

Abstract
Vaccinia virus is the prototypical orthopoxvirus of Poxviridae, a family of viruses that includes the human pathogens Variola (smallpox) and Monkeypox. Core viral functions are conserved among orthopoxviruses, and consequently Vaccinia is routinely used to study poxvirus biology and screen for novel antiviral compounds. Here we describe the development of a series of fluorescent protein-based reporter Vaccinia viruses that provide unprecedented resolution for tracking viral function. The reporter viruses are divided into two sets: (1) single reporter viruses that utilize temporally regulated early, intermediate, or late viral promoters; and (2) multi-reporter viruses that utilize multiple temporally regulated promoters. Promoter and reporter combinations were chosen that yielded high signal-to-background for stage-specific viral outputs. We provide examples for how these viruses can be used in the rapid and accurate monitoring of Vaccinia function and drug action.
Keywords: Orthopoxvirus, Antiviral, Gene expression, High-throughput, Drug discovery
1. Introduction
The Poxvirus family of enveloped DNA viruses includes several known human pathogens (Dhar et al., 2004, McFadden, 2005), against which population immunity has declined following the discontinuation of routine smallpox vaccination (Hammarlund et al., 2005, Hammarlund et al., 2003, Slifka, 2005). The potential for poxvirus bioterrorism, and a rise in reports of individuals infected with naturally occurring Monkeypox in Central and Western Africa (Parker et al., 2007, Whitley, 2003), are emerging concerns. Additionally, in 2003 there was an outbreak of 71 cases of human Monkeypox spanning six states in the Midwestern United States, an area without prior reported cases (Reed et al., 2004). Despite the existing and potential threat from these viruses, our understanding of how to develop better and safer vaccines is incomplete (Hammarlund et al., 2003, Slifka, 2005). There are also currently no drugs licensed by the US Food and Drug Administration to treat poxvirus-infected individuals.
An important tool in the development of new poxvirus therapies is Vaccinia virus. Vaccinia, the prototypical orthopoxvirus, was used in world-wide vaccination to eradicate naturally occurring smallpox. Vaccinia is a genetically tractable Biosafety Level 2 virus that is routinely used in the laboratory to study poxvirus function and search for novel antivirals. For example, recent work with Vaccinia identified ST-246, which shows demonstrable antiviral activity against Variola and Monkeypox (Smith et al., 2009).
Vaccinia entry into cells is followed by the orchestrated expression of viral genes, classically categorized as early, intermediate, and late, that precede viral assembly (Broyles, 2003, Moss, 2007). The virus is predominantly intracellular, and while capable of dissemination, infection of adjacent cells is a major mode of viral spread. Currently, the comprehensive, quantitative analysis of the cellular progression of Vaccinia infection commonly involves either (1) Western or Northern blotting for viral genes or (2) use of Vaccinia viruses harboring enzymatic reporters under control of canonical promoter elements (Satheshkumar et al., 2009). We were interested in developing a series of fluorescent protein-based Vaccinia viruses for rapid, homogenous assays in higher throughput applications. Fluorescent protein-based reporters have been applied with success in other virus systems, and have previously been incorporated into Vaccinia for various purposes (Hansen et al., 2002, Johnson et al., 2008, Popov et al., 2009, Villa et al., 2010, Ward and Moss, 2001). However, a systematic incorporation of fluorescent reporters to query each step of the virus life-cycle has not been undertaken.
Here we describe the development of reporter Vaccinia viruses that provide high resolution, real-time monitoring of viral function. Our first approach was to construct single reporter viruses, each of which contains a single fluorescent reporter under control of temporally regulated viral promoters. Our second approach was to construct single viruses with multiple fluorescent reporters for simultaneous monitoring of different phases of viral gene expression. When coupled to nucleic acid analysis, we believe that these systems provide unprecedented resolution in the accurate and quantitative monitoring of Vaccinia function.
2. Methods
2.1. Constructs and plasmids
Plasmids encoding fluorescent proteins were obtained from the following sources: Addgene 15214 (Cerulean), Clontech 6085-1 (EGFP), Addgene 11931 (Venus), Clontech 632523 (Cherry), and Axxora EVN-FP174 (TagBFP). Gateway entry vectors for Cerulean, EGFP, Venus, and Cherry were generated by PCR and cloning into pENTR-D/TOPO (Invitrogen K240020). CMV-promoter versions were generated by Gateway cloning into pcDNA DEST-40 (Invitrogen 12274-015).
2.2. Cell lines, infections, and viability assays
The following cell lines were obtained from ATCC: HEK293T (CRL-11268), BHK-21 (CCL-10), HeLa (CCL-2), A549 (CCL-185), and Vero (CCL-81). Cell lines were maintained in recommended media plus 10% FBS except in kinetic fluorescent plate reader experiments, where OptiMEM was used (see below). Virus infections were performed in 2% FBS-containing media and cells were maintained in 2% FBS-containing media for the duration of infection. Virus addition was designated as time 0 in all experiments. Cell viability was determined in a colorimetric MTT assay (Invitrogen V-13154).
2.3. Viral promoters and insertion sites
Promoter/leader sequences, with ATG reporter start codon underlined were as follows: early C11R, AGTTTATATTACTGAATTAATAATATAAAATTCCCAATCTTGTCATAAACACACACTGAGAAACAGCATAAACACAAAATCCATCAAAAATG; intermediate G8R, CATTTAACTTTAAATAATTTACAAAAATTTAAAATG; late F17R, GAATTTCATTTTGTTTTTTTCTATGCTATAAATG. Promoter/leader sequences were introduced as PCR primer overhangs upstream of fluorescent protein coding sequences. Reporter insertion sites, with the exception of late TagBFP in the triple reporter virus (TrpV; see below), were between J4L and J5R in the forward direction in the following sequence (∗ marks the insertion site): TAATCAATTAGTAGAGATGAGATAAGAACATTATAATAATCAATAATATATCTTATATCTC∗GTTTA. Late TagBFP was inserted between K7R and F1L in the forward direction in following sequence: TGATGATATAGGGGTCTTCATAACGCATAATTATTACGTTAGCATTCTATATCCGTGTTAAAAAA∗ AATTA, with AA used for the TAA stop codon for TagBFP. Promoter-less Venus (PLV) was generated from LV reporter virus using an insertion cassette designed to recombine out the F17R promoter (see below).
