

Abstract
Titin (also known as connectin) is an intrasarcomeric muscle protein that functions as a molecular spring and generates passive tension upon muscle stretch. The N2B element is a cardiac-specific spring element within titin's extensible region. Our goal was to study the contribution of the N2B element to the mechanical properties of titin, particularly its hypothesized role in limiting energy loss during repeated stretch (diastole)-shortening (systole) cycles of the heart. We studied energy loss by measuring hysteresis from the area between the stretch and release passive force-sarcomere length curves and used both wild-type (WT) mice and N2B knockout (KO) mice in which the N2B element has been deleted. A range of protocols was used, including those that mimic physiological loading conditions. KO mice showed significant increases in hysteresis. Most prominently, in tissue that had been preconditioned with a physiological stretch-release protocol, hysteresis increased significantly from 320 ± 46 pJ/mm2/sarcomere in WT to 650 ± 94 pJ/mm2/sarcomere in N2B KO myocardium. These results are supported by experiments in which oxidative stress was used to mechanically inactivate portions of the N2B-Us of WT titin through cysteine cross-linking. Studies on muscle from which the thin filaments had been extracted (using the actin severing protein gelsolin) showed that the difference in hysteresis between WT and KO tissue cannot be explained by filament sliding-based viscosity. Instead the results suggest that hysteresis arises from within titin and most likely involves unfolding of immunoglobulin-like domains. These studies support that the mechanical function of the N2B element of titin includes reducing hysteresis and increasing the efficiency of the heart.
Introduction
During each heart beat the sarcomeres (contractile units of muscle) shorten during systole (the contraction phase of the heart cycle) and elongate during diastole (the filling phase). The cardiac sarcomere contains the giant protein titin that maintains the structural integrity of the sarcomere and that is responsible for the passive forces that determine the filling characteristics of the ventricle during the cardiac cycle (1–3). Single titin molecules span the half-sarcomeric distance, from the Z-disk to the middle of the A-band (4). The I-band region of titin is extensible and acts as an entropic spring that extends and provides passive tension to muscle (5,6). Titin's extensible I-band region is composed of two distinct elements that are found in all cardiac and skeletal muscle types: the tandem Ig segments that contain serially linked immunoglobulin-like (Ig-like) domains and the PEVK element (rich in proline (P), glutamate (E), valine (V), and lysine (K) residues) (7). In addition to these two elements that are found in all striated muscles, cardiac muscle expresses a specialized titin isoform, containing the N2B element. This isoform is called the N2B cardiac titin isoform (8) and corresponds to a splice isoform in which titin's exon 49 (also known as the N2B exon) is included, whereas exon 49 is skipped in skeletal muscles (8). This heart-specific exon 49 codes for 3 Ig domains (I24 and I25 at the N-terminus; I26 at the C-terminus) that flank a central large unique sequence (N2B-Us) that contains 530 residues in the mouse (9). In stretched cardiac muscle, the N2B-Us extends to a maximal length of ∼215 nm (10). Thus, the I-band region of the cardiac titin N2B isoform contains three spring elements: tandem Ig segments, the PEVK element, and the N2B element (8). In slack sarcomeres, titin's extensible region is in a compact and high conformational entropy state, and sarcomere stretch initially extends the tandem Ig segment (10); such stretch is thought to be due to straightening of the sequences that link the Ig domains while the domains themselves are thought to remain folded as β-barrel structures (10,11). Next, the N2B-Us and the PEVK region extend, largely due to straightening of random coil structures (12,13). Each of titin's three extensible regions has distinct properties and is thought to serve different functions during sarcomeric stretch (13,14).
In addition to the roles already known for the N2B region of titin, such as in protein kinase signaling (15,16) and anchoring signaling molecules (17,18), the N2B element has been hypothesized to limit energy loss during repeated stretch (diastole)-shortening (systole) cycles of the heart (12). Passive energy loss arises from differences in energy required to stretch passive restoring force elements during diastole and the energy retrieved from these elements when sarcomeres shorten during systole. This difference in loading and unloading energies is known as hysteresis. In this study we investigated the functional role of the N2B element with respect to hysteresis. We hypothesized that cardiac tissue has incorporated the N2B-Us to reduce hysteresis. To test this we used the N2B knockout (N2B KO) mouse model in which the N2B element has been excised (19), while leaving the remainder of the molecule intact. We used a variety of precision mechanical assays to elucidate the possible changes in hysteresis caused by the removal of the N2B element. We also incorporated a thin filament extraction technique (using the F-actin severing protein gelsolin (20)) to differentiate intrinsic titin hysteresis from extrinsic hysteresis arising from thin filament-titin interactions and thin filament-thick filament interactions. Finally, oxidative conditions were used to induce intramolecular disulfide cross-links between cysteine groups in the N2B-Us (21). It has been shown in single molecule studies that these links mechanically hide the protein sequences between two linked cysteines, effectively reducing the length of the N2B unique sequence (21), providing a complementary approach to our genetic N2B KO model.
