

Abstract
Objective
Enduring, abnormal expression and function of the ion channel hyperpolarization-activated cyclic-AMP gated channel type 1 (HCN1) occurs in temporal lobe epilepsy (TLE). We examined the underlying mechanisms, and queried if interfering with these mechanisms could modify disease course.
Methods
Experimental TLE was provoked by kainic acid-induced status epilepticus (SE), HCN1 channel repression was examined at mRNA, protein and functional levels. Chromatin immunoprecipitation was employed to identify the transcriptional mechanism of repressed hcn1 expression, and the basis for their endurance. Physical interaction of the repressor, NRSF, was abolished using decoy oligodeoxynucleotides (ODNs). Video-EEG recordings were performed to assess the onset and initial pattern of spontaneous seizures.
Results
Levels of NRSF and its physical binding to the hcn1 gene were augmented after SE, resulting in repression of hcn1 expression and HCN1-mediated currents (Ih), and reduced Ih-dependent resonance in hippocampal CA1 pyramidal cell dendrites. Chromatin changes typical of enduring, epigenetic gene repression were apparent at the hcn1 gene within a week after SE. Administration of decoy ODNs comprising the NRSF DNA-binding sequence (NRSE) in vitro and in vivo, reduced NRSF binding to hcn1, prevented its repression and restored Ih function. In vivo, decoy NRSE-ODN treatment restored theta rhythm and altered the initial pattern of spontaneous seizures.
Interpretation
Acquired HCN1 channelopathy derives from NRSF-mediated transcriptional repression that endures via chromatin modification and may provide insight into the mechanisms of a number of channelopathies that co-exist with, and may contribute to, the conversion of a normal brain into an epileptic one.
Introduction
Temporal lobe epilepsy (TLE), the most common and severe form of epilepsy in adults, is often triggered by an initial insult, which results in altered expression of hundreds of genes1-3. The factors orchestrating such transcriptional changes remain unknown. Their identification is important, because targeting them may ameliorate the epileptic disease process4.
Ion channels are a class of gene products that influence neuronal and network excitability. Channelopathies occur in pathological conditions, such as epilepsy4, leading to disrupted neuronal and network function5. We focused on the hyperpolarization-activated cyclic nucleotide gated (HCN) ion channels, because reduced expression of the major isoform, HCN1, has been reported to accompany, and perhaps contribute to, the epileptogenic process in several TLE models6-10. Thus, mice lacking HCN channels are vulnerable to seizures and epilepsy11,12, and a mutation in HCN2 has been identified in individuals with epilepsy13. The current conducted by HCN channels, Ih, plays a key role in the filtering properties of CA1 pyramidal cell dendrites14. The high density of Ih in distal dendrites 15 efficiently attenuates the summation of excitatory inputs,16 potentially dampening neuronal excitability17. Additionally, Ih tunes the membrane to respond optimally to inputs in the theta frequency band18,19, a brain rhythm central to numerous cognitive processes20. Therefore, we used the HCN channelopathy in TLE as a model system to study transcriptional regulatory mechanisms, and tested if these mechanisms were amenable to therapeutic intervention.
The transcriptional repressor NRSF (neuron-restrictive silencer factor REST) binds its cognate sequences (NRSEs) that are present on several hundred neuronal genes21,22. NRSF recruits specific cofactors and histone deacetylases, leading to often enduring epigenetic alterations of chromatin structure and thus persistent repression of gene expression that may eventually be autonomous of the repressor’s binding22. NRSF expression is low in naive adult hippocampus and increases after seizures that promote epilepsy23,24. Additionally, the regulatory region of the hcn1 gene contains a highly conserved sequence (ttCAGCACCacGGAcAGcgcC) that can bind NRSF21. Therefore, we tested if NRSF regulates the expression and thus function of HCN1 channels after a proepileptogenic insult, if this resulted in chromatin changes, and if interfering with the ability of NRSF to regulate target genes affected the outcome of a proepileptogenic insult.
Material and Methods
A detailed description of the methods used can be found in the online supplementary materials.
Male Wistar-Han rats (n = 20) were implanted with cannulae and electrodes, and a second set of rats (n = 16) were implanted with cannulae. Experimental protocols conformed to NIH guidelines, and were approved by INSERM and by the IACUC of the University of California-Irvine. Continuous video/EEG monitoring was performed.
To induce status epilepticus (SE), kainic acid (KA) was given by intraperitoneal injection once per hour (5 mg/kg), and pilocarpine hydrochloride (310 mg / kg) was injected 30 minutes after a preliminary scopolamine injection (1mg/kg).
To assess molecular changes and in vitro physiology, rats were infused with ordered or scrambled oligonucleotides (ODNs) on days 1 (10 nmol), and 2 (5nmol) after the SE. Electrophysiological and biochemical studies were performed on the day following the 2nd infusion. For long-term effects of the ODN in vivo, repeated infusions alternated full dose (10 nmol/hemisphere) and half dose the following post-SE days: day 1 (full dose), day 2 (half dose), day 3 (half dose), day 6 (full dose), day 8 (half dose), day 10 (full dose). Recordings were discontinued on day 13 post-SE.
Analysis of seizures, interictal activities and theta rhythm were performed as described previously 25.
Organotypic hippocampal slice cultures were prepared as described before26,27 using P8 rats. Phosphorothioate oligodeoxynucleotides, were added in the culture medium (1 μM) 3 hours after treatment with kainic acid that provoked seizure-like events28. Cultures were assessed for HCN expression 2-days after KA, and for NRSF at 4h-7d.
