

Abstract
To improve the effectiveness of anti-cancer therapies, it is necessary to identify molecular targets that are essential to a tumor cell but dispensable in a normal cell. Increasing evidence indicates that the transcription factor STAT3, which regulates the expression of genes controlling proliferation, survival, and self-renewal, constitutes such a target. Recently it has been found that STAT3 can associate with the cytoskeleton. Since many of the tumors in which STAT3 is activated, such as breast cancer and ovarian cancer, are responsive to drugs that target microtubules, we examined the effect of these compounds on STAT3. We found that microtubule stabilizers, such as paclitaxel, or microtubule inhibitors, such as vinorelbine, decrease the activating tyrosine phosphorylation of STAT3 in tumor cells and inhibit the expression of STAT3 target genes. Paclitaxel decreases the association between STAT3 and microtubules, and appears to decrease STAT3 phosphorylation through induction of a negative feedback regulator. The cytotoxic activity of paclitaxel in breast cancer cell lines correlates with its ability to decrease STAT3 phosphorylation. However, consistent with the necessity for expression of a negative regulator, treatment of resistant MDA-MB-231 cells with the DNA demethylating agent 5-azacytidine restores the ability of paclitaxel to block STAT3-dependent gene expression. Finally, the combination of paclitaxel and agents that directly target STAT3 has beneficial effects in killing STAT3-dependent cell lines. Thus, microtubule-targeted agents may exert some of their effects by inhibiting STAT3, and understanding this interaction may be important for optimizing rational targeted cancer therapies.
Keywords: Cancer, Signal transduction, Transcription factors, Phosphorylation, Chemotherapy, Targeted therapy
Introduction
Although cancer claims the lives of over half a million people annually in the United States, the cornerstone of therapy for patients with advanced cancer remains non-specific cytotoxic therapies such as chemotherapy and radiation. In recent years, great strides have been made in understanding the molecular abnormalities that occur in tumor cells. This knowledge of the molecular pathogenesis of cancer has suggested new approaches to developing targeted therapies, and has also provided insight into important alternative mechanisms by which current therapies may be exerting their effects. Ultimately, this understanding can lead to enhanced strategies by which these therapies can be harnessed to improve patient outcomes.
Transcription Factors in Cancer Pathogenesis
Since the neoplastic phenotype of a cell is largely driven by its pattern of gene expression, increasing attention has been focused on the proteins that regulate gene expression. Many of the first oncogenes identified were mutated or overexpressed versions of transcription factors such as myc, and a variety of mutations have been described in transcription factors and transcriptional modulators in human tumors. It has also become clear that transcription factors which are not themselves mutated may be key intermediaries that are driving the malignant phenotype of a cancer cell. These oncogenic transcription factors have escaped the normal negative feedback pathways that tightly control gene expression, driving high levels of transcription of genes promoting cell cycle progression, inappropriate survival, self-renewal, and invasion. Loss of function of transcription factors in normal non-transformed cells often has little consequence in gene expression and cellular function due to redundancies in signaling pathways that regulate expression of any given gene. However, in a cancer cell, the continued activation of a transcription factor is often necessary for maintaining cell survival and function. This type of dependence has been described for many oncogenes, genes which are mutated and thereby contribute to malignant behavior (1). However, a similar dependence may also apply to so-called non-oncogenes, such as oncogenic transcription factors, which though not mutated themselves are still critical components for maintaining the malignant state. Thus, in recent years, increasing attention has focused on the role of these proteins in cancer pathogenesis and as targets for molecular therapy (2).
STAT3 as an Oncogenic Transcription Factor
STATs are latent transcription factors that reside in the cytoplasm until activated by tyrosine phosphorylation (3, 4). The seven STAT family members can be phosphorylated by the Jak family of kinases and by other receptor and non-receptor tyrosine kinases. Tyrosine phosphorylated STAT dimers translocate to the nucleus, bind to specific nine base pair sequences in regulatory genomic regions, and modulate transcription of target genes involved in a variety of cellular processes. One STAT family member, STAT3, is expressed fairly ubiquitously, and plays a prominent role of transducing signals from cytokines and growth factors by modulating genes regulating proliferation, survival, and differentiation (Figure 1).