2.4. Reporter virus recovery
Promoter/reporter sequences were appended on either end with ∼400–500 bp of sequences flanking the insertion site by overlap PCR. No additional or intervening sequences were introduced with the exception of early C11R-directed reporters, which included an early terminator sequence (TATTATATTTTTTAT) immediately following the stop codon. Loss-less insertion cassettes were generated for Late Venus (LV), Late Red (LR), Early Venus (EV), Intermediate Venus (IV), Late Red Early EGFP (LREG), Late Red Early Venus (LREV), and Intermediate Red Early Venus (IREV) reporter viruses. Early, intermediate, and late refer to the viral promoter sequences described above, and Red refers to mCherry. Dual reporter viruses were constructed with promoter/reporter inserted in the order they appear in the name with no intervening sequence. The IREV virus was used in sequential recombination to generate TrpV, which contains Late TagBFP. All PCR was performed using KOD Hot Start DNA Polymerase (Novagen 71086-3). Homologous recombination cassettes were generated from PCR of gel-purified PCR fragments (Qiagen 20051) and were column-purified (Qiagen 28106) prior to transfection.
Homologous recombination was performed in HeLa cells by sequential infection/transfection in 6-well plates. Following 1 h infection with MOI 0.1 Vaccinia strain Western Reserve, infection media was removed and replaced with DMEM/10% FBS. Cells were then transfected with 1 μg of homologous recombination cassette using Lipofectamine 2000 (Invitrogen 11668-019). Transfection media was removed after 6 h and replaced with DMEM/10% FBS. After 24 h of post-infection, crude virus preparations were made and serially diluted to infect Vero cells, which were overlaid with DMEM/2.5% FBS/1% agarose. Fluorescent foci were identified by microscopy, picked, and purified through serial plaque purification. Plaque clonality, insertion site, and insert sequence were verified by sequencing PCR product from infected HeLa cell lysates that were heat-inactivated for 60 °C for 1 h. PCR primers outside and flanking the recombination cassette were used, and this entire PCR product was sequenced. For Promoter-less Venus (PLV), non-fluorescent plaques from LV infection/transfection were picked, purified, and validated.
2.5. Fluorescent plate reader assays
Well fluorescence following reporter virus infection was measured on a Tecan Infinite M1000 fluorescent plate reader (Tecan Group Ltd.). Cells were seeded at approximately 20,000 cells/well in 96-well plates (Corning 3603) or 10,000 cells/well in 384-well plates (Corning 3712). A high MOI of 5–10 was used for all fluorescent plate reader experiments. For endpoint assays, plates were incubated in a 37 °C, 5% CO2 incubator for the indicated time and sealed with optical film (USA Scientific 2978-2700) prior to fluorescence measurement. For time-course experiments, cells were seeded and infected in 2% FBS-containing OptiMEM (GIBCO 31985). OptiMEM was used because it is buffered and does not require 5% CO2 incubation. Plates were sealed with optical film and placed in the fluorescent plate reader with chamber set to 37 °C. A kinetic plate reader cycle was used to measure fluorescence hourly. The following settings were used: bottom read, flash frequency 100 Hz, optimal gain for endpoint assays, and manual gain for kinetic cycles. Manual gain setting was chosen based on previous experience with endpoint assays to avoid “over” readings at late time-points. The following excitation/emission wavelengths were used (in nm, with 5 nm bandpass): 448/473 for Cerulean, 495/510 for EGFP, 515/530 for Venus, 587/610 for Cherry, and 415/457 for TagBFP. Optimum wavelength settings were determined by comparing excitation and emission sweeps of non-reporter- and reporter-containing cells. Fold-inductions were determined from time 0 or uninfected cells, which gave indistinguishable readings when using sucrose purified reporter viruses.
2.6. Microscopy
Images were acquired using a Zeiss inverted microscope and Axiovision software (Zeiss). Additional image processing was performed in Volocity LE software (Perkin–Elmer). The following filters were used for microscopy: DAPI, CFP (TagBFP), FITC (EGFP), YFP (Venus), and TRITC (Cherry).
2.7. Nucleic acid analysis
mRNA analysis was performed by direct RT-qPCR of A549 cell lysates obtained in 96-well format. A549 cells were seeded to achieve a density approximately 10,000 cells/well the day of infection. Cell lysates were obtained using a Cells-to-Ct kit (Ambion AM1728) with two modifications to the supplied protocol: (1) cells were lysed in 25 μl lysis solution consisting of 24 μl of supplied Lysis Buffer and 1 μl RNase-free DNase I (Ambion AM2222); and (2) cell lysis and DNA digestion were allowed to proceed for 30 min at room temperature to completely remove viral DNA prior to addition of 2.5 μl of the supplied Stop Solution. Lysates were transferred to 96-well PCR plates, sealed, and stored at −80 °C for further processing. cDNA was generated from 2 μl of lysate in a 20 μl RT reaction (random and oligo dT primed), and 2 μl of this RT reaction was used in a 20 μl qPCR reaction using kit-supplied reagents. Duplex Taqman was performed for Venus and 18S rRNA (Invitrogen 4310893E) using a CFX96 Real-Time System and CFX Manager software (BioRad). The following Taqman primer/probe set was used for Venus (IDT): AAAGACCCCAACGAGAAGC (forward), GTCCATGCCGAGAGTGATC (reverse), 6-FAM-TGCTGGAGTTCGTGACCGCC-IBFQ (probe). Venus Cts were adjusted for deviations in 18S rRNA Cts for each time-point, although these deviations were negligible within and across time-points. Infection media was removed after 1 h and wells were washed once with PBS prior to media re-addition, with the exception of time 0 which was harvested similarly but without 1 h incubation. Infection start times were staggered to allow for simultaneous lysis of all samples.