Animals used for NRSF, HCN1, HCN2 and Kv4.2 measurements were decapitated and the hippocampi were rapidly dissected. Organotypic slice culture tissues were harvested directly from the membranes. All tissues were processed for Western Blot analyses as described in the supplementary methods.
Chromatin immunoprecipitation was performed, as described in the supplementary methods, to detect the physical binding of transcription factors and histones to DNA.
Hippocampal slices were prepared from the dorsal hippocampus and recordings performed as previously described 10.
SPSS software was used for statistical analysis. We used non parametric Mann-Whitney test for samples lower than 6 in size, and t-test otherwise. Results are expressed as mean ± s.e.m, with p<0.05 considered significant.
Results
Attenuation of Ih and resonance in CA1 pyramidal cell dendrites in KA-treated rats
We first characterized the downregulation of HCN1 on distal dendrites of dorsal CA1 pyramidal cells in KA-treated animals, an experimental model of TLE29. Distal dendrites (>280 μm from the soma, Supplementary Table 1) had large Ih currents in control animals (Fig 1A to B, Supplementary Table 1)10. Three days after KA, the amplitude of Ih was reduced by 50% and the membrane potential for half-maximal activation (V1/2) was more hyperpolarized, further limiting the availability of Ih at physiological potentials (Fig 1B, Supplementary Table 1). The current’s kinetics were slowed by 30% (Supplementary Table 1), consistent with a reduced contribution of the fast-kinetics HCN1 isoform to the total Ih30. Reduced Ih resulted in increased membrane resistance and a more hyperpolarized resting membrane potential (Supplementary Table 1). A key functional read-out of Ih, theta resonance, was compromised in KA-SE rats (Fig 1C, Supplementary Table 1, Supplementary Figs 1-2): resonance frequency (Fres) was shifted toward lower values, and the amplification ratio (Q) was decreased by 20%. In addition to its function as a band-pass filter in the theta range in the dendrites19, Ih introduces an apparent negative time delay to theta inputs18. This function was altered in KA-treated animals, as the apparent negative time delays were shifted toward lower frequencies (Fig 1C, Supplementary Table 1, Supplementary Figs 1-2). Theta resonance and temporal coding were abolished by the Ih antagonist ZD7288 (Supplementary Figs 1-2), suggesting that the loss of resonance after KA was due to decreased Ih.
Fig 1.
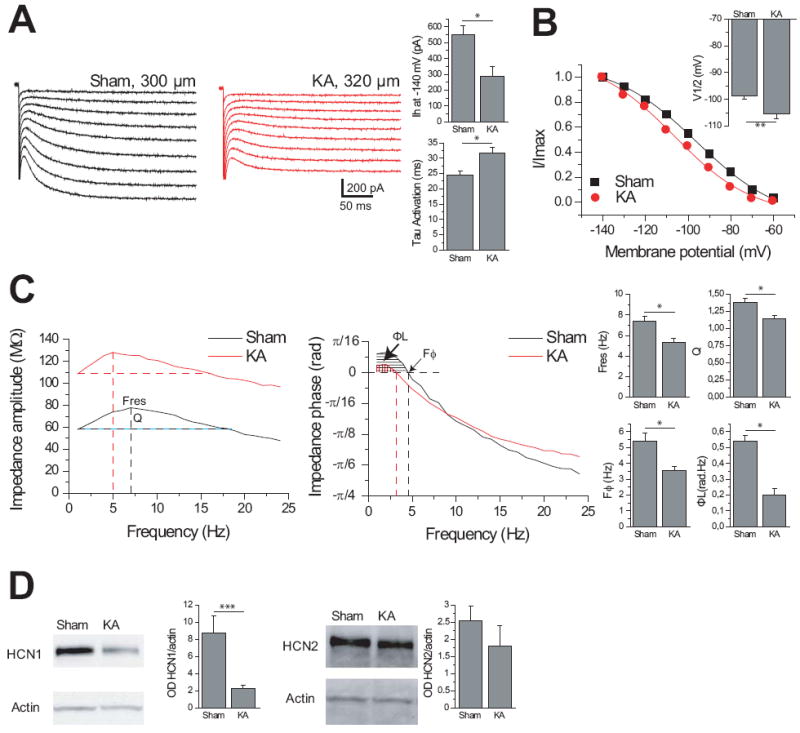
Loss of HCN1 protein in hippocampal CA1, and of Ih current, resonance and temporal coding in CA1 pyramidal cell distal dendrites in KA-treated animals. Ih current was decreased and the activation time constant was slower (A). The left panel shows typical examples of Ih activation in distal dendrites. The membrane potential for half activation was shifted to hyperpolarized values in KA-treated animals (B). The displayed activation curves correspond to the dendrites recorded in A. (C) Left panel: The impedance magnitude profile as a function of input frequency shows the decrease in resonance in KA-treated animals. The same dendrites as in (A and B) were hyperpolarized to -70 mV by steady current injection. The vertical dotted lines indicate the resonance frequency (Fres) at which the impedance reaches a maximum. Q is the amplification ratio between the impedance at Fres and at 1 Hz. Q and Fres were decreased in KA-treated animals (right panels). The impedance at 1 Hz was larger in KA-treated than in sham because of the increase in input resistance due to loss of Ih (Supplementary Table 1). Middle panel: Impedance phase profiles. Each curve is characterized by two regions, one with a positive impedance phase (phase lead, shaded regions), followed by a negative impedance phase (phase lag) as the frequency increases. In the phase lead (lag) regions, the membrane response appears to precede (follow) the current inputs (Supplementary Fig 1). Vertical lines indicate the crossover frequency, Fϕ. The shaded regions represent the total inductive phase ФL. Fϕ and ФL were decreased in KA-treated animals (right panels). (D) Western blots from hippocampal CA1 homogenates collected 48 hours after seizure initiation from Sham and KA treated rats, demonstrate a reduction in HCN1 protein expression (Sham 8.75 ± 2.02 normalized OD, n=8; KA 2.23 ± 0.37 normalized OD, n=6; p=0.02; normalized with actin levels), but not HCN2 protein expression, in KA-treated animals (Sham 2.53 ± 0.59 normalized OD, n=4; KA 1.79 ± 0.43 normalized OD, n=3; p=0.34; normalized with actin levels). (single asterisk, P < 0.01; double asterisk, P < 0.05; triple asterisk, P < 0.0001).