Methods
Skinned muscle
Papillary muscles were harvested from male 5-month-old N2B KO and WT mice on a C57BL/6 background. All experiments were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of The University of Arizona. Muscle preparations were dissected with details and solution compositions as previously described (22). Briefly, skinned muscles were dissected into fiber bundles (0.026 ± 0.001 mm2 cross-sectional area) and secured to a servo-motor (308B, Aurora Scientific, Aurora, Ontario, Canada) and force transducer (SensorOne AE801, Kronex Technologies, Oakland, CA) on either end with aluminum T-clips. Tension and sarcomere length (SL) were recorded simultaneously with the force transducer and a calibrated laser diffraction system. (The resolution of the laser diffraction system was measured by using small amplitude sinusoidal oscillations in muscle length and was found to be ∼10 nm/sarcomere.) Muscles were set to slack SL, defined as SL with zero passive stress. Active tension was used as a measure of tissue health by activating the skinned fibers with pCa 4.0 activating solution (22). Total passive tension was measured with a variety of mechanical protocols used to probe the passive energetic differences between WT and KO skinned tissue (described in detail below). After total passive tension had been collected, high molar 0.6 M KCl and 1.0 M KI solutions were used to depolymerize thick and thin filaments, respectively, thereby removing anchoring sites for titin (23). With anchoring sites removed, titin no longer contributes to passive tension within the sarcomere, the mechanical protocols were then repeated and the remaining passive tensions were attributed to the extracellular matrix (ECM). By subtracting the extraction insensitive tensions from total tension, extraction sensitive tension was calculated, which is attributed to titin (23). All reported passive tensions (except where noted) represent titin tension.
The slack SL was determined by shortening the passive muscle until it clearly buckled, holding it there for 10 min, and then stretching the muscle to the minimal length at which the buckle was taken up and where no detectable passive force was developed, at which point the SL was measured by laser diffraction. After slack length had been determined and the tissue had been activated and relaxed once, one of three different mechanical protocols were run. The first protocol investigated peak tension and hysteresis in rested muscle and consisted of single triangle stretches from slack to specified SLs (2.1, 2.2, and 2.3 μm) at each of three different strain rates (10%, 100%, and 1000% base length/s), for a total of nine stretches. Between each stretch a period of rest was allotted (a minimum of 5 min, with longer rest periods for larger stretches) to allow the tissue to fully recover. A second mechanical protocol evaluated the time it takes to fully recover from an initial stretch. This protocol consisted of a series of stretch-release pairs, initially rested muscle was stretched from slack to 2.2 μm at 10%/s. After this stretch the tissue was returned to slack at 1000%/s and allowed to rest for a period between 0 and 120 s. After resting a set amount of time a second triangle stretch-release from slack to 2.2 μm was performed to evaluate recovery progression. After each stretch pair the tissue was allowed to rest for 20 min to ensure full recovery before evaluating recovery again. A third protocol mimicked the cardiac cycle. In this protocol a series of tandem triangle stretches from slack to an SL of 2.2 μm at 300%/s (this combination of stretch amplitude and stretch speed provides a cyclical rate of ∼600 beats/min) were repeated up to 12,000 cycles. The stretch speed and maximal SL approximate the physiological range during diastole (3). The physiological SL range in the mouse heart was recently estimated between 1.8 μm at the end of the ejection phase and 2.2 μm at the end of diastole (3). The end-systolic SL value is likely to be a lower-end estimate because it was based on studying barium contractions of unloaded ventricles. Using a whole animal x-ray diffraction technique Toh et al (24) deduced an end-systolic SL of 1.9 μm for the in situ beating mouse heart. Considering that in Toh et al (24) anesthetized mice were studied, which depresses contractility, it seems likely that the SL range was slightly underestimated in that study. Thus, the in vivo end-systolic SL in the mouse is ∼1.8–1.9 μm, the end-diastolic SL is ∼2.2 μm, and the SL range that we studied (1.86–2.2 μm for N2B KO and 1.89–2.2 μm for WT muscle; see also Results) reflects well the physiological SL range.
Disulfide bond formation
To preclude cross-linking between actin and either myosin or titin, we studied tissue from which the thin filaments had been extracted with gelsolin (for details, see (20)). Tandem triangle stretches, from slack to 2.2 μm at 600 cycles/min, were imposed on the tissue as described previously. Initially preparations were bathed in regular relaxing solution for the first 10 min of mechanical perturbations. Once fully preconditioned (i.e., stretch-released by >400 cycles and having a steady-state peak tension and steady-state hysteresis), the solution was switched to relaxing solution without the reducing agent DTT (dithiothreitol), after which the oxidant H2O2 (5 mM) was added to the relaxing solution. This concentration is within values used to induce oxidative states in other studies (25,26). After cross-linking, the solution was switched to a relaxing solution free of DTT and H2O2. Finally, relaxing solution with 10 mM DTT was used to dissociate cross-links.
Calculations
Hysteresis values were calculated in a custom program written in the open source language Ruby with a link to MATLAB (The MathWorks, Natick, MA), where area under the loading and unloading curves was totaled with trapezoidal integration using the trapz subroutine.
Statistics
Data are presented as mean ± SE. Significant differences were probed using student's t-test. Probability values <0.05 were taken as significant.
Results
The N2B KO expresses a mutant titin that migrates slightly faster than WT titin on high-resolution sodium dodecyl sulfate-agarose gels (which is especially apparent in heterozygous samples, see Fig. 1 A), consistent with the deletion of the ∼80 kDa N2B element from the ∼3000 kDa WT protein (19). We first measured the maximal active tension (pCa 4.0) at an SL of 2.0 μm (Fig. 1 B) and found that the KO and WT myocardium generate similar active stress levels: 48 ± 4 mN/mm2 (KO) vs. 42 ± 3 mN/mm2 (WT). The slack SL was carefully established as explained in the Methods. Consistent with previous work on N2B KO single cardiac myocytes (19) the slack SL was significantly shorter in KO than WT muscle (1.86 ± 0.01 μm (KO) vs.1.89 ± 0.01 μm (WT), Fig. 1 C).
Figure 1.