DNA analysis was performed by direct qPCR of A549 cell lysates obtained in 96-well format. A549 cells were seeded into wells to achieve a density approximately 10,000 cells/well the day of infection, and infections were with MOI 5 LV reporter virus. To remove excess virus and viral genomes, infection media was removed after 1 h and cells were washed once with PBS prior to adding compound- or vehicle-containing media. Cell lysates were obtained by media removal and 10 min room temperature incubation with 40 μl lysis buffer (10 mM glycine, 1% Triton X-100, pH 2.5 with HCl, filtered) per well. Samples were transferred to 96-well PCR plates, sealed, and stored at −20 °C for further processing. Two microlitre of cell lysate (500 cell equivalents, or c.e.) was directly analyzed in a 20 μl qPCR reaction using reagents supplied in the Cells-to-Ct kit (see above). Duplex Taqman was performed for Venus (see above for primer/probe sequences) and endogenous RNase P (Invitrogen 4403326). Venus Cts were adjusted for deviations in RNase P Cts, which were negligible.
2.8. Compounds and screens
Compound- or vehicle-control treated infections were initiated by removing cell culture media, adding a 2× solution of compound or vehicle in 2% FBS-containing media, followed shortly by adding an equal volume of 2× solution of virus in 2% FBS-containing media to dilute compound to 1×. In experiments where infection media was removed, media was replaced with media containing 1× compound or vehicle.
A panel of 18 cellular stress-related compounds (Fig. 5 A) was kindly provided by Susan Lindquist (Whitehead Institute, Cambridge, MA). The screening library (Fig. 5B) was generated by the Chemical Methodology and Library Development group at Boston University (CMLD; Boston University, Boston, MA). Screening of approximately 2000 compounds was performed using the LREV reporter virus. A549 cells were seeded at 20,000 cells/well in 96-well plates (Corning 3603) the previous day and infected with MOI 10 LREV, and infections were allowed to proceed in the presence of compound. Final compound concentrations were estimated to be approximately 10 μM. At 12 hpi, media was removed and cells were fixed using PBS containing 4% formaldehyde to enable fixed endpoints for next day scoring.
Fig. 5.
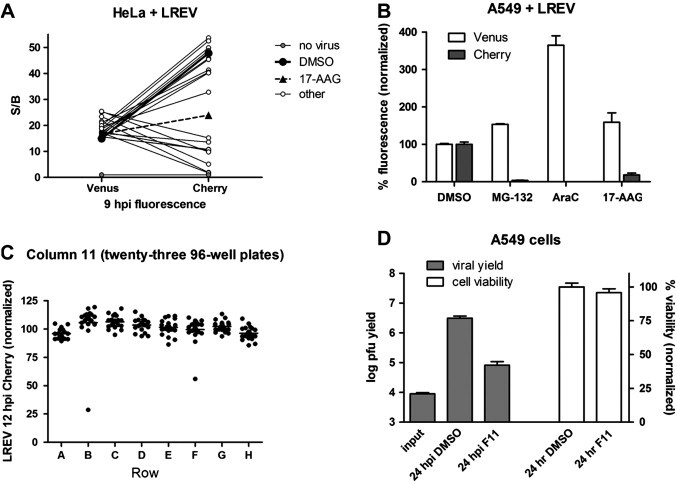
(A) Testing of 18 compounds, including 17-AAG (geldanaycin), for an effect on early and late reporter expression following high MOI LREV infection of HeLa cells. Compounds with cellular stress-modulating activity were pre-selected and tested at a range of of 1–20 μM. 17-AAG was tested at a concentration of 2 μM. S/B ratios at 9 hpi over uninfected cells are plotted. Lines connect the Venus and Cherry data points for each compound. (B) The effects of 5 μM MG-132, 1 μg/ml AraC, or 5 μM 17-AAG on reporter expression following overnight infection of A549 cells with LREV. Venus or Cherry fluorescence is normalized to values obtained in the DMSO-treated infection. (C) Representative data from screening of approximately 2000 compounds. Compound concentrations were estimated to be 10 μM. Data from Column 11 for each of the twenty-three screening plates are shown, with rows on the x-axis. A549 cells were infected with high MOI LREV and Venus and Cherry fluorescence were measured at 12 hpi. Plotted is Cherry S/B in each of 184 wells. (D) The effect of 20 μM compound F11, shown in Fig. 1C, on viral replication and A549 cell viability. Plaque forming unit (pfu) yield from a 24 h single-cycle growth assay is plotted on the left y-axis, in log-scale. A549 cell viability following 24 h incubation with DMSO or 20 μM compound F11 is plotted on the right y-axis.
3. Results
3.1. Fluorescent reporter proteins
We initially determined the signal-to-background (S/B) ratios of several different fluorescent proteins in transient transfection assays. CMV promoter-driven constructs for Cerulean, EGFP, Venus, or Cherry were transfected into HEK 293T cells and well fluorescence was determined 24 h post-transfection using a fluorescent plate reader. Optimum signal-to-background (S/B) was determined by comparison to mock transfected cells using excitation and emission sweeps surrounding the reported peaks for each fluorescent protein (see Section 2 for details). Venus yielded a S/B of 154.9, while the other proteins had good but substantially lower S/B in the order Cherry > Cerulean > EGFP (13.5, 12.1, 8.1, respectively; Fig. 1 A). Given that the quantum yields of these fluorescent proteins are expected to be similar, this was likely due to differences in cell auto-fluorescence in the relevant wavelength regions used for detection.
Fig. 1.