Figure 1. STAT3 controls critical cellular processes through both transcriptional and non-transcriptional mechanisms.
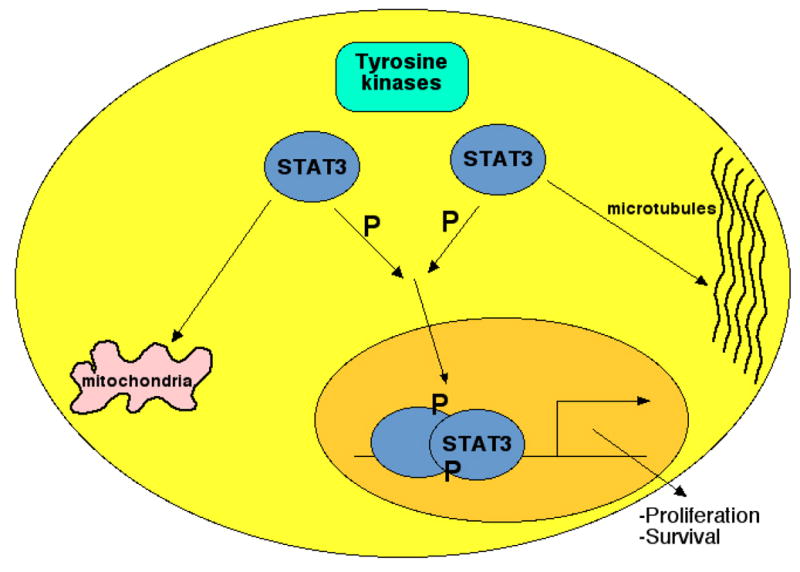
STAT3 is found in the cytoplasm under resting conditions. When phosphorylated by Jaks or other kinases on a single tyrosine residue, STAT3 dimers translocate to the nucleus, and activate transcription of genes regulating cell survival and proliferation. STAT3 can also associate with microtubules and mitochondria, and may also regulate cellular behavior through these mechanisms.
In normal cells, after modulating gene expression, STATs become dephosphorylated by tyrosine phosphatases and are thus free for subsequent rounds of stimulation (5, 6). In fact, cytokine-induced phosphorylation of STAT3 generally peaks 15 to 30 minutes following stimulation, and then declines to baseline levels over one to two hours. Many STAT3 target genes are themselves negative regulators of STAT activation, and this ensures that regulation of the key STAT3- target genes is tightly controlled. Although tyrosine phosphorylation plays a central role in STAT3 function, STAT3 can also be phosphorylated on a specific serine residue, serine 727. STAT3 serine phosphorylation may enhance STAT3-mediated transcription (7), though it may also inhibit the tyrosine phosphorylation of STAT3 (8). It is also possible that STAT3 phosphorylated solely on serine residues may play a transcriptional function (9).
STAT3 regulates genes that control a variety of key cellular processes such as proliferation, survival, and self-renewal (10). Thus, tight regulation of STAT3 activity is essential for maintaining homeostasis. However, in contrast to the rapid but transient activation of STAT3 that occurs with physiological stimuli, STAT3 is activated constitutively in a wide array of human cancers. For example, STAT3 is activated in 70% of breast tumors and is often associated with aggressive and invasive tumors (11, 12). Furthermore, inhibition of STAT3 using genetic or pharmacological approaches leads to a reversion of the malignant phenotype of these cells, indicating that it is a key mediator of cancer pathogenesis. A constitutively active form of STAT3 is sufficient to induce malignant transformation in cell culture (13), further emphasizing the central role played by this protein in cancer pathogenesis. STAT3 is largely dispensable in normal cells, and thus identifying methods to inhibit STAT3 holds great promise for developing rational therapies for cancer.