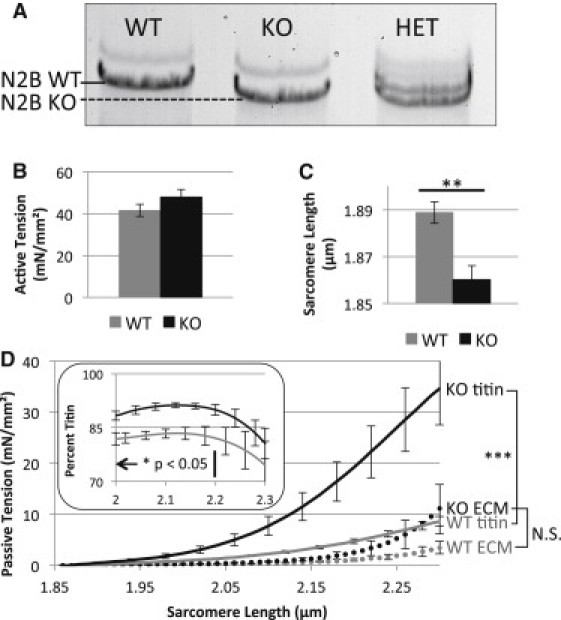
Basic characterization of N2B KO skinned myocardium. (A) Gel electrophoresis of WT, N2B KO, and N2B heterozygous papillary muscle. KO titin has a slightly higher mobility compared to WT titin. (B) Active tension in KO and WT are not significantly different. (C) Slack sarcomere length of KO is significantly less than in WT tissue. (D) Passive tension—SL curves for ECM and titin. Although mean ECM-based tension is slightly elevated in N2B KO mice, this increase is not significant. Titin tension is significantly increased at SLs ≥ 1.9 μm. (Inset) Relative contribution of titin to total passive tension in WT (gray) and KO (black).
The passive tension-SL relation was characterized by stretching rested muscle from its slack length to a final SL of 2.3 μm with a velocity of 10%/s in relaxing solution. Total, KCl/KI ECM, and KCl/KI extraction sensitive (titin) passive tensions were calculated (see Methods) and are shown in Fig. 1 D. Although the ECM-based tension trended higher at longer SL in N2B KO tissue, the difference was not significant. Titin-based passive tension, on the other hand, was significantly higher in KO tissue, and for example at SL 2.3 μm, developed 35 ± 7 mN/mm2 of passive tension vs. 9 ± 1 mN/mm2 in WT (Fig. 1 D). Dividing titin tension by total tension, the percent contribution of titin was evaluated. Titin contribution to total passive tension was significantly greater in KO compared to WT tissue at SL < 2.2 μm (Fig. 1 D, inset).
To evaluate the passive energetic changes due to removal of the N2B region, we first stretched rested fiber bundles from slack to three different SLs (2.1, 2.2, and 2.3 μm) at three different speeds (10%, 100%, and 1000%/s). We measured peak passive tension and calculated hysteresis by integrating the area between the tension-SL curves of the loading and unloading portions of a stretch-release triangle stretch protocol (Fig. 2 A). Peak tension was significantly larger in the KO compared to WT tissue in all nine stretch patterns (Fig. 2 B). Hysteresis was also significantly larger in KO muscles at all strain amplitudes and strain rate combinations (Fig. 2 C). The result at SL 2.2 μm is displayed in Fig. 2 D and reveals an ∼5-fold higher hysteresis in KO tissues.
Figure 2.
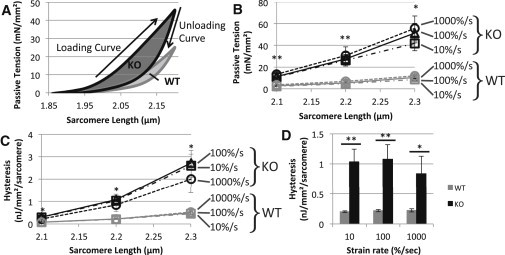
Speed dependence of passive tension and hysteresis in rested WT and N2B KO skinned myocardium. (A) Protocol. The skinned muscle was first rested below the slack length (for >5 min), set to slack, and then stretched with a predetermined speed to a predetermined peak SL (in this example 100%/s and 2.3 μm, respectively) followed by a release back to slack with the same speed. We measured peak tension at the end of the stretch and hysteresis (area between the loading and unloading curves, highlighted in gray). (B) Peak titin tension was significantly higher in skinned muscle fibers of N2B-KO mice at every speed and at every peak SL tested. (C) Titin hysteresis increased significantly in skinned muscle fibers of N2B-KO mice at every speed when stretched from rest at slack length to every SL tested. (D) Bar graphs showing hysteresis differences in WT and KO tissue stretched at a variety of stretch speeds to a final SL of 2.2μm.
To evaluate whether similar mechanisms underlie hysteresis in WT and KO muscle, we studied hysteresis recovery and reasoned that if the recovery time constants were greatly different the mechanisms would be different as well. Muscles were stretched from slack length to 2.2 μm at 10%/s followed by a rapid release back to the original length in 10 ms; following a rest period of variable duration a second stretch was performed at 10%/s to an SL of 2.2 μm (protocol further explained in Fig. 3 A). The force-SL trace was similar between the initial stretch and the stretch following rest in both the WT and KO for long rest periods (>30 s) but differed substantially at short rest periods (<∼10 s). The difference was quantified as percent recovery by dividing the area under the loading curve of the second stretch by that of the initial stretch (Fig. 3 B). Time constants of 0.16 and 0.14 s−1, WT and KO, respectively, were derived from a single exponential time relationship fit with a nonlinear least squares method to the aggregate data (Nelder-Mead simplex algorithm, R2 > 0.8). No significant differences were found between WT and KO muscles.
Figure 3.