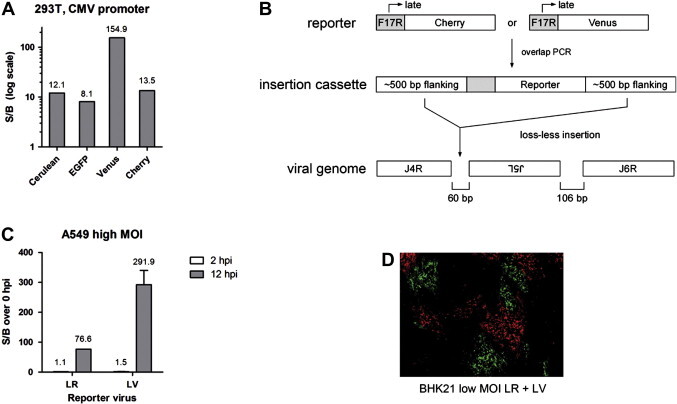
(A) Fluorescent protein performance in plate reader assays. HEK 293T cells were transiently transfected with CMV-promoter driven Cerulean, EGFP, Venus, or Cherry plasmids. Fluorescence was measured 24 h post-transfection and normalized to mock-transfected cells. (B) Schematic of the strategy used to generate the single reporter viruses late Cherry (or Red; LR) and late Venus (LV). The canonical late F17R viral promoter was used. The recombination cassette was generated by overlap PCR for homologous, loss-less insertion in the 60 bp intergenic region between Vaccinia J4R and J5L genes. (C) Plate reader fluorescence for Cherry following LR infection, or Venus following LV infection, at 2 and 12 hpi of A549 cells. (D) 10× image of a confluent BHK21 cell monolayer 24 hpi after low MOI co-infection with LR and LV viruses, showing easily identifiable foci of infected cells originating from infection centers with either virus.
3.2. Single reporter viruses
We chose Venus and Cherry for initial construction of recombinant Vaccinia viruses. Venus or Cherry under control of the canonical late F17R Vaccinia promoter (Bertholet et al., 1987, Schwer et al., 1987) was inserted into the genome of Vaccinia strain Western Reserve between the J4L and J5R genes, as previous work had shown that such insertions are well tolerated by the virus (Fig. 1B; (Satheshkumar et al., 2009). We used standard methods for generating recombinant virus through homologous, loss-less recombination, with the exception that the procedure was streamlined to eliminate any cloning (see Section 2). An added benefit to this modification was routinely higher recombination frequency with linear PCR product when compared to plasmid (data not shown). The resulting reporter viruses, Late Red (LR) and Late Venus (LV), strongly expressed fluorescent protein in infected cells. Fluorescence measurement following single cycle, high multiplicity-of-infection (MOI) of A549 cells with LR or LV showed no fluorescence 2 h post-infection (hpi), prior to the expected onset of late gene expression, and high fluorescence at 12 hpi (Fig. 1C). S/B ratios were 76.6 for Cherry and 291.9 for Venus, consistent with the transient transfection results in Fig. 1A. Reporter expression allowed for easy identification of recombinant virus during plaque purification and in pooled format, as has been noted earlier (Popov et al., 2009). This is illustrated in Fig. 1D, where distinct Venus- and Cherry-expressing foci are clearly visible following low MOI co-infection and spread of LR and LV reporter viruses on BHK-21 cells.
3.3. Stage-specific single reporter viruses
Based on the superior plate reader performance of Venus, we constructed additional viruses designed to express this fluorescent protein at each of the three classical stages of viral gene expression. Using the strategy described above, Venus was inserted with the canonical G8R intermediate promoter (Baldick et al., 1992, Baldick and Moss, 1993) to generate Intermediate Venus (IV). In a separate virus, Venus was inserted with the canonical C11R early promoter (Broyles et al., 1991) to generate Early Venus (EV). This virus contained an early terminator sequence immediately downstream of the Venus stop codon (Broyles, 2003, Yuen and Moss, 1987). We also generated Promoter-less Venus (PLV), which contains Venus but lacks a promoter element. The insertions in these four viruses (LV, IV, EV, and PLV) are depicted schematically in Fig. 2 A. Together, the viruses were designed to independently track stages of viral gene expression through the analysis of a single reporter protein.
Fig. 2.
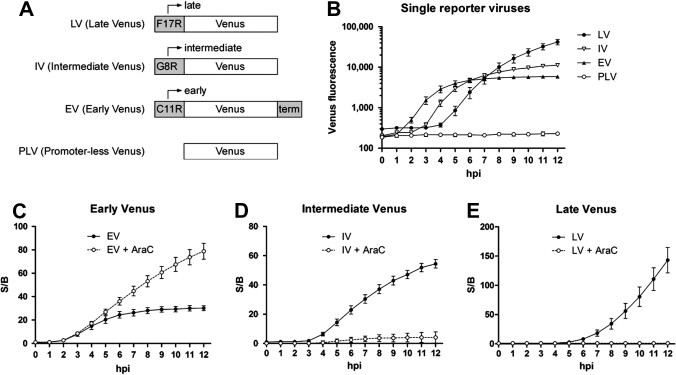
(A) Schematic of the inserts for Late Venus (LV), Intermediate Venus (IV), Early Venus (EV), and Promoter-less Venus (PLV) single reporter viruses. Inserts were placed between the Vaccinia J4R and J5L genes, as shown in Fig. 1. The canonical late F17R, intermediate G8R, and early C11R viral promoters were used. EV contains an early terminator sequence (term) immediately after the Venus stop codon. PLV contains the Venus open reading frame with no preceding promoter element. (B) Kinetics of Venus fluorescence following high MOI infection of A549 cells with either LV, IV, EV, or PLV. Raw fluorescence is plotted in log-scale. (C–E) Effect of the DNA replication inhibitor AraC on reporter expression following EV infection (C), IV infection (D), or LV infection (E), in the same experiment as (B). Open circles are untreated infections and filled circles are infections in the presence of 1 μg/ml AraC. S/B ratios are plotted.