Non-Transcriptional Roles of STAT3
In addition to its transcriptional function, two other “non-canonical” roles for STAT3 have been proposed. First, increasing evidence suggests that STAT3 can localize to mitochondria, and directly alter cellular metabolism and bioenergetics (14). This effect requires serine, though not tyrosine, phosphorylation of STAT3, and can mediate neoplastic transformation in some models. Second, STAT3 has been found to interact with cytoskeletal structures, such as focal adhesions, and may thereby modulate microtubule function and cellular motility through non-transcriptional means (15). For example, loss of functional STAT3 impairs growth factor dependent migration in keratinocytes (16), and STAT3 inhibition decreases the motility of ovarian cancer cells in vitro (17). These findings have a number of interesting implications for understanding cancer pathogenesis and in devising targeted therapeutic strategies. For example, the activation status of STAT3 in a cell, particularly in clinical samples, is assessed based on its tyrosine phosphorylation (as measured by immunoblotting or immunohistochemistry with phosphorylation-specific antibodies), its ability to bind DNA (as assessed by electrophoretic mobility shift assay (EMSA) or chromatin immunoprecipitation (ChIP)), or its nuclear localization; however, if STAT3 were contributing to cancer pathogenesis through a non-transcriptional mechanism it would not be detected by any of these approaches. Similarly, drugs that target STAT3 may show an on-target therapeutic effect even in cells that do not display constitutive tyrosine phosphorylation of STAT3. Conversely, drugs that inhibit STAT3 tyrosine phosphorylation or that target the transcriptional function of STAT3 may not show an effect on cells that are functionally dependent on STAT3 through one of these other mechanisms. Finally, it suggests that drugs known to have anticancer properties, though which do not obviously interact with STAT3, may be exerting some or all of their effects by modulating these non- transcriptional functions of this protein.
Microtubule targeted therapy
Both microtubule inhibitors, like vinca alkaloids, and microtubule stabilizers, such as the taxanes, have been among the most active chemotherapeutic drugs in treating human cancer (18). Microtubules are highly dynamic structures, undergoing polymerization and de-polymerization through the addition or removal of tubulin subunits at their plus ends. Microtubules play an important role in a range of cellular events including mitosis, intracellular transport, and cytoskeletal dynamics, and the central role of microtubules in axonal transport underlies the sometimes debilitating neurotoxicity of tubulin-binding agents in the clinic. When used as anti-cancer agents, microtubule-targeted agents generally cause cell cycle arrest and subsequent apoptosis. Given that taxanes are among the most active drugs in both breast cancer and ovarian cancer, two tumors with a high prevalence of constitutive STAT3 activation, we considered the hypothesis that microtubule-targeted agents may modulate STAT3 function, and that this may underlie some of the effects of these compounds.
Microtubule-Targeting Drugs Inhibit STAT3
When breast cancer cell lines such as MDA-MB-468 or BT549, which are dependent on constitutive STAT3 activation, are treated with the microtubule stabilizer paclitaxel a dose-dependent decrease in STAT3 tyrosine phosphorylation can be observed (19). In addition, cell lines derived from other tumor types that are treated with taxanes, such as ovarian cancer (OVCAR8 and SKOV3) and prostate cancer (DU145) also display inhibition of STAT3 tyrosine phosphorylation following paclitaxel treatment. This effect on STAT3 phosphorylation is not confined to the constitutive activation seen in tumor cells, as cytokine-induced STAT3 phosphorylation is also inhibited by paclitaxel. This effect is not related to potential cytotoxic effects of paclitaxel, as it can be detected within six hours of paclitaxel treatment, well before any evidence of cytotoxicity can be observed. Similarly, inhibition of STAT3 phosphorylation is not part of a broader inhibition of signaling pathways, as other key signaling molecules such as Src, Erk MAP kinases, and Akt show no change in phosphorylation with paclitaxel treatment. A similar effect is seen with treatment with the microtubule inhibitor vinorelbine, but not with other drugs that are active in breast cancer through non-microtubule-dependent mechanisms such as the anthracycline doxorubicin.