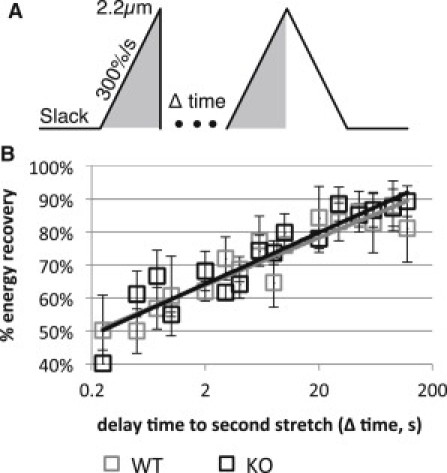
Hysteresis recovery in WT and N2B KO skinned myocardium. (A) Protocol. After an initial stretch to SL 2.2 μm at 300%/s, the preparation was returned within ∼10 msec to slack length and allowed to rest for a variable amount of time. A second stretch was then imposed with the same characteristics as the first. (B) Recovery with respect to rest time did not show a differentiation between KO and WT tissue. Line shows best fits through the aggregate data.
Results shown above were obtained on muscles that had been rested at their slack length. This is very different from in the living mouse where titin is continuously stretch-released in rapid succession at a rate of ∼600 beats/min. To more accurately represent physiology, a repeated stretch-release protocol was used from slack SL to 2.2 μm at a speed of 300%/s, approximating physiological conditions (Fig. 4 A). During this protocol, the initial stretch had tensions consistent with rested tissues. However, as these triangle stretches were repeated the peak tension decreased in an exponential manner to ∼60% of its rested value. Additionally, in both WT and KO tissue, hysteresis was highest in the first stretch-release cycle, then rapidly decreased during the subsequent five cycles, after which the decrease was more slowly to reach a steady value after ∼200 cycles (Fig. 4 B). A 20-min rest at slack fully recovered both peak tension and hysteresis, indicating that the decay in hysteresis and passive tension during the repeated stretches is a reversible process and is not due to permanent damage. Hysteresis started off much higher in the KO tissue and leveled off at a significantly higher level as well. After 200 cycles hysteresis were 650 ± 94 pJ/mm2/sarcomere in KO and 320 ± 46 pJ/mm2/sarcomere in WT muscle (p-value <0.05).
Figure 4.
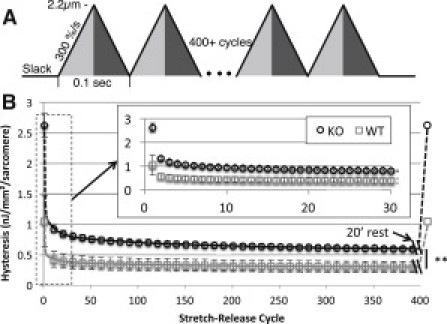
Hysteresis in WT and N2B KO skinned myocardium under physiological loading conditions. (A) Protocol. Triangle stretch patterns were repeated in tandem to simulate the cyclical loading of the heart. These stretches imposed a displacement from slack to 2.2 μm (light gray) and back to slack (dark gray) at a rate of 10Hz. (B) Titin hysteresis versus stretch-release cycle count. Passive hysteresis is elevated significantly in skinned muscle fibers of N2B-KO mice at all cycles. Main graph, every 10th cycle is shown up to 400 cycles. (Inset) The first 30 cycles are shown. After the full stretch-release pattern, the tissue was allowed to rest at slack for 20 min after which the stretch-release cycles were started again. Results showed that hysteresis was fully recovered (last data point in main graph).
We also studied hysteresis in skinned muscle in which the N2B-Us contour length was reduced by using oxidative cross-linking. The large unique sequence that comprises the extensible region of the N2B element contains cysteine residues that form disulfide cross-links under oxidative states (21). These cross-links functionally hide part of the N2B-Us by removing the spring-like properties of the amino acid chain between the two cysteines (see schematic of Fig. 5 left). Oxidative states were induced in WT and KO tissue while undergoing the physiological stretch-release protocol, triangle stretches from slack to 2.2 μm at 300%/s. Before oxidative conditions were induced, thin filaments were extracted with gelsolin to minimize cross-links other than in the N2B-Us, for instance myosin cross-links to actin or titin cross-links to the thin filament. H2O2 increased passive tension in WT muscle by 41 ± 5%, which is significantly more than the 11 ± 4% in KO tissue, p-value <0.01 (Fig. 5 A). Similarly, the percent increase in hysteresis response was 32 ± 5% in WT tissue, which is significantly greater than 13 ± 2% increase seen in KO tissue, p-value <0.05 (Fig. 5 B). Both peak tension and hysteresis returned to pre-H2O2 levels in WT and KO tissue after the addition of DTT, showing the reversibility of this effect (Fig. S1 in the Supporting Material).
Figure 5.
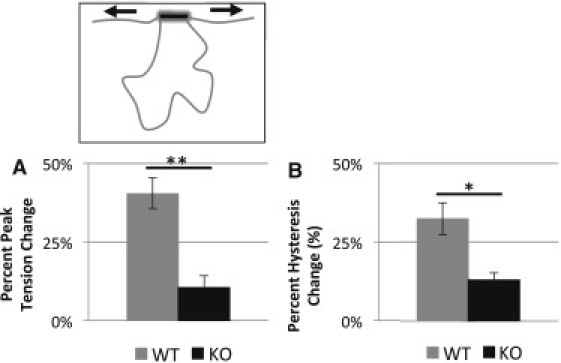
Effect of oxidation on passive tension and hysteresis. Oxidative stress response while using a physiological cyclical loading pattern. When the tissue had been fully preconditioned, 5 mM hydrogen peroxide was added to form cysteine disulfide cross-links (schematically shown above). (A and B) Bar graphs showing the increases in peak passive tension (A) and hysteresis (B) in response to oxidation. KO tissue has a significantly smaller increase in passive tension and hysteresis compared to WT.