At the molecular level, there was a 75% decrease in HCN1 protein levels, with no significant change in HCN2 (Fig 1D). Thus, the 50% reduction of Ih amplitude likely resulted from selective downregulation of the HCN1 channel isoform which contributes an estimated 66% of the current30, whereas the changes in Ih kinetics are best explained by a larger contribution of HCN2 channels to the Ih current or by augmented HCN1/HCN2 heteromerization31.
NRSF-dependent downregulation of HCN1 in vitro
The regulatory region of the hcn1 gene contains several NRSF-binding sequences21, including a highly conserved sequence residing in the first hcn1 intron (Fig 2A)21. Since NRSF expression is enhanced by seizures23,24, NRSF could be responsible for HCN1 downregulation. To test this hypothesis, we first used hippocampal organotypic slice cultures. Application of KA generated seizure-like events28 that resulted in reduced HCN1 protein levels (Fig 2B), whilst HCN2 remained unchanged (Fig 2B) consistent with previous results (6,31 and Fig 1D). Concurrent with repression of HCN1 expression, NRSF expression was strongly increased (Fig 2C). If NRSF binds to hcn1 to repress its transcription, then using an excess amount of a decoy NRSF-binding oligodeoxynucloetide (ODN) should prevent NRSF from binding the NRSE sequence on the hcn1 gene and prevent the transcriptional repression of HCN1 (Fig 2D). Application of NRSE-ODNs following 3 hours of KA-induced seizure-like activity abrogated the reduction of HCN1 mRNA (Fig 2E) and protein (Fig 2F). Because basal levels of NRSF in naive hippocampus were low, there was little effect of the NRSE-ODN on HCN1 expression in the controls. Control ODNs with a random nucleotide sequence (‘scrambled’-SCRLD, Fig 2D) had little effect on seizure-induced HCN1 mRNA and protein downregulation (Fig 2E to F). Neither NRSE- nor SCRLD-ODNs changed HCN2 mRNA levels (Supplementary Fig 3). Together, these results suggest that the seizure-like activity-dependent upregulation of NRSF represses HCN1 mRNA and protein expression in vitro, likely via binding of the repressor to its cognate NRSE silencer element on the hcn1 gene.
Fig 2.

NRSE-sequence oligonucleotides (ODNs) block the downregulation of HCN1 channels by KA-induced seizure-like events in hippocampal organotypic slice cultures. (A) The hcn1 gene contains a highly conserved NRSF recognizing element (NRSE) within its first intron, as apparent from the aligned element in three species. Numbers refer to the location of a nucleotide from the gene origin; upper case letters indicate nucleotide bases considered important for NRSF binding; and stars indicate matches to the putative left and right half-site binding motifs for NRSF. (B) Western blots of organotypic hippocampal slice culture tissue homogenates collected 48 hours after KA treatment and the resulting seizure-like network activity, compared with control cultures (CTL). A significant reduction (KA 77.33 ± 2.96 % of CTL OD, n=3 per group; p=0.01) of HCN1 protein expression (normalized for actin) is apparent in the KA group, but there is no significant change in HCN2 expression (KA 106.90 ± 4.27 % of CTL OD, n=3 per group; p=0.25). (C) Western blots of nuclear protein extracts of similarly treated organotypic slice cultures demonstrated a significant increase (CTL 2.14 ± 0.03 OD, n=3; KA 3.98 ± 0.51 OD, n=6; p=0.04) in the protein levels of the transcription factor NRSF as a result of the KA-induced seizure-like events. (D) Schematic of the intervention strategy. Left panel. NRSF binds to the NRSE sequence of hcn1, resulting in its repression, and this binding is not influenced by the presence of random-sequence (scrambled; SCRLD) ODNs with a modified backbone to enhance their stability. Right panel. Decoy’ deoxyoligonucleotides (ODNs) consisting of the NRSE sequence bind to available nuclear NRSF and consequently limit the interaction of this repressor with NRSE sequences within the DNA of target genes. (E, F) Application of NRSE-sequence ODNs to hippocampal organotypic slice cultures after a three-hour KA treatment prevented the reduction in HCN1 mRNA (CTL+SCRLD 281.0 ± 9.6 nCi/g, n=5; CTL+NRSE 292.4 ± 13.1 nCi/g, n=8; KA+SCRLD 203.6 ± 15.5 nCi/g, n=7; KA+NRSE 260.3 ± 12.4 nCi/g, n=12; p=0.03 KA+SCRLD compared with CTL+SCRLD) (E) and protein (CTL+SCRLD 0.50 ± 0.06 normalized OD, n=4; CTL+NRSE 0.58 ± 0.03 normalized OD, n=3; KA+SCRLD 0.35 ± 0.04 normalized OD, n=4; KA+NRSE 0.50 ± 0.03 normalized OD, n=3; p=0.04 KA+SCRLD compared with CTL+SCRLD; all normalized with actin levels) (F) expression. The KA-induced repression was still apparent in the presence of SCRLD ODNs. Application of NRSE and SCRLD ODNs had no significant effect on the basal expression of HCN1 mRNA and protein in control cultures.