We validated the performance of these viruses in kinetic fluorescent plate reader experiments. For these 96-well experiments, confluent A549 cells were infected with a high MOI of PLV, EV, IV, or LV and Venus fluorescence was measured hourly. To simplify data acquisition, buffered Opti-MEM media was used and plates were sealed with optical film and maintained at 37 °C in the fluorescent plate reader chamber (with no CO2). This allowed for automated fluorescence measurement at designated intervals. Raw fluorescence units up to 12 hpi are shown Fig. 2B. No fluorescence was observed with PLV infection, and Venus fluorescence arose with the anticipated order and kinetics for EV, IV, and LV infections (Baldick and Moss, 1993, Black and Condit, 1996, Xiang et al., 1998). At 12 hpi under these conditions, the S/B ratios of Venus for PLV, EV, IV, and LV were 1, 31, 55, and 141, respectively, with LV fluorescence continuing to increase. Because fluorescent proteins such as Venus have long half-lives of over 24 h (Corish and Tyler-Smith, 1999, Li et al., 1998), no decline after peak fluorescence was observed in these experiments.
To further validate these viruses, we tested the effect of cytosine arabinoside (AraC) in the same experiment. AraC is a potent inhibitor of viral DNA replication, which is required to reset the transcriptional landscape for post-early gene expression (Keck et al., 1990). The data are distributed across separate panels for clarity, with Venus S/B plotted (Fig. 2C–E). The expected effect of AraC on the different reporter viruses was seen. There was a failure to terminate EV reporter expression (Fig. 2C), and a concomitant loss of IV and LV reporter expression (Fig. 2D and E, respectively). Taken together, the timing, magnitude, and AraC sensitivity of fluorescence originating from these single reporter viruses faithfully recapitulate the canonical stages of viral gene expression.
3.4. Single reporter mRNA and DNA analysis
A potential advantage of a single-reporter system is the ability to use a single RT-PCR approach to correlate mRNA and protein expression. We used the four viruses described in Fig. 2 to assess the kinetics of Venus mRNA accumulation. For this assay, two-step RT-qPCR was performed directly from cell lysates generated from 96-well cell culture plates. The PLV virus was included as a control for any non-specific, read-through, and opposing strand transcription, which cannot be distinguished from promoter-dependent RNA accumulation by this RT-qPCR analysis. Fig. 3 A shows a typical result of these experiments. For each reporter virus, including PLV, the threshold cycle (Ct) value dropped with increasing time, indicating the accumulation of RNA with Venus primer/probe sequence. Viral DNA replication did not contribute to declining Ct values, as no change was observed over the time-course in the absence of RT using these DNase-treated lysates (Fig. 3A, no RT).
Fig. 3.
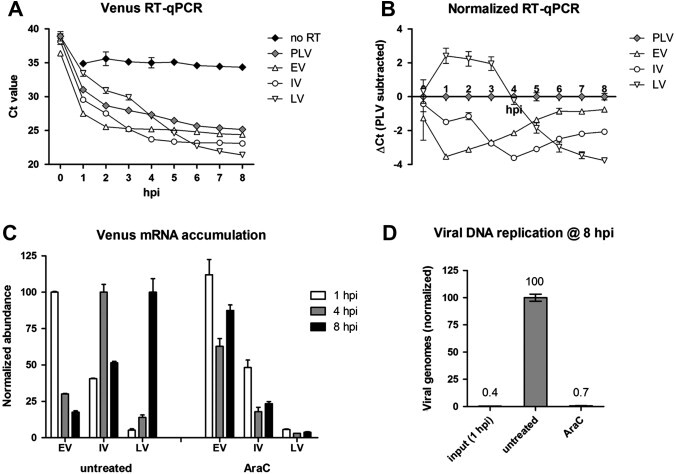
(A) Venus RNA analysis following high MOI infection of A549 cells with PLV, EV, IV, or LV reporter viruses. The assay was performed entirely in 96-well format and directly from cell lysates. Infection start times were staggered to allow simultaneous cell lysis. Ct values from Venus RT-qPCR are plotted, starting from Ct 20. “no RT” samples were from the LV infected cell lysates with the RT enzyme omitted from Venus RT-qPCR. (B) Ct values for EV, IV, and LV infections normalized against the Ct value for PLV infection at each time-point. This normalization uncovers promoter-dependent accumulation of Venus RNA (i.e., Venus mRNA). (C) Venus mRNA accumulation at 1, 4, and 8 hpi following EV, IV, or LV infection of A549 cells, in the absence (untreated) or presence of 1 μg/ml AraC. Normalized abundance is plotted. EV data were normalized to 1 hpi EV untreated, IV data were normalized to 4 hpi IV untreated, and LV data were normalized to 8 hpi LV untreated. (D) DNA analysis for LV viral genomes following infection of A549 cells. Shown are normalized qPCR signals for Venus DNA for input (1 hpi) and 8 hpi in the absence or presence of 1 μg/ml AraC.
The expression kinetics of Venus mRNA were not entirely clear until Ct values for the EV, IV, and LV infections were normalized against the PLV infection at each time-point (ΔCt; Ct EV, IV, or LV minus Ct PLV). This normalization step revealed the contribution of each individual promoter element (early, intermediate, or late) to the Venus RT-PCR signal, and was taken as a measure of Venus mRNA abundance. The ΔCt values for Venus mRNA following EV, IV, or LV infection relative to the PLV infection are shown in Fig. 3B, where a negative ΔCt indicates mRNA accumulation. Over the 8 h time-course, the peak in Venus mRNA abundance occurred at 1 hpi for EV, 4 hpi for IV, and 8 hpi for LV. These results are consistent with the Venus expression patterns in Fig. 2, and reveal an approximately 1 h lag between detectable mRNA induction and fluorescence.