To elucidate the contribution of interfilament viscosity to hysteresis, a series of thin filament extraction experiments using gelsolin were conducted to remove potential interactions between the thin and thick filament as well as the thin filament and titin. By removing these potential interactions we isolated passive stress and hysteresis intrinsic to titin. We tested both gelsolin extracted WT and KO muscle against their respective genotype matched nongelsolin extracted controls with the triangle physiological stretch described in Fig. 4 A. In both WT and KO tissues the removal of the thin filaments reduced initial and steady-state peak passive tensions equally by ∼50% compared to nongelsolin extracted control muscle. On the other hand, hysteresis was significantly reduced in the initial rested triangle stretch by 60% (Fig. 6, A and B), but was not significantly different at steady state (Fig. 6 C); again, this was consistent in both WT and KO tissue. Importantly, steady-state hysteresis in thin-filament extracted KO muscle was significantly higher compared to thin-filament extracted WT tissue (Fig. 6 A, bottom right asterisks), indicating that the increased passive hysteresis in N2B KO tissue is due to a mechanism that is intrinsic to titin.
Figure 6.
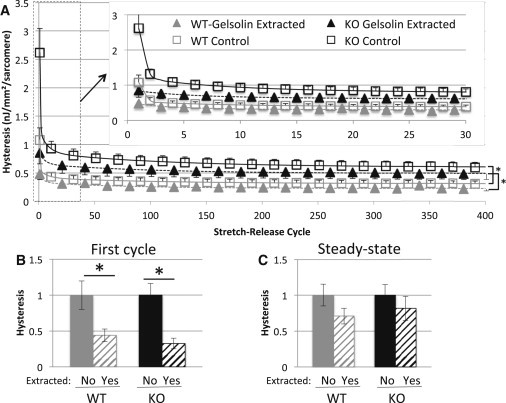
Physiological hysteresis in thin filament extracted myocardium. (A) Hysteresis as a function of cycle number. WT tissue is shown in gray, KO tissue is shown in black. Gelsolin-extracted tissue is indicated by solid triangles while control (nongelsolin extracted) tissue is shown by open square boxes. For the larger graph, every 20th point is shown with the selected gelsolin extracted points offset from nonextracted points by 10 cycles (for clarity reasons). The inset shows an expanded version of the first 30 cycles. (B and C) Hysteresis of the initial stretch-release cycle (B) and the steady state after 400 cycles (C). (Hysteresis is normalized to that of the unextracted tissue.) Hysteresis is decreased by ∼60% for the initial triangle stretch in gelsolin-extracted rested muscle compared to genotype-matched controls. However, this difference quickly diminishes and steady-state hysteresis values are not significantly different in extracted versus nonextracted tissues.
Discussion
Deficiency in the N2B region of titin significantly increases passive tension and energy loss in loading-unloading cycles. Using repeated stretch-release cycles on rested myocardium, hysteresis starts out large, but quickly decreases to settle at a steady-state value where the KO has an ∼2-fold increase in hysteresis over WT tissue. Thin-filament extraction established that filament sliding viscosity greatly contributes to the initial hysteresis of rested muscle but cannot explain the increased steady-state hysteresis in physiological stretch-release protocols of KO myocardium relative to WT. Below we discuss our results in detail.
The N2B element functions as an extensible spring within titin that contributes up to ∼215 nm to titin's extensibility (10). Myocardium deficient in the N2B region of titin develops increased passive tension. This finding can be understood by the increase in strain that the remaining elements (tandem Ig and PEVK) experience for the same sarcomere deformation in KO compared to WT tissue. Removing a large portion of titin's extensible region is also expected to reduce the slack SL in the KO model by the distance contributed by the N2B element under no force. This distance is approximated by the unloaded mean square separation for a wormlike chain: (27). (LC is the contour length of the chain (215 nm) and LP is the persistence length (0.65 nm) (13)). This equates to ∼17 nm/half sarcomere, or ∼34 nm/sarcomere. This value is close to the measured difference in slack SL of 30 nm (1.89–1.86 μm, WT and KO, respectively). The measured SL reduction is slightly smaller than previously reported for single cardiac myocytes (19) and this may arise because myocytes experience a residual amount of active tension during diastole (28), or, alternatively, it might indicate that the extracellular matrix exerts a restoring force below SL 1.86 μm.
Oxidative conditions were used to induce intramolecular cross-links within the N2B-Us, creating disulfide bonds between cysteine groups in the sequence. Single molecule studies have shown that these links mechanically hide the protein sequence demarcated by two linked cysteines, effectively reducing the contour length, LC, of the unique sequence (21). Aligning the human N2B-Us used in the Grützner et al. (9) study with the mouse N2B-Us, 4 of the 6 predicted cysteine-bonding residues are conserved. On average, 242 residues separate the conserved residues; with a maximum residue spacing of 0.38 nm for an unfolded polypeptide, this reduces the LC of the N2B-Us by on average 92 nm, a little under half of the 201 nm (530 residues × 0.38 nm) LC of the N2B-Us in the mouse. (Note that the PEVK region does not contain cysteines.) Thus, the oxidative cross-linking of the N2B-Us provides a reversible mechanism to complement the genetic N2B full length KO model for the study of the mechanical role of the N2B-Us. Oxidation increases both passive tension and hysteresis (Fig. 5) and these results support that the removal of the N2B is the cause of increased passive tension and energy loss in the N2B KO.