Blocking the ability of NRSF to bind the hcn1 gene abrogates HCN1 repression and restores its function in rats exposed to an epileptogenic insult
To test if the mechanisms identified in vitro were operational in vivo during epileptogenesis, we first measured NRSF expression and binding to the hcn1 gene. Two days following KA treatment in vivo, NRSF expression was increased over threefold in hippocampal CA1 compared to sham controls (Fig 3A). Similar changes were found in the pilocarpine model of TLE (Fig 3B), which also results in HCN1 downregulation10. Using chromatin immunoprecipitation (ChIP) followed by quantitative PCR (Fig 3C-F) to examine if the physical binding of NRSF to the regulatory region of the hcn1 gene was augmented in hippocampi from KA rats, and if this augmented binding was selective, we detected NRSF binding at the NRSE site of the hcn1 gene (Fig 3C to D), but not at a downstream region lacking an NRSE sequence (Fig 3C to E). In KA-treated rats, NRSF binding to the hcn1 gene (but not the hcn2 gene21; Sham vs. KA p>0.05, not shown) was significantly augmented (Fig 3F). We then asked if blocking NRSF-NRSE binding in vivo would prevent the repression of the hcn1 gene. Scrambled or ordered NRSE-ODNs were infused into the cerebral ventricles between days 1 and 10 post KA. In sham rats treated with scrambled or NRSE-ordered ODN, NRSF binding to the hcn1-NRSE was modest (Fig 4A). A striking increase of NRSF-NRSE binding was found in the KA group treated with scrambled ODN. This increase was largely abrogated by NRSE-ODN treatment (Fig 4A and Supplementary Fig 4). As a result, HCN1 levels were reduced in KA rats treated with scrambled ODNs, but were similar to sham in KA rats treated with NRSE-ODNs (Fig 4B and Supplementary Fig 4). In contrast to HCN1, HCN2 levels were not significantly different in any experimental group (Supplementary Fig 5). The ‘rescue’ of HCN1 protein was functionally significant, because it was accompanied by the full restoration of Ih current amplitude and kinetics. The known functions of Ih on theta resonance and temporal coding were also normalized (Fig 5A, 5B, Supplementary Table 1).
Fig 3.

NRSF levels increase following epilepsy-provoking seizures, and promote binding of the repressor to the hcn1 gene. (A) When measured two days later, KA-induced seizures resulted in a large increase of NRSF protein levels in nuclear extracts of hippocampi (sham 1.95 ± 0.07 OD, n=4; KA 6.39 ± 0.11 OD, n=4). (B) When measured two days later, pilocarpine-induced seizures also resulted in a two-fold increase of NRSF protein in nuclear extracts of hippocampi (sham 2.03 ± 0.42 OD, n=6; pilocarpine 3.96 ± 0.52 OD, n=5). (C) Schematic of the hcn1 gene indicating the location of the NRSE and of the primer sets for PCR used in chromatin immunoprecipitation. (D, E) Chromatin immunoprecipitation (ChIP) using an antiserum to NRSF (H290, Santa Cruz) compared to IgG for precipitating the DNA-protein complex. Tissue was obtained from rats two days after the KA treatment, and quantitative PCR of input and immunoprecipitated DNA was used to calculate the percent of input DNA recovered. (D) Augmented specific immunoprecipitation of DNA comprising the HCN1-NRSE region is apparent when the NRSF antiserum rather than IgG, is used: (IgG 0.207 ± 0.021 %, n=4; NRSF 0.342 ± 0.016 %, n=4). (E) This augmented binding of NRSF, detected via anti NRSF precipitation, was selective to DNA of NRSE-containing regions, and was not found for an NRSE-lacking region 1600bp downstream of the HCN1-NRSE (IgG 0.237 ± .039 %, n=7; NRSF 0.230 ± 0.059 %, n=7). (F) KA-induced seizures resulted in a large increase in NRSF binding to the HCN1-NRSE region when measured three days later (Sham 100 ± 16, n=4; KA 233 ± 18, n=4). (single asterisk, P < 0.05).
Fig 4.