To further validate this system, the experiment was repeated in a more limited time-course of 1, 4 and 8 hpi in the absence or presence of AraC. In the untreated infections, EV mRNA peaked at 1 hpi, IV mRNA at 4 hpi, and LV mRNA at 8 hpi, as before (Fig. 3C). In AraC-treated infections, EV mRNA accumulation failed to decline normally and IV and LV mRNA accumulation was dampened. This was consistent with Venus expression patterns, and with the established effect of a DNA replication inhibitor on Vaccinia transcription. Taken together, the results support the use of this system for stage-specific mRNA analysis where reasonably high throughput is desired, with the critical inclusion of the PLV virus for normalization. Moreover, the system is readily adapted to reporter virus DNA analysis using non-DNase treated lysates and Venus PCR (see Section 2 for details). Using this approach, LV viral genomes were found to increase ∼250-fold from 1 hpi to 8 hpi, an increase that was eliminated in the presence of the DNA replication inhibitor AraC (Fig. 3C).
3.5. Multi-reporter viruses
In addition to these single reporter viruses, we constructed multi-reporter viruses to simultaneously monitor multiple stages of gene expression. To do this we designed a dual-reporter insertion cassette with two sequential promoter/fluorescent reporters for homologous recombination between J4L and J5R. Cherry was placed under control of the late F17R promoter, and EGFP or Venus was placed under control of the early C11R promoter and positioned immediately downstream of Cherry. This resulted in the Late Red Early Green (LREG) and Late Red Early Venus (LREV) viruses (Fig. 4 A). Fig. 4B shows a single focus of infected Vero cells 48 hpi following LREG infection. As the infection radiates outward, the outer, newly infected cells express early GFP, while cells located more centrally express both early GFP and late Cherry. The performance of LREG and LREV dual reporter viruses in fluorescent plate reader experiments is shown in Fig. 4C, with hourly reads up to 10 hpi following high MOI infection of A549 cells. As before, Venus is superior to EGFP in these assays, although EGFP is adequate for microscopic analysis. At 10 hpi following LREV or LREG infection, early Venus and early EGFP S/B ratios were 26.8 and 3.2, respectively. Late Cherry S/B ratios at 10 hpi were comparable between the two viruses (87 and 77 for LREG and LREV, respectively).
Fig. 4.
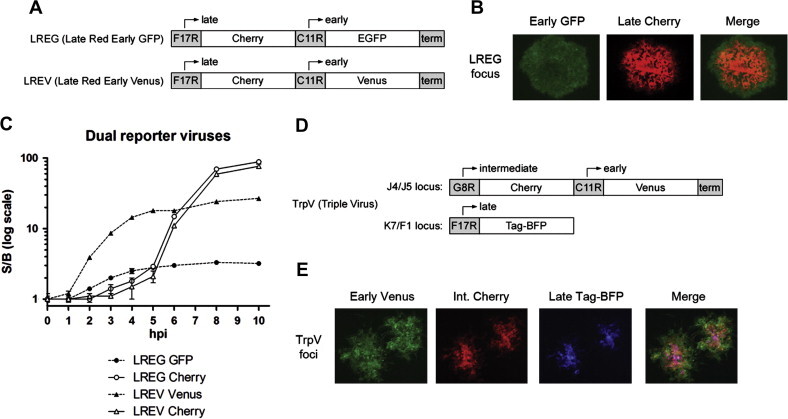
(A) Schematic of the inserts for Late Cherry Early EGFP (LREG) and Late Cherry Early Venus (LREV) multi-reporter viruses. There are no intervening sequences between promoter and reporter elements. Inserts were placed between the Vaccinia J4R and J5L genes, as shown in Fig. 1. (B) EGFP and Cherry fluorescence in a focus of Vero cells 48 hpi following low MOI LREG infection. 10× magnification. (C) Schematic of the inserts for Intermediate Cherry, Early Venus, and Late Tag-BFP in the multi-reporter Triple Virus (TrpV). Intergenic insertion sites are noted (J4/J5 and K7/F1). (E) Venus, Cherry, and Tag-BFP fluorescence in two foci of A549 cells 24 hpi following low MOI TrpV infection. 10× magnification.
We also generated a triple reporter virus for simultaneous monitoring of early, intermediate, and late reporter expression. Initial efforts using a combination of Cerulean, Venus, and Cherry, which have sufficient spectral separation for simultaneous use, were complicated by the high similarity of Cerulean and Venus (see Section 4). We therefore turned to the more divergent blue fluorescent protein TagBFP to use in combination with Venus and Cherry. Venus, Cherry, and TagBFP were designed under control of early C11R, intermediate G8R, and late F17R promoters, respectively. Intermediate Cherry and early Venus were inserted between the J4R and J5L genes, as before, and late TagBFP was inserted between the K7R and F1L genes (Fig. 4D). This combination resulted in a stable Triple Virus (TrpV). We found that while early Venus and intermediate Cherry expression arose with the expected kinetics and magnitude, the performance of TagBFP in fluorescent plate reader experiments was poor (data not shown). This precluded the use of this virus for high confidence, quantitative plate reader analysis. However, TrpV was suitable for use in microscopy. Fig. 4E shows two foci of infected A549 cells 24 hpi following low MOI TrpV infection. As in Fig. 4B, regions of reporter expression mark the different maturation stages in infected cells as the infection radiates outward.
3.6. Examples of application
A critical use for reporter viruses is to identify and gain mechanistic insight into antiviral compounds. Examples of such use, using some of the reporter viruses described here, are shown in Fig. 5. It was previously shown that 17-AAG (geldanamycin), an inhibitor of the ATPase activity of heat shock protein 90 (Hsp90), can attenuate Vaccinia replication (Hung et al., 2002). We were therefore interested in testing a panel of selected compounds, including 17-AAG, that were independently found to have cellular stress modulating activity (unpublished observations). HeLa cells were infected with high MOI LREV in the absence and presence of 18 selected compounds, and early Venus and late Cherry fluorescence were measured at 9 hpi (Fig. 5A). A high percentage of these pre-selected compounds reduced late Cherry expression, while none reduced early Venus expression. 17-AAG (at 2 μM) and 9 of the other 17 compounds (at a range of concentrations between 1 and 20 μM) reduced late Cherry expression by ⩾50%. This reduction was often accompanied by slightly elevated early Venus expression. In the case of 17-AAG, the data are consistent with reports that it affects late but not early viral gene expression (Hung et al., 2002). The proteasome inhibitor MG-132 is another compound with reported antiviral activity (Satheshkumar et al., 2009, Teale et al., 2009). A comparison of the effects of 5 μM MG-132, 1 μg/ml AraC, or 5 μM 17-AAG on reporter expression following LREV infection is shown in Fig. 5B. The results indicate that a simple experiment with a dual reporter virus faithfully reproduces the established effects on viral gene expression of several compounds with known antiviral activity.