Given the key role of tyrosine phosphorylation in the transcriptional function of STAT3, we examined the effect of paclitaxel on gene regulation mediated by this protein. We first employed chimeric reporter constructs in which a luciferase reporter gene under the control of a promoter responsive to a specific transcription factor was introduced into cells. Using this approach, we found that paclitaxel led to a prominent decrease in STAT3-dependent gene expression. Again this was not related to cytotoxicity or non-specific effects, as an analogous reporter dependent on the transcription factor NF-κB was unaffected by paclitaxel. Similarly, cytokine induced STAT3-dependent gene transcription was also suppressed by paclitaxel, though activity of the homologous transcription factor STAT1 was not similarly affected. Finally, bona fide endogenous STAT3 target genes including BCL3, BCL6, and SMAD7 were all inhibited by 60 to 90% in MDA-MB-468 cells by either paclitaxel or vinorelbine.
The finding that microtubule-targeted drugs lead to prominent and specific inhibition of STAT3 transcriptional activity raises three key questions: How much of the cytotoxic effect of these drugs is due to inhibition of STAT3 function? What is the mechanism by which STAT3 inhibition occurs? and, How can these findings be exploited therapeutically?
Does STAT3 Inhibition Contribute to the Cytotoxic Activity of Paclitaxel?
The ability of paclitaxel to inhibit STAT3 phosphorylation in breast cancer cell lines correlates with its cytotoxic effect in these cells. This could reflect the fact that STAT3 inhibition is a critical mechanism of this drug’s effects, or it can relate to a common mechanism of resistance, such as increased expression of drug efflux pumps, that would reduce all of the cellular effects of this agent. To examine more critically the role that STAT3 inhibition plays in the therapeutic effects of paclitaxel and related drugs, it is necessary to separate the STAT3-inhibitory effect from the microtubule-targeting effect of these agents. To try to accomplish this, we employed a constitutively active form of STAT3 that contains two cysteine residues, allowing it to form dimers stabilized by disulfide bonds (13). However, presumably because tyrosine phosphorylation is still required for the activity of this mutant (20), it was still inhibited by paclitaxel. Thus, this issue remains unresolved.
How does Paclitaxel Modulate STAT3?
While the mechanism by which paclitaxel inhibits STAT3 phosphorylation has not been completely elucidated, a number of mechanistic insights have arisen thus far. Given that reciprocal relationships have been observed between the serine phosphorylation of STAT3 and its tyrosine phosphorylation, we examined the effect of this drug on the phosphorylation of STAT3 on serine 727. In fact, paclitaxel induces a prominent increase in phosphorylation of this serine residue, raising the possibility that this was the mechanism for the decreased tyrosine phosphorylation. To test this hypothesis, we examined the effect of paclitaxel on tyrosine phosphorylation of STAT3 on a mutated form of STAT3 in which serine 727 was mutated to an alanine residue. If the decrease in tyrosine phosphorylation was triggered by enhanced serine phosphorylation, it would be expected that the effect would be lost in this STAT3 mutant lacking the critical serine residue. However, paclitaxel was equally effective at decreasing tyrosine phosphorylation in the mutant as in wildtype STAT3. While the effect of the increased phosphorylation of serine 727 remains to be determined, it is not the trigger for the decrease in tyrosine phosphorylation.
Nonetheless, two interesting observations do provide some insight into potential mechanisms by which microtubule-targeted agents exert their STAT3 inhibitory effects. First, STAT3 physically associates with tubulin, as can be shown by the co-immunoprecipitation of these two proteins. Following treatment of MDA-MB-468 cells with paclitaxel for five hours, this association was nearly completely lost. It could be argued that the loss of association between STAT3 and tubulin could be mediated by the decrease in STAT3 tyrosine phosphorylation. However, when these cells are treated with an inhibitor of Jak family kinases, Jak inhibitor 1, the tyrosine phosphorylation of STAT3 is nearly completely inhibited. However, the association between STAT3 and tubulin is unaffected. This suggests that a loss of tyrosine phosphorylation alone is insufficient to disrupt the interaction between STAT3 and tubulin, and that microtubule-targeting drugs may mediate this effect directly.