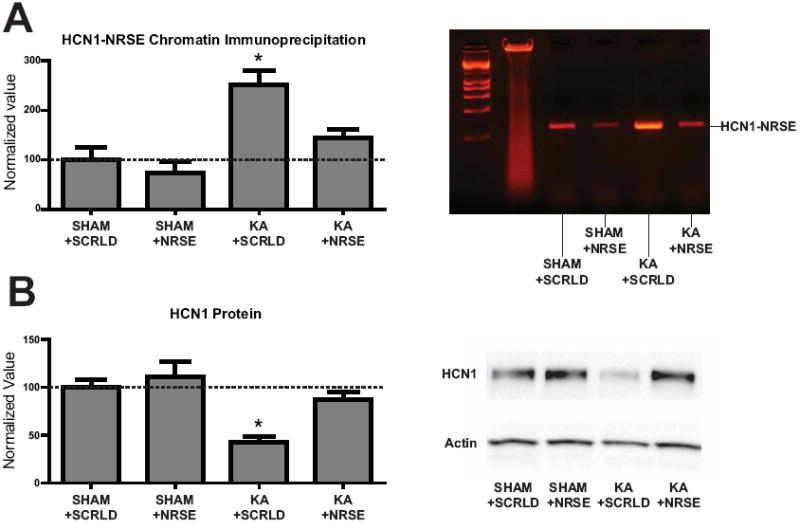
Abrogation of NRSF binding and recovery of HCN1 expression following NRSE-ODN infusion in vivo (A), left panel: Infusion of NRSE-ODNs abrogated the KA-induced increase of NRSF binding to the hcn1 gene. A large increase in NRSF binding near the HCN1-NRSE region was found only in KA treated rats receiving scrambled-ODN (SHAM+SCRLD 100 ± 25 n=3, SHAM+NRSE-ODN 73 ± 25, n=4; KA+SCRLD 251 ± 28, n=5; KA+NRSE-ODN 145 ± 16, n=4). An example of a complete gel showing PCR products from the ChIP is shown in the second panel, illustrating these differences. (B) NRSE-ODN, but not scrambled-ODN treatment, prevented the KA-induced repression of HCN1 expression. Normalized values of SHAM+SCRLD: 100 ± 8 (n=5); SHAM+NRSE-ODN: 111 ± 16 (n=6); KA+SCRLD: 43 ± 6 (n=7); KA+NRSE-ODN: 87 ± 9 (n=7). A representative western blot including all experimental groups is shown in the second panel. (single asterisk, P < 0.05)
Fig 5.

Recovery of Ih function following NRSE-ODN injection in vivo and specificity of NRSE-ODN treatment. (A) NRSE-ODN, but not scrambled-ODN treatment, restored Ih amplitude and channel kinetics (for both activation time constant and membrane potential for half-maximal activation, V1/2). Averaged activation curves are shown for KA+NRSE (n=7) and KA+SCRLD (n=8) experiments. Errors bars (s.e.m) are hidden in the symbols. (B) NRSE-ODN, but not scrambled-ODN treatment, restored theta resonance and temporal coding. Resonance and phase response were voltage-dependent, and were reduced at -60 mV as compared to -70 mV. There were no significant differences between Sham and KA+NRSE values, as well as between KA+saline and KA+SCRLD values. (C) Time course of NRSF protein levels in hippocampus of KA-SE rats. Western blot analysis demonstrated that NRSF levels were still significantly elevated at one week (p<0.05) after the insult, Sham 1.96 ± 0.18, n=3; KA+72h 6.75 ± 0.54, n=3; KA+1w 5.42 ± 0.36, n=3. (D). Augmented binding of an antiserum directed against the dimethylated form of histone 3 (at lysine 9), a general indicator of epigenetic gene repression. Chromatin immunoprecipitation demonstrates selective augmentation of dimethylated H3K9 in the NRSE-containing regulatory region of the hcn1 gene (p<0.05), but not in a non-seizure regulated gene (HCN2) or a non-NRSF-regulated gene (Kv4.2) (normalized values, HCN1: Sham 100 ± 5, n=6; KA+72h 152 ± 21, n=3; KA+1w 133 ± 15, n=3; HCN2: Sham 100 ± 6, n=6; KA+72h 95 ± 9, n=3; KA+1w 93 ± 4, n=3; Kv4.2: Sham 100 ± 8, n=6; KA+72h 109 ± 9, n=3; KA+1w 90 ± 4, n=3). (E) Downregulation of Kv4.2 is NRSF-independent in experimental TLE. KA treatment resulted in a large decrease of Kv4.2 protein levels in the presence of both NRSE and scrambled ODNs (normalized values, SHAM+NRSE 1.00 ± 0.19, n=2; KA+NRSE 0.07 ± 0.05, n=2, p<0.05; SHAM+SCRLD 1.00 ± 0.16, n=2; KA+SCRLD 0.08 ± 0.01, n=2, p<0.05). (F) The amplitude of back-propagating action potentials (bAPs) was still increased in KA+NRSE and KA+SCRLD treated rats. Left panel. Typical examples of bAPs recorded in CA1 pyramidal cell dendrites in a Sham (290 μm from the soma, black trace), a KA+NRSE treated (340 μm from the soma, red trace), and a KA+SCRLD (320 μm from the soma, green trace) animal. Note that bAPs have much larger amplitude in KA treated animals. Right panel. Summary of bAPs amplitude measured in Sham (15.1 ± 1.9 mV, n=8), KA+NRSE (40.2 ± 3.9 mV, n=5) and KA+SCRLD (38.7 ± 4.5 mV, n=5) animals. All recordings were performed at the same distance (around 300 μm) from the soma. * P<0.05.