Because the fluorescent plate reader format can be used in high-throughput format, we screened roughly 2000 compounds from a diversity oriented synthesis library for LREV reporter expression following high MOI infection of A549 cells. A sampling of the data obtained are provided in Fig. 5C, which shows 12 hpi normalized fluorescence in a single column (column 11) of the twenty-three 96-well plates screened (23 columns × 8 rows/column = 184 data points). While none of these compounds affected early Venus expression (data not shown), two compounds reduced late Cherry expression and were easily identified. The hit-rate of this naïve library was substantially lower than that observed for the set of pre-selected compounds shown in Fig. 5A. The effect of one of these compounds, F11 from well F11 in Fig. 5C, in a single-cycle growth assay using Vaccinia strain Western Reserve is shown in Fig. 5D. A plaque forming unit (pfu) burst size of 2.6-log was reduced to 1.0-log in the presence of 20 μM compound F11. Cell viability of A549 cells was unaffected in metabolic activity assays after 24 h treatment with 20 μM compound F11 (Fig. 5D). The results demonstrate that these systems can be used to rapidly identify compounds with anti-viral activity, while simultaneously providing stage-specificity of inhibitory effects.
4. Discussion
We present here the development and performance of a set of replication-competent reporter Vaccinia viruses for tracking viral function. A key practical finding was that of the fluorescent proteins tested, Venus (Nagai et al., 2002) provided an exceptionally high S/B ratio. While this increase may not be important where expression is strong (i.e., with a late Vaccinia promoter at late time-points), it could be pivotal in systems where only weaker expression can be achieved (i.e., with an early Vaccinia promoter). Although the high Venus S/B ratio could be due to something intrinsic to the protein or its expression in cells, excitation and emission sweeps indicated that it was more likely due to low cell auto-fluorescence in the Venus measurement region (Dower, unpublished). The performance of Venus allowed us to generate stage-specific reporter Viruses with high outputs. Furthermore, the use of these single reporter viruses enabled coupling to a semi-high throughput RT-qPCR assay that provided complimentary and reinforcing data on mRNA expression.
When designing multi-reporter viruses that utilized different fluorescent proteins at unique stages of the virus life cycle, the choice of fluorescent reporters was not routine. EGFP and its variants Cerulean and Venus are derived from the jellyfish Aequorea victoria, and are highly similar, while the dsRed variant Cherry is derived from the sea anemone Discosoma sp., and is divergent from EGFP. As mentioned, our attempts to incorporate both Venus and Cerulean into a single virus were initially successful but ultimately failed upon passaging. This appeared to be due to extraordinarily high recombination rates leading to reporter loss and/or mis-expression (Dower, unpublished). As a result, a combination of more dissimilar fluorescent proteins such as Venus, Cherry, and TagBFP (Subach et al., 2008), derived from the sea anemone Entacmaea quadricolor, was required for stable, simultaneous incorporation. A useful aspect of the multi-reporter viruses was that early and intermediate reporter proteins showed saturating expression kinetics under standard conditions due to long reporter protein half-life. Consequently, a single endpoint measurement at late stages of infection provided an accurate depiction of earlier stages of the virus life-cycle. Slowed reporter accumulation, however, is readily detected through automated real-time fluorescence measurement.
The reporter viruses described here are well suited to drug screening, and provide leverage to narrow the drug mechanism of action. In addition, the use of multi-reporter viruses can potentially provide tiered information in a single screen. In our screening of roughly 2000 compounds against LREV infection, the late Cherry S/B ratio was on average ∼120. We have subsequently transitioned the assay to the LV virus in 384-well format, which yielded a S/B ratio of ∼300 following high MOI infection (Dower, unpublished). In addition, a multi-cycle, low MOI LV infection yielded a S/B ratio of ∼800 at 48 hpi (Dower, unpublished). Z′ values were 0.5 in these experiments and are expected to increase with continued development and automation. A low MOI format screen of this fitness is of particular interest since it would additionally score for defects in viral assembly and transmission.
In summary, we have developed a series of reporter viruses that provide, to our knowledge, unparalleled resolution of the Vaccinia life-cycle in a homogenous system. The systems described here allow for rapid and comprehensive probing of viral function in high-throughput applications such as new drug identification, and are also likely to be useful for tracking basic aspects of virus replication such as viral spread, host-range effects, and the role of host factors in viral replication.
Acknowledgements
We would like to thank Mohamed R. Mohamed (McFadden lab) for expert technical advice on the generation of recombinant Vaccinia viruses, as well as Sue Lindquist and Sandro Santagata for kindly providing compounds. We also thank Claire Marie Filone for helpful manuscript comments and members of the Rubins, Hensley and Connor labs for comments and advice during the development of these reagents.