The second insight into potential mechanisms by which paclitaxel inhibits STAT3 tyrosine phosphorylation concerns the fact that the kinetics by which paclitaxel leads to a decrease in STAT3 phosphorylation is slower than that occurring following treatment with kinase inhibitors. This argues against a direct role of paclitaxel as a kinase inhibitor, and also raises the possibility that paclitaxel is inducing expression of one of the many negative feedback mediators that block STAT3 phosphorylation or enhance its dephosphorylation. This hypothesis is further supported by the finding that pretreating breast cancer cells with the protein synthesis inhibitor cycloheximide completely abrogates the ability of paclitaxel to decrease STAT3 phosphorylation. However, when expression of 17 distinct known negative feedback regulators of STAT3 were examined by quantitative RT-PCR, none showed prominent induction following paclitaxel treatment. Thus, the exact mechanism by which paclitaxel induces a loss of STAT3 tyrosine phosphorylation remains unclear, though the protein synthesis-dependent expression of a negative regulator seems most likely (Figure 2).
Figure 2. Paclitaxel blocks STAT3 tyrosine phosphorylation and STAT3-dependent gene transcription.

In many common cancers, including 70% of breast cancers, STAT3 is constitutively tyrosine phosphorylated, thereby driving expression of genes promoting malignant cellular behavior. STAT3 is also associated with tubulin (left panel). Following treatment with paclitaxel, or other microtubule-targeted drugs, STAT3 phosphorylation is inhibited, as is STAT3-driven gene expression. This likely results from expression of a negative regulator of STAT3 phosphorylation, and is also associated with an inhibition of the association between STAT3 and tubulin (right panel).
Therapeutic Implications of Interactions Between STAT3 and Microtubule-Targeted Drugs
Reversing resistance to paclitaxel
One breast cancer cell line that is resistant to both the cytotoxic effects of paclitaxel and its effects on STAT3 tyrosine phosphorylation is MDA-MB-231. Given the suggestion that paclitaxel inhibits STAT3 phosphorylation through new gene expression, we considered the possibility that the absence of an effect on STAT3 might reflect the repression of gene expression. It is known that epigenetic gene silencing plays an important function in cancer by inhibiting the expression of many tumor-suppressor genes (21). The methylation of CpG islands in the promoter region of a gene, mediated by members of the DNA methyltransferase (DNMT) family, can lead to stable though reversible gene silencing. Inactivation of DNMT by small-molecule inhibitors such as 5-azacytidine can lead to reexpession of silenced genes. To test the hypothesis that the lack of responsiveness to MDA-MB-231 cells to the STAT3 inhibitory effect of paclitaxel was mediated by DNA methylation, these cells were treated with either vehicle or 5-azacytidine for five days. Vehicle treated cells showed no change in expression of two key STAT3 target genes, BCL6 and SMAD7, following treatment with paclitaxel. By contrast, following treatment with 5-azacytidine, paclitaxel could induce a 50 to 70% decrease in expression of these genes (Figure 3). Thus, although the direct mechanism by which paclitaxel inhibits STAT3 transcriptional function remains unknown, these findings raise the possibility that epigenetic modulation of tumor cells with a clinically used DNA demethylating agent may restore this effect of paclitaxel.
Figure 3. Treatment of MDA-MB-231 cells with 5-azacytidine restores the ability of paclitaxel to inhibit STAT3 function.