NRSF augmentation results in selective epigenetic modification of the chromatin
HCN1 channelopathy is characterized by protracted dysregulation of channel expression. The mechanisms for this enduring repression might involve persistent upregulation of NRSF and its binding to the hcn1 gene. Alternatively, augmented levels of NRSF might be relatively short-lasting, yet result in long-lasting changes of the chromatin structure that promote repression of HCN1 expression. Thus, NRSF together with co-repressors, has been shown to augment histone-3 dimethylation in local regions of target genes 32, with consequent gene repression. Looking at the time-course of NRSF protein levels, we found that the repressor was still upregulated a week following KA-SE (Fig. 5C). In addition, the binding of the NRSE-containing regulatory region of the HCN1 gene to dimethylated histone H3K9 was augmented 72 hours and one week after KA-SE compared with sham controls (Fig. 5D). Dimethylation of Histone 3 at the K9 position is generally considered a marker of persistent repression that may be autonomous from the binding of repressors or co-factors. Thus, whereas elevation of NRSF levels, still present at a week after the KA-SE, may not persist, it suffices to set in motion an epigenetic process thatmodifies gene expression in an enduring manner.
Blocking the ability of NRSF to bind target genes does not influence an ion channel in which the gene lacks an NRSE sequence
In addition to HCN1, disturbances in several ion channels, termed acquired channelopathies, have been reported in experimental TLE5. In particular, decreased expression of Kv4.2 proteins results in reduced A type potassium current, leading to enhanced back-propagation of action potentials in CA1 pyramidal cell dendrites25. The Kv4.2 gene does not contain a functional NRSE within its regulatory region21. In accordance, we found that NRSE-sequence ODNs did not influence the KA-provoked reduction of Kv4.2 protein levels in hippocampal CA1 (Fig 5E), and had no effect on the consequent augmentation of back-propagation (Fig 5F). These data suggest a different, NRSF-independent mechanism for the Kv4.2 channelopathy in epilepsy.
Prevention of NRSF interaction with target genes influences the initial pattern of spontaneous seizures after KA-SE
HCN1 is an important ion channel, and reduction of HCN1 expression and/or function has been implicated in experimental and human epilepsy6-10,12,33. In addition, hcn1 is one of several hundred neuronal genes that contain an NRSE sequence and might thus be targets for NRSF-mediated epigenetic modulation21,22. Therefore, we examined whether the decoy ODN treatment, blocking the interaction of NRSF with hcn1 and other target genes, influenced the outcome of KA-SE. During continuous video-EEG recordings, spontaneous seizures (Fig 6A) emerged concurrently in both KA-SE groups, and seizure duration and Racine scores did not distinguish the NRSE ODN- and scrambled ODN-treated rats (not shown). However, during the two continuous recording weeks, the average number of seizures in NRSE ODN-treated KA animals was 70% lower, associated with a two-fold seizure frequency reduction (Fig 6C). In addition, the trend to increased cumulative seizure number with time was attenuated in NRSE ODN-treated KA rats (Fig 6D). In experimental TLE, interictal-like activity typically precedes the first spontaneous seizure1,34,35, and the number, frequency and duration of interictal events and seizures provide measures of the natural history of the resulting epilepsy36,37. In both sets of animals, interictal activity occurred mostly in bursts (Fig 6B). Treatment with NRSE-ODNs decreased the number of interictal bursts and the cumulative time spent in interictal activity by 90% (Fig 6E). Another hallmark of abnormal network electrophysiological activity is the degradation of theta oscillations38. Treatment with NRSE-ODNs abrogated the degradation of theta rhythm in KA rats (Fig 6F), an effect apparent already on post-seizure day 5 and persisting throughout the recording period.
Fig 6.

Preventing the interaction of NRSF with target genes alters the outcome of KA-SE. (A) Example of a spontaneous seizure recorded in an animal treated with NRSE-ODN (top trace). The bottom trace shows an expansion of the seizure. (B) Example of a burst of interictal spikes (top trace) recorded in the same animals as in (A). Note the difference in time scale between the burst of interictal spikes, which lasts roughly 30 min as compared to the seizure, which lasts around 35 s. The bottom trace shows an expansion of the burst of interictal spikes using the same time scale as the expanded portion of the seizure. Interictal activity occurred within a background of theta rhythm. (C) NRSE-ODN treatment (n=5) significantly reduced the mean number of seizures per day (top panel, 2.7 ± 0.8) and the total number of seizures over the 14 day-long continuous recording period, (bottom panel, 25 ± 2) as compared to SCRLD-ODN treatment (n=5, 5.2 ± 0.5 and 82 ± 2, respectively). (D) Cumulative number of seizures recorded in NRSE-ODN and SCRLD-ODN animals. Note the slow evolution in NRSE-ODN animals. (E) NRSE-ODN reduced the number of interictal bursts (NRSE, 1.8 ± 1.5; SCRLD, 22.7 ± 11.7) and the cumulative time spent in interictal activity (NRSE, 26 ± 24 min; SCRLD, 244 ± 92 min). (F) KA-induced seizures resulted in a progressive decline of theta rhythm power in the EEGs, as found in KA animals treated with scrambled ODN. This decline was prevented by NRSE-ODN treatment (there was no difference between the pre KA values and the post KA values). (single asterisk, P < 0.01; triple asterisks P < 0.05).
Discussion
Taken together, the above data indicate that: (1) HCN1 channelopathy, found in models of epileptogenesis and in human epilepsy, is a direct consequence of NRSF-mediated repression of the hcn1 gene, (2) NRSF initiates selective alterations of chromatin structure that may enable protracted repression of target genes, and (3) the use of ‘decoy’ ODNs comprising the NRSE sequence leads to short-term modulation of the pattern of spontaneous seizures that follow kainic-acid induced status epilepticus.