References
- Baldick C.J., Jr., Keck J.G., Moss B. Mutational analysis of the core, spacer, and initiator regions of vaccinia virus intermediate-class promoters. J. Virol. 1992;66:4710–4719. doi: 10.1128/jvi.66.8.4710-4719.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldick C.J., Jr., Moss B. Characterization and temporal regulation of mRNAs encoded by vaccinia virus intermediate-stage genes. J. Virol. 1993;67:3515–3527. doi: 10.1128/jvi.67.6.3515-3527.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet C., Van Meir E., ten Heggeler-Bordier B., Wittek R. Vaccinia virus produces late mRNAs by discontinuous synthesis. Cell. 1987;50:153–162. doi: 10.1016/0092-8674(87)90211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black E.P., Condit R.C. Phenotypic characterization of mutants in vaccinia virus gene G2R, a putative transcription elongation factor. J. Virol. 1996;70:47–54. doi: 10.1128/jvi.70.1.47-54.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broyles S.S. Vaccinia virus transcription. J. Gen. Virol. 2003;84:2293–2303. doi: 10.1099/vir.0.18942-0. [DOI] [PubMed] [Google Scholar]
- Broyles S.S., Li J., Moss B. Promoter DNA contacts made by the vaccinia virus early transcription factor. J. Biol. Chem. 1991;266:15539–15544. [PubMed] [Google Scholar]
- Corish P., Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12:1035–1040. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- Dhar A.D., Werchniak A.E., Li Y., Brennick J.B., Goldsmith C.S., Kline R., Damon I., Klaus S.N. Tanapox infection in a college student. N. Engl. J. Med. 2004;350:361–366. doi: 10.1056/NEJMoa031467. [DOI] [PubMed] [Google Scholar]
- Hammarlund E., Lewis M.W., Carter S.V., Amanna I., Hansen S.G., Strelow L.I., Wong S.W., Yoshihara P., Hanifin J.M., Slifka M.K. Multiple diagnostic techniques identify previously vaccinated individuals with protective immunity against monkeypox. Nat. Med. 2005;11:1005–1011. doi: 10.1038/nm1273. [DOI] [PubMed] [Google Scholar]
- Hammarlund E., Lewis M.W., Hansen S.G., Strelow L.I., Nelson J.A., Sexton G.J., Hanifin J.M., Slifka M.K. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 2003;9:1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- Hansen S.G., Cope T.A., Hruby D.E. BiZyme: a novel fusion protein-mediating selection of vaccinia virus recombinants by fluorescence and antibiotic resistance. Biotechniques. 2002;32:1182–1187. doi: 10.2144/02325dd08. pp. 1178, 1180. [DOI] [PubMed] [Google Scholar]
- Hung J.J., Chung C.S., Chang W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J. Virol. 2002;76:1379–1390. doi: 10.1128/JVI.76.3.1379-1390.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.C., Damon I.K., Karem K.L. A rapid, high-throughput vaccinia virus neutralization assay for testing smallpox vaccine efficacy based on detection of green fluorescent protein. J. Virol. Methods. 2008;150:14–20. doi: 10.1016/j.jviromet.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Keck J.G., Baldick C.J., Jr., Moss B. Role of DNA replication in vaccinia virus gene expression: a naked template is required for transcription of three late trans-activator genes. Cell. 1990;61:801–809. doi: 10.1016/0092-8674(90)90190-p. [DOI] [PubMed] [Google Scholar]
- Li X., Zhao X., Fang Y., Jiang X., Duong T., Fan C., Huang C.C., Kain S.R. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 1998;273:34970–34975. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- McFadden G. Poxvirus tropism. Nat. Rev. Microbiol. 2005;3:201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. (Poxviridae: The Viruses and Their Replication). [Google Scholar]
- Nagai T., Ibata K., Park E.S., Kubota M., Mikoshiba K., Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Parker S., Nuara A., Buller R.M., Schultz D.A. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 2007;2:17–34. doi: 10.2217/17460913.2.1.17. [DOI] [PubMed] [Google Scholar]
- Popov S., Mirshahidi S., Essono S., Song R., Wang X., Ruprecht R.M. Generation of recombinant vaccinia viruses via green fluorescent protein selection. DNA Cell Biol. 2009;28:103–108. doi: 10.1089/dna.2008.0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed K.D., Melski J.W., Graham M.B., Regnery R.L., Sotir M.J., Wegner M.V., Kazmierczak J.J., Stratman E.J., Li Y., Fairley J.A., Swain G.R., Olson V.A., Sargent E.K., Kehl S.C., Frace M.A., Kline R., Foldy S.L., Davis J.P., Damon I.K. The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 2004;350:342–350. doi: 10.1056/NEJMoa032299. [DOI] [PubMed] [Google Scholar]
- Satheshkumar P.S., Anton L.C., Sanz P., Moss B. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 2009;83:2469–2479. doi: 10.1128/JVI.01986-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B., Visca P., Vos J.C., Stunnenberg H.G. Discontinuous transcription or RNA processing of vaccinia virus late messengers results in a 5′ poly(A) leader. Cell. 1987;50:163–169. doi: 10.1016/0092-8674(87)90212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka M.K. The future of smallpox vaccination: is MVA the key? Med. Immunol. 2005;4:2. doi: 10.1186/1476-9433-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.K., Olson V.A., Karem K.L., Jordan R., Hruby D.E., Damon I.K. In vitro efficacy of ST246 against smallpox and monkeypox. Antimicrob. Agents Chemother. 2009;53:1007–1012. doi: 10.1128/AAC.01044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach O.M., Gundorov I.S., Yoshimura M., Subach F.V., Zhang J., Gruenwald D., Souslova E.A., Chudakov D.M., Verkhusha V.V. Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 2008;15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teale A., Campbell S., Van Buuren N., Magee W.C., Watmough K., Couturier B., Shipclark R., Barry M. Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J. Virol. 2009;83:2099–2108. doi: 10.1128/JVI.01753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa N.Y., Bartee E., Mohamed M.R., Rahman M.M., Barrett J.W., McFadden G. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology. 2010;401:266–279. doi: 10.1016/j.virol.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward B.M., Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J. Virol. 2001;75:4802–4813. doi: 10.1128/JVI.75.10.4802-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley R.J. Smallpox: a potential agent of bioterrorism. Antiviral Res. 2003;57:7–12. doi: 10.1016/s0166-3542(02)00195-x. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Simpson D.A., Spiegel J., Zhou A., Silverman R.H., Condit R.C. The vaccinia virus A18R DNA helicase is a postreplicative negative transcription elongation factor. J. Virol. 1998;72:7012–7023. doi: 10.1128/jvi.72.9.7012-7023.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen L., Moss B. Oligonucleotide sequence signaling transcriptional termination of vaccinia