.S., Ríos E. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 1999;113:469–489. doi: 10.1085/jgp.113.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran K., Smith N.P., Crampin E.J. A thermodynamic model of the cardiac sarcoplasmic/endoplasmic Ca2+ (SERCA) pump. Biophys. J. 2009;96:2029–2042. doi: 10.1016/j.bpj.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andrienko T.N., Picht E., Bers D.M. Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J. Mol. Cell. Cardiol. 2009;46:1027–1036. doi: 10.1016/j.yjmcc.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marx S.O., Gaburjakova J., Marks A.R. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ. Res. 2001;88:1151–1158. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 24.Williams G.S.B., Huertas M.A., Smith G.D. A probability density approach to modeling local control of calcium-induced calcium release in cardiac myocytes. Biophys. J. 2007;92:2311–2328. doi: 10.1529/biophysj.106.099861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams G.S., Huertas M.A., Smith G.D. Moment closure for local control models of calcium-induced calcium release in cardiac myocytes. Biophys. J. 2008;95:1689–1703. doi: 10.1529/biophysj.107.125948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukyanenko V., Viatchenko-Karpinski S., Györke S. Dynamic regulation of sarcoplasmic reticulum Ca2+ content and release by luminal Ca2+-sensitive leak in rat ventricular myocytes. Biophys. J. 2001;81:785–798. doi: 10.1016/S0006-3495(01)75741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin J., Valle G., Fill M. Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys. J. 2009;97:1961–1970. doi: 10.1016/j.bpj.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng H., Lederer W.J. Calcium sparks. Physiol. Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 29.Poláková E., Zahradníková A., Jr., Zahradníková A. Local calcium release activation by DHPR calcium channel openings in rat cardiac myocytes. J. Physiol. 2008;586:3839–3854. doi: 10.1113/jphysiol.2007.149989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobie E.A., Ramay H.R. Excitation-contraction coupling gain in ventricular myocytes: insights from a parsimonious model. J. Physiol. 2009;587:1293–1299. doi: 10.1113/jphysiol.2008.163915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Györke I., Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gómez A.M., Schuster I., Richard S. FKBP12.6 overexpression decreases Ca2+ spark amplitude but enhances [Ca2+]i transient in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1987–H1993. doi: 10.1152/ajpheart.00409.2004. [DOI] [PubMed] [Google Scholar]
- 33.Wang S.Q., Song L.S., Cheng H. Ca2+ signaling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 34.Picht E., Zima A., Bers D. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ. Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domeier T.L., Blatter L.A., Zima A.V. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J. Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens S.C.W., Terentyev D., Györke S. Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. J. Physiol. 2009;587:4863–4872. doi: 10.1113/jphysiol.2009.175547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanford C. Steady state of an ATP-driven calcium pump: limitations on kinetic and thermodynamic parameters. Proc. Natl. Acad. Sci. USA. 1982;79:6161–6165. doi: 10.1073/pnas.79.20.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bers D. 2nd Ed. Springer; New York: 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. [Google Scholar]
- 39.Bers D.M., Stiffel V.M. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am. J. Physiol. 1993;264:C1587–C1593. doi: 10.1152/ajpcell.1993.264.6.C1587. [DOI] [PubMed] [Google Scholar]
- 40.Smith G.D., Keizer J.E., Cheng H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys. J. 1998;75:15–32. doi: 10.1016/S0006-3495(98)77491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.