Of the possible sources of passive hysteresis in muscle, the two most relevant types are 1), viscosity that arises from filament sliding and 2), structural transitions within titin, particularly Ig domain unfolding. Filament sliding viscosity occurs during the sarcomere stretch/release cycle and might arise from thin and thick filament sliding and/or thin and titin filament sliding. Viscosity opposes the sliding motion on both the ascending and descending limbs of the triangle stretch, increasing tension during the loading cycle and decreasing it during the unloading cycle. To differentiate between interfilament hysteresis and hysteresis arising from within titin, thin filaments were extracted with gelsolin. Most notable from these experiments is the difference between thin filament extracted and nonthin filament extracted hysteresis of the initial stretch imposed on rested tissue (Fig. 6 B). Hysteresis is greatly reduced initially in gelsolin-extracted muscle, relative to unextracted controls, but as it is preconditioned and the tissue enters steady state, this difference decreases to nonsignificant levels (Fig. 6 C). This suggests that the initial well-rested hysteresis has a large interfilament interaction component. A likely source is PEVK-actin interaction that is known to take place in cardiac muscle (29,30). Our work suggests that this is most prominent in rested muscle and that it weakens during preconditioning. Because steady-state hysteresis is insensitive to thin filament removal, its source is likely to reside within titin.
In the case of the N2B KO, the source of hysteresis within titin has to be the PEVK and/or the tandem Ig segment. The PEVK consists of mostly random coil with interspersed short polyproline type II (PPII) helices (31). During stress, these secondary structures may unfold causing hysteresis. For cardiac N2B titin a total of only 7 PEVK residues out of 188 are predicted to participate in PPII helix folding (13) and the contour length (LC) gain from their unfolding, estimated at 0.5 nm (13), would produce very little hysteresis. Indeed, stretch-release curves of single PEVK molecules largely overlap with little evidence for hysteresis (13). On the other hand, LC gain from Ig domain unfolding is ∼30 nm/unfolding event (13), large enough to cause significant amounts of hysteresis. Although it has been argued that Ig unfolding is unlikely to take place in WT titin (10,32,33), unfolding of a few domains during stretch cannot be excluded and several studies have given support to this idea (34,35). Additionally, considering that deletion of the N2B element abolishes ∼215 nm of extensibility within titin, unfolding is more likely to occur in the N2B deficient titin and, thus, Ig unfolding is a strong candidate to account for the increase in hysteresis of N2B KO mice. In fact, the maximum extension possible without Ig unfolding in the N2B KO is ∼2.26 μm. In the KO the total contour length (LC) of titin's extensible region is 70 nm (PEVK) plus 160 nm (36 Ig domains in the tandem Ig segment × 4.5 nm/domain) = 230 nm; 1.8 μm is the SL at which the extensible region has an end-to-end length of ∼0 nm (10). Thus, in the KO, titin's extensibility will be exhausted at SL 2.26 μm (1.8 μm + 2 × 0.23 μm) and unfolding must occur at or before this SL. Because passive tension approximates a linear relationship to SL starting at ∼2.15 μm (Fig. 1 D), and does not progressively increase in steepness as expected if unfolding were to be absent, we consider it likely that unfolding occurs at a wide SL range.
Another indication that Ig domain unfolding contributes to hysteresis can be obtained from the ratio between hysteresis and tension determined at different strain rates. Ig domain unfolding during stretch reduces peak tension (due to the abrupt length increase of the domain from ∼4.5 to ∼35 nm (36) and this decreases tension) and it increases hysteresis of the stretch-release cycle. Thus, increased unfolding during stretch will increase the hysteresis/tension ratio. Another insight that is needed is the stretch-speed dependence of unfolding. Because of the kinetic nature of unfolding, when stretch speed is increased, less time is spent at lower forces, reducing the probability that domains will unfold at those forces and allowing greater passive tensions to develop (13). Combined, this leads to the prediction that if hysteresis is due to unfolding of Ig domains, the hysteresis/tension ratio will decrease with stretch speed. Note that hysteresis based on interfilament sliding viscosity will give rise to a hysteresis/tension ratio that increases in proportion to stretch speed (this is because the viscous forces acts on both the loading and unloading curves, increasing hysteresis more than peak tension). Results shown in Fig. 7 indicate that in both WT and KO muscle, at all three strain amplitudes, as stretch speed increases the hysteresis/tension is reduced. Knowing that in KO myocardium Ig domain unfolding is required in triangle stretches to 2.3 μm, the relationship between the hysteresis/tension ratio and stretch speed is used as a positive control for Ig domain unfolding. The similarity between KO and WT tissue in the strain-rate dependence against normalized hysteresis (Fig. 7) suggests that Ig domain unfolding contributes to hysteresis in both KO and WT muscle.
Figure 7.
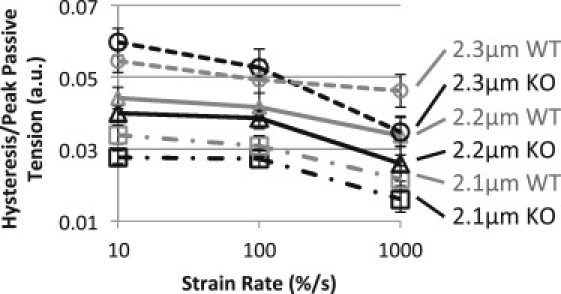
Hysteresis peak tension ratio. Hysteresis normalized by peak passive tension for each stretch. This ratio increases as tissue is stretched to longer SLs and decreases with increasing strain rate. See Discussion for details.