MDAMB-231 breast cancer cells, which are resistant to both the cytotoxic and the STAT3-inhibitory effects of paclitaxel, were treated for five days with vehicle or the demethylating agent 5-azacytidine (500 nM). The cells were then exposed to vehicle or paclitaxel for 6 hours, after which RNA was harvested. 5-azacytidine treatment conferred sensitivity to the STAT3 inhibitory effect of paclitaxel as assessed by quantitative RTPCR assessment of expression of two STAT3 target genes, SMAD7 and BCL6.
Combinations of microtubule-targeted drugs and STAT3 inhibitors
Since constitutively activated STAT3 is necessary for maintaining the malignant state of many tumor cells, and this transcription factor is largely dispensable in normal cells, inhibiting STAT3 has been a major therapeutic goal (22, 23). In fact, a number of different strategies are being utilized to specifically inhibit this transcription factor, and clinical trials utilizing this approach for a number of malignancies, including squamous cell carcinoma of the head and neck and chronic lymphocytic leukemia, are being initiated. Since microtubule targeted agents can inhibit STAT3 transcriptional activity, this raises the question as to whether the combination of a drug like paclitaxel with a direct STAT3 inhibitor might show favorable combinatorial effects. To identify STAT modulators, we have generated reporter cell lines in which a luciferase reporter construct whose activity reflects the activation status of a single STAT family member has been stably integrated (24–26). These systems have proven to be an efficient and powerful way to screen large chemical libraries. Utilizing this approach, we have identified the anti-microbial drug nifuroxazide as a specific STAT3 inhibitor. To test the interaction between nifuroxazide and paclitaxel on a STAT3-dependent cell line, we utilized the ovarian cancer cell line OVCAR8. In initial experiments, both paclitaxel and nifuroxazide were able to decrease viability of these cells, and the combination of the two led to enhanced killing (Figure 4). Further mechanistic and pharmacological studies will be necessary to dissect this effect further, but it raises the possibility that the combination of STAT3 inhibitors and microtubule-targeted drugs will show therapeutic benefit.
Figure 4. The combination of paclitaxel with a STAT3 inhibitor leads to increased killing of OVCAR8 ovarian cancer cells.
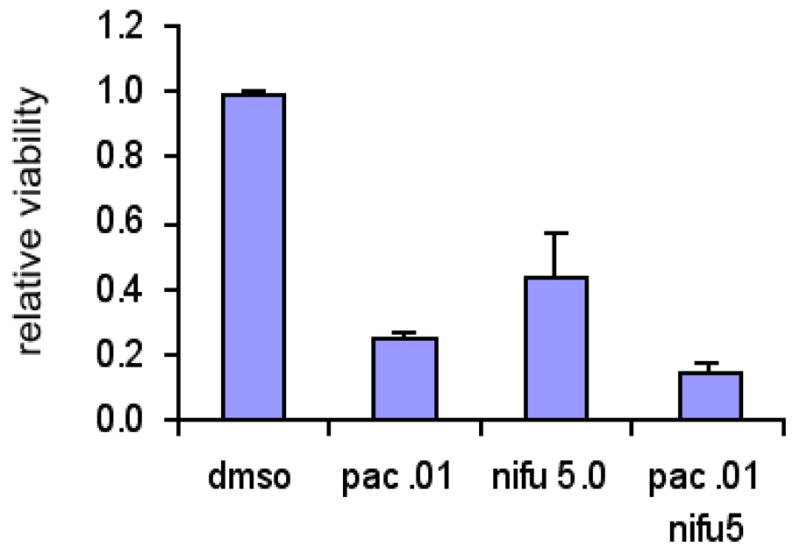
Cells were treated with either paclitaxel (10 nM) or the STAT3 inhibitor nifuroxazide (5 μM) alone or in combination for 48 hours, and surviving cells were quantitated using ATP-dependent bioluminescence.