Epileptogenesis is associated with altered expression of hundreds of genes. Discovering the key genes that contribute to the disease process, and identifying the common mechanisms that might coordinately regulate these genes, and that underlie the persistent changes in their expression, would provide therapeutic opportunities. We chose to initiate our studies looking at the hcn1 gene because dysfunction of Ih, initially reported in an experimental model of febrile seizures39 has now been strongly associated with the epileptogenic process in several models of TLE7-10,40-42, and altered expression of HCN1 has been found in hippocampi resected from patients with severe TLE27. Here, we obtained evidence that the HCN1 channelopathy is a direct consequence of NRSF upregulation following the initial KA-induced status epilepticus. We found that NRSF physically bound the NRSE site on the hcn1 gene, and that this binding was augmented after KA. In addition, the prevention of NRSF-NRSE binding by decoy NRSE-ODNs restored HCN1 mRNA and protein levels, as well as Ih current properties and functions, to control levels. It is conceivable that NRSE-ODNs also influenced HCN1 expression indirectly. For example, our previous study in hippocampal slice cultures suggested that downregulation of the GluR2 AMPA receptor subunit, favoring the expression of Ca2+ permeable AMPA receptors and the activation of CaMKII, resulted in HCN1 mRNA downregulation after in vitro seizures28. Interestingly, the GluR2 gene also possesses a functional NRSE sequence43,44. However, although this mechanism might contribute to the initial repression of HCN1 expression during the first 24h following the KA-seizures, the transient reduction of GluR2, which dissipated by 72 hours (Richichi et al., Soc. Neurosci abstract 377.16, 2005) prevents this GluR2-mediated mechanism from contributing significantly to the persistent repression of the hcn1 gene found in vivo in the current study.
NRSF has binding sites within the regulatory regions of around 300 hippocampally-expressed genes22, including the delta GABAA- and GluR2 receptor subunits, which are downregulated in experimental TLE45,46. The probability of enriched binding of NRSF to a given NRSE, a measure of the functionality of the binding site, varies widely among genes. The relative value of NRSF binding to the hcn1 gene NRSE was 86, whereas the corresponding value for the NRSE site on the hcn2 gene was much lower (31), suggesting a weaker, less functional regulation of HCN2 expression by NRSF21. Accordingly, HCN2 expression was not significantly altered in our experimental conditions (but see 8,41), despite augmented levels of NRSF. These findings indicate that the presence of an NRSE site may be required but is not sufficient for functional regulation of the expression of a given gene by NRSF. NRSF can also bind to the brain-derived neurotrophic factor (BDNF), and increased expression of this factor has been linked to the progression of kindling43. Treatment with 2-deoxy-D-glucose reduced kindling progression by recruiting NRSF and preventing BDNF upregulation43.
The mechanisms of the upregulation of NRSF and the persistent repression of HCN1 expression during the epileptic state merit further investigation. Pathologically increased network activity manifest as interictal discharges and seizures34,35,47 have been found to induce NRSF expression. Here, we found NRSF-mediated epigenetic changes of the chromatin, consisting of changes in methylation of histones that typically lead to enduring repression that might no longer require the presence of NRSF. Thus, NRSF levels might fluctuate after an epilepsy-provoking insult, and might be upregulated repeatedly by interictal activity or spontaneous seizures during the latent and active phases of epilepsy, but may not be required for persistent repression of target genes including HCN1. For this channelopathy and others, post-translational mechanisms have also been demonstrated. These include phosphorylation48 or altered channel trafficking9,49. They may also contribute to Ih downregulation.
Supplementary Material
Acknowledgments
This work was supported by NIH R37 NS35439, INSERM, Fondation Française pour la Recherche sur l’Epilepsie (FFRE), Fondation pour la Recherche sur le Cerveau (FRC), Agence Nationale pour la Recherche (ANR, ANTARES and MINOS projects) and CURE. S.M. Was supported by a T32 Training grant, NS45540, and C.F. was supported by an Alberta Heritage Foundation for Medical Research (AHFMR) Fellowship. We thank M-P. Nesa and Jackie Yang for technical help.