Additionally, hysteresis recovery experiments also indicate that the mechanism behind hysteresis might be the same in WT and KO tissue. Hysteresis recovery time constants were similar in WT and KO tissue, 0.16 and 0.14 s−1, respectively. These values are somewhat lower than obtained in recovery experiments in single molecule studies, ∼1 s−1 (37). This could arise from the differences in stretch patterns. We used an asymmetric triangle stretch with a very rapid release, whereas the single molecule work used symmetrical triangles, and the refolding process could have already begun during the slow release, shortening the rest time in between stretches required for full recovery. Additionally, in single molecule experiments, one molecule is allowed to refold in a huge volume of space. However, in the sarcomere molecular crowding may influence refolding of titin domains, possibly explaining why the hysteresis recovery rates are slower in the tissue than in the single molecule experiments.
The results of our studies indicate that both interfilament viscous forces and Ig domain unfolding-refolding cause the initial rested hysteresis. During subsequent stretch-release cycles interfilament forces contribute less while the majority of hysteresis is likely to be contributed by Ig domain unfolding. It is interesting to highlight that our data suggest that a limited degree of Ig domain unfolding occurs in WT myocardium. We speculate that having domains that are not fully resistant to unfolding might be advantageous under some conditions, such as to accommodate an increase in end-diastolic sarcomere length that occurs for example in response to increased preload. The removal of the large compliant N2B-Us from titin causes a significant increase in hysteresis, most likely due to additional Ig unfolding. To evaluate this increase in hysteresis on a physiological scale, we calculated the percent of total energy consumption saved by incorporating the N2B-Us into cardiac titin. Total energy consumption by the left ventricle (LV) per beat was calculated by multiplying stroke volume, 31 μL (38), by mean arterial pressure, 112 mmHg (39), which after unit conversion is 463 μJ (3.1 × 10−9 m3 × 14,900 N/m2) of mechanical energy exerted per beat. This value is compared against the increased hysteresis in KO over WT tissue. First, measured hysteresis/mm2/sarcomere was converted to hysteresis/mm3 of LV. LV wall muscle volume was approximated from the known LV mass of WT and N2B KO mice (19) divided by muscle density (1.06 g/cm3). This results in an approximate energy loss due to hysteresis in preconditioned tissue, of 18.8 μJ/beat and 26.2 μJ/beat for WT and KO tissue, respectively. Thus, including the N2B element in WT titin lowers the energy loss due to hysteresis by 7.9 μJ/beat, which corresponds to ∼2% of the 463 μJ pump energy needed per beat. Considering that the mouse heart rhythmically beats at 10Hz, this savings is likely to be functionally advantageous.
In summary, mechanical studies that mimic the physiological loading conditions of the heart reveal that the N2B region of titin significantly lowers the energy loss through hysteresis by shielding the tandem Ig domains from high stress and reducing the probability of unfolding events. Thus, the cardiac-specific N2B element minimizes energy loss, and we propose that due to its rhythmic activity pattern, this is an important aspect of cardiac function. Our work also supports that oxidative conditions result in increased passive tension and hysteresis, most likely through cross-linking of cysteine residues within the N2B region titin. Considering the prevailing oxidative conditions in heart failure patients (40), this might contribute to the increased diastolic wall stress and reduced energetic efficiency of these patients and warrants future investigation.
Acknowledgments
We thank the members of the Granzier laboratory for help and assistance.
The National Institutes of Health grant HL62881 and the Biomedical Engineering National Institutes of Health Cardiovascular Training Grant (T32HL007955) funded this work.
Supporting Material
References
- 1.LeWinter M.M., Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krüger M., Linke W.A. Titin-based mechanical signaling in normal and failing myocardium. J. Mol. Cell. Cardiol. 2009;46:490–498. doi: 10.1016/j.yjmcc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Chung C., Granzier H. Contribution of titin and extracellular matrix to passive pressure and measurement of sarcomere length in the mouse left ventricle. J. Mol. Cell. Cardiol. 2011;50:731–739. doi: 10.1016/j.yjmcc.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fürst D.O., Osborn M., Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell Biol. 1988;106:1563–1572. doi: 10.1083/jcb.106.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kellermayer M.S., Smith S.B., Granzier H.L. Complete unfolding of the titin molecule under external force. J. Struct. Biol. 1998;122:197–205. doi: 10.1006/jsbi.1998.3988. [DOI] [PubMed] [Google Scholar]
- 6.Trombitás K., Redkar A., Granzier H. Extensibility of isoforms of cardiac titin: variation in contour length of molecular subsegments provides a basis for cellular passive stiffness diversity. Biophys. J. 2000;79:3226–3234. doi: 10.1016/S0006-3495(00)76555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labeit S., Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 8.Bang M.L., Centner T., Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 9.Granzier H., Radke M., Labeit S. Functional genomics of chicken, mouse, and human titin supports splice diversity as an important mechanism for regulating biomechanics of striated muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R557–R567. doi: 10.1152/ajpregu.00001.2007. [DOI] [PubMed] [Google Scholar]