Conclusion
The finding that paclitaxel and other microtubule-targeted drugs can inhibit STAT3 tyrosine phosphorylation and transcriptional function has raised a number of interesting mechanistic issues. Questions remain as to how these drugs mediate this effect, how much of their therapeutic action depends on their ability to inhibit STAT3, and how cells become resistant to this particular aspect of their function. However, this finding has suggested potentially useful therapeutic strategies such as combining DNA demethylating agents or STAT3 inhibitors with paclitaxel, vinorelbine, or related drugs. Although further research is needed to elucidate these issues, these observations highlight a potentially important convergence point between nonspecific cytotoxic drugs and a key molecular mediator that plays a central role in the pathogenesis of many common human cancers.
Acknowledgments
This work was supported by the Breast Cancer Research Foundation, the American Association for Cancer Research, Susan G. Komen for the Cure, the Friends of Dana-Farber Cancer Institute, and the Brent Leahey Fund.
Footnotes
PharmSight on Walker SR, et al. Microtubule-targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol 2010;78:903-8.
Conflicts of Interest
No potential conflicts of interest to disclose.
References
- 1.Weinstein IB. Addiction to oncogenes-the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 2.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–9. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 3.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 4.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–7. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 5.David M, Grimely PM, Finbloom DS. A nuclear tyrosine phosphatase downregulates interferon-induced gene expression. Mol Cell Biol. 1993;13:5715–21. doi: 10.1128/mcb.13.12.7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haque SJ, Flati V, Deb A, Williams BR. Roles of protein-tyrosine phosphatases in Stat1 alpha-mediated cell signaling. J Biol Chem. 1995;270:25709–14. doi: 10.1074/jbc.270.43.25709. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–4. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- 8.Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and - independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–16. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hazan-Halevy I, Harris D, Liu Z, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–63. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez JV, Frank DA. Genome-wide analysis of STAT target genes: Elucidating the mechanism of STAT- mediated oncogenesis. Cancer Biol Ther. 2004;3:1045–50. doi: 10.4161/cbt.3.11.1172. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–62. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 12.Walker SR, Nelson EA, Zou L, Chaudhury M, et al. Reciprocal effects of STAT5 and STAT3 in breast cancer. Mol Cancer Res. 2009;7:966–76. doi: 10.1158/1541-7786.MCR-08-0238. [DOI] [PubMed] [Google Scholar]
- 13.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 14.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras- dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of STAT3 for cancer therapy. Clin Cancer Res. 2007;13:5665–9. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- 16.Sano S, Itami S, Takeda K, et al. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999;18:4657–68. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64:3550–8. doi: 10.1158/0008-5472.CAN-03-3959. [DOI] [PubMed] [Google Scholar]
- 18.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 19.Walker SR, Chaudhury M, Nelson EA, Frank DA. Microtubule-targeted chemotherapeutic agents Inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol. 2010;78:903–8. doi: 10.1124/mol.110.066316. [DOI] [PubMed] [Google Scholar]
- 20.Liddle FJ, Alvarez JV, Poli V, Frank DA. Tyrosine phosphorylation is required for functional activation of disulfide-containing constitutively active STAT mutants. Biochemistry. 2006;45:5599–605. doi: 10.1021/bi0525674. [DOI] [PubMed] [Google Scholar]
- 21.Baylin SB. DNA methylation and gene silencing in cancer. Nature Clinical Practice Oncology. 2005;2:S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 22.Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–26. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- 23.Frank DA. STAT inhibition in the treatment of cancer: Transcription factors as targets for molecular therapy. Curr Cancer Ther Rev. 2006;2:57–65. [Google Scholar]
- 24.Lynch RA, Etchin J, Battle TE, Frank DA. A small- molecule enhancer of signal transducer and activator of transcription 1 transcriptional activity accentuates the antiproliferative effects of IFN-gamma in human cancer cells. Cancer Res. 2007;67(3):1254–61. doi: 10.1158/0008-5472.CAN-06-2439. [DOI] [PubMed] [Google Scholar]
- 25.Nelson EA, Walker SR, Kepich A, et al. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008;112:5095–102. doi: 10.1182/blood-2007-12-129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson EA, Walker SR, Weisberg E, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2010 doi: 10.1182/blood-2009-11-255232. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]