References
- 1.Pitkanen A, Kharatishvili I, Karhunen H, et al. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl 2):13–20. doi: 10.1111/j.1528-1167.2007.01063.x. [DOI] [PubMed] [Google Scholar]
- 2.Dube CM, Brewster AL, Richichi C, et al. Fever, febrile seizures and epilepsy. Trends Neurosci. 2007;30:490–496. doi: 10.1016/j.tins.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukasiuk K, Kontula L, Pitkanen A. cDNA profiling of epileptogenesis in the rat brain. Eur J Neurosci. 2003;17:271–279. doi: 10.1046/j.1460-9568.2003.02461.x. [DOI] [PubMed] [Google Scholar]
- 4.Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- 5.Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- 6.Brewster A, Bender RA, Chen Y, et al. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah MM, Anderson AE, Leung V, et al. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jung S, Jones TD, Lugo JN, Jr, et al. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin M, Brager D, Jaramillo TC, et al. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol Dis. 2008;32:26–36. doi: 10.1016/j.nbd.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcelin B, Chauviere L, Becker A, et al. h channel-dependent deficit of theta oscillation resonance and phase shift in temporal lobe epilepsy. Neurobiol Dis. 2009;33:436–447. doi: 10.1016/j.nbd.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 11.Ludwig A, Budde T, Stieber J, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santoro B, Lee JY, Englot DJ, et al. Increased seizure severity and seizure-related death in mice lacking HCN1 channels. Epilepsia. 2010;51:1624–1627. doi: 10.1111/j.1528-1167.2010.02554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dibbens LM, Reid CA, Hodgson B, et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol. 2010;67:542–546. doi: 10.1002/ana.21909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- 15.Brewster AL, Chen Y, Bender RA, et al. Quantitative Analysis and Subcellular Distribution of mRNA and Protein Expression of the Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels throughout Development in Rat Hippocampus. Cereb Cortex. 2007;17:702–712. doi: 10.1093/cercor/bhk021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:848. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- 17.George MS, Abbott LF, Siegelbaum SA. HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K(+) channels. Nat Neurosci. 2009;12:577–584. doi: 10.1038/nn.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narayanan R, Johnston D. The h channel mediates location dependence and plasticity of intrinsic phase response in rat hippocampal neurons. J Neurosci. 2008;28:5846–5850. doi: 10.1523/JNEUROSCI.0835-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buzsaki G. Rhythms of the Brain. New York: Oxford University Press Inc; 2006. [Google Scholar]
- 21.Johnson DS, Mortazavi A, Myers RM, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 22.Roopra A, Huang Y, Dingledine R. Neurological disease: listening to gene silencers. Mol Interv. 2001;1:219–228. [PubMed] [Google Scholar]
- 23.Palm K, Belluardo N, Metsis M, et al. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spencer EM, Chandler KE, Haddley K, et al. Regulation and role of REST and REST4 variants in modulation of gene expression in in vivo and in vitro in epilepsy models. Neurobiol Dis. 2006;24:41–52. doi: 10.1016/j.nbd.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 25.Bernard C, Anderson A, Becker A, et al. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- 26.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 27.Bender RA, Kirschstein T, Kretz O, et al. Localization of HCN1 channels to presynaptic compartments: novel plasticity that may contribute to hippocampal maturation. J Neurosci. 2007;27:4697–4706. doi: 10.1523/JNEUROSCI.4699-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richichi C, Brewster AL, Bender RA, et al. Mechanisms of seizure-induced ‘transcriptional channelopathy’ of hyperpolarization-activated cyclic nucleotide gated (HCN) channels. Neurobiol Dis. 2008;29:297–305. doi: 10.1016/j.nbd.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 30.Surges R, Brewster AL, Bender RA, et al. Regulated expression of HCN channels and cAMP levels shape the properties of the h current in developing rat hippocampus. Eur J Neurosci. 2006;24:94–104. doi: 10.1111/j.1460-9568.2006.04880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brewster AL, Bernard JA, Gall CM, et al. Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol Dis. 2005;19:200–207. doi: 10.1016/j.nbd.2004.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roopra A, Qazi R, Schoenike B, et al. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol Cell. 2004;14:727–738. doi: 10.1016/j.molcel.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 33.Bender RA, Soleymani SV, Brewster AL, et al. Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci. 2003;23:6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staley K, Hellier JL, Dudek FE. Do interictal spikes drive epileptogenesis? Neuroscientist. 2005;11:272–276. doi: 10.1177/1073858405278239. [DOI] [PubMed] [Google Scholar]
- 35.El-Hassar L, Milh M, Wendling F, et al. Cell domain-dependent changes in the glutamatergic and GABAergic drives during epileptogenesis in the rat CA1 region. J Physiol. 2007;578:193–211. doi: 10.1113/jphysiol.2006.119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams PA, White AM, Clark S, et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White A, Williams PA, Hellier JL, et al. EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia. 2009 doi: 10.1111/j.1528-1167.2009.02339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chauviere L, Rafrafi N, Thinus-Blanc C, et al. Early deficits in spatial memory and theta rhythm in experimental temporal lobe epilepsy. J Neurosci. 2009;29:5402–5410. doi: 10.1523/JNEUROSCI.4699-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen K, Aradi I, Thon N, et al. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang K, Peng BW, Sanchez RM. Decreased IH in hippocampal area CA1 pyramidal neurons after perinatal seizure-inducing hypoxia. Epilepsia. 2006;47:1023–1028. doi: 10.1111/j.1528-1167.2006.00574.x. [DOI] [PubMed] [Google Scholar]
- 41.Powell KL, Ng C, O’Brien TJ, et al. Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats. Epilepsia. 2008;49:1686–1695. doi: 10.1111/j.1528-1167.2008.01593.x. [DOI] [PubMed] [Google Scholar]
- 42.van Gassen KL, Hessel EV, Ramakers GM, et al. Characterization of febrile seizures and febrile seizure susceptibility in mouse inbred strains. Genes Brain Behav. 2008;7:578–586. doi: 10.1111/j.1601-183X.2008.00393.x. [DOI] [PubMed] [Google Scholar]
- 43.Garriga-Canut M, Schoenike B, Qazi R, et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9:1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- 44.Jia YH, Zhu X, Li SY, et al. Kainate exposure suppresses activation of GluR2 subunit promoter in primary cultured cerebral cortical neurons through induction of RE1-silencing transcription factor. Neurosci Lett. 2006;403:103–108. doi: 10.1016/j.neulet.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 45.Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng Z, Huang CS, Stell BM, et al. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams PA, White AM, Clark S, et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poolos NP, Bullis JB, Roth MK. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci. 2006;26:7995–8003. doi: 10.1523/JNEUROSCI.2069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noam Y, Zha Q, Phan L, et al. Trafficking and surface expression of hyperpolarization-activated cyclic nucleotide-gated channels in hippocampal neurons. J Biol Chem. 2010;285:14724–14736. doi: 10.1074/jbc.M109.070391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.