- 10.Trombitás K., Freiburg A., Granzier H. Molecular dissection of N2B cardiac titin's extensibility. Biophys. J. 1999;77:3189–3196. doi: 10.1016/S0006-3495(99)77149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trombitás K., Wu Y., Granzier H. Molecular basis of passive stress relaxation in human soleus fibers: assessment of the role of immunoglobulin-like domain unfolding. Biophys. J. 2003;85:3142–3153. doi: 10.1016/S0006-3495(03)74732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helmes M., Trombitás K., Granzier H. Mechanically driven contour-length adjustment in rat cardiac titin's unique N2B sequence: titin is an adjustable spring. Circ. Res. 1999;84:1339–1352. doi: 10.1161/01.res.84.11.1339. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe K., Nair P., Granzier H. Molecular mechanics of cardiac titin's PEVK and N2B spring elements. J. Biol. Chem. 2002;277:11549–11558. doi: 10.1074/jbc.M200356200. [DOI] [PubMed] [Google Scholar]
- 14.Li H., Linke W.A., Fernandez J.M. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki R., Wu Y., Granzier H. Protein kinase A phosphorylates titin's cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 2002;90:1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 16.Krüger M., Kötter S., Linke W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 17.Sheikh F., Raskin A., Chen J. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lange S., Auerbach D., Ehler E. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J. Cell Sci. 2002;115:4925–4936. doi: 10.1242/jcs.00181. [DOI] [PubMed] [Google Scholar]
- 19.Radke M.H., Peng J., Gotthardt M. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc. Natl. Acad. Sci. USA. 2007;104:3444–3449. doi: 10.1073/pnas.0608543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granzier H., Kellermayer M., Trombitás K. Titin elasticity and mechanism of passive force development in rat cardiac myocytes probed by thin-filament extraction. Biophys. J. 1997;73:2043–2053. doi: 10.1016/S0006-3495(97)78234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grützner A., Garcia-Manyes S., Linke W.A. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys. J. 2009;97:825–834. doi: 10.1016/j.bpj.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee E.J., Peng J., Granzier H.L. Calcium sensitivity and the Frank-Starling mechanism of the heart are increased in titin N2B region-deficient mice. J. Mol. Cell. Cardiol. 2010;49:449–458. doi: 10.1016/j.yjmcc.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Y., Cazorla O., Granzier H. Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J. Mol. Cell. Cardiol. 2000;32:2151–2162. doi: 10.1006/jmcc.2000.1281. [DOI] [PubMed] [Google Scholar]
- 24.Toh R., Shinohara M., Yagi N. An x-ray diffraction study on mouse cardiac cross-bridge function in vivo: effects of adrenergic beta-stimulation. Biophys. J. 2006;90:1723–1728. doi: 10.1529/biophysj.105.074062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slezak J., Tribulova N., Singal P.K. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and x-ray microanalysis. Am. J. Pathol. 1995;147:772–781. [PMC free article] [PubMed] [Google Scholar]
- 26.Avner B.S., Hinken A.C., Solaro R.J. H2O2 alters rat cardiac sarcomere function and protein phosphorylation through redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H723–H730. doi: 10.1152/ajpheart.00050.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rivetti C., Guthold M., Bustamante C. Scanning force microscopy of DNA deposited onto mica: equilibration versus kinetic trapping studied by statistical polymer chain analysis. J. Mol. Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 28.King N.M., Methawasin M., Granzier H. Mouse intact cardiac myocyte mechanics: cross-bridge and titin-based stress in unactivated cells. J. Gen. Physiol. 2011;137:81–91. doi: 10.1085/jgp.201010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamasaki R., Berri M., Granzier H. Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys. J. 2001;81:2297–2313. doi: 10.1016/S0006-3495(01)75876-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulke M., Fujita-Becker S., Linke W.A. Interaction between PEVK-titin and actin filaments: origin of a viscous force component in cardiac myofibrils. Circ. Res. 2001;89:874–881. doi: 10.1161/hh2201.099453. [DOI] [PubMed] [Google Scholar]
- 31.Ma K., Kan L., Wang K. Polyproline II helix is a key structural motif of the elastic PEVK segment of titin. Biochemistry. 2001;40:3427–3438. doi: 10.1021/bi0022792. [DOI] [PubMed] [Google Scholar]
- 32.Linke W.A., Granzier H. A spring tale: new facts on titin elasticity. Biophys. J. 1998;75:2613–2614. doi: 10.1016/S0006-3495(98)77706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witt C.C., Ono Y., Labeit S. Induction and myofibrillar targeting of CARP, and suppression of the Nkx2.5 pathway in the MDM mouse with impaired titin-based signaling. J. Mol. Biol. 2004;336:145–154. doi: 10.1016/j.jmb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 34.Minajeva A., Kulke M., Linke W.A. Unfolding of titin domains explains the viscoelastic behavior of skeletal myofibrils. Biophys. J. 2001;80:1442–1451. doi: 10.1016/S0006-3495(01)76116-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soteriou A., Clarke A., Trinick J. Titin folding energy and elasticity. Proc. Biol. Sci. 1993;254:83–86. doi: 10.1098/rspb.1993.0130. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe K., Muhle-Goll C., Granzier H. Different molecular mechanics displayed by titin's constitutively and differentially expressed tandem Ig segments. J. Struct. Biol. 2002;137:248–258. doi: 10.1006/jsbi.2002.4458. [DOI] [PubMed] [Google Scholar]
- 37.Carrion-Vazquez M., Oberhauser A.F., Fernandez J.M. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janssen B., Debets J., Smits J. Chronic measurement of cardiac output in conscious mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;282:R928–R935. doi: 10.1152/ajpregu.00406.2001. [DOI] [PubMed] [Google Scholar]
- 39.Mattson D.L. Comparison of arterial blood pressure in different strains of mice. Am. J. Hypertens. 2001;14:405–408. doi: 10.1016/s0895-7061(00)01285-1. [DOI] [PubMed] [Google Scholar]
- 40.Grieve D.J., Shah A.M. Oxidative stress in heart failure. More than just damage. Eur. Heart J. 2003;24:2161–2163. doi: 10.1016/j.ehj.2003.10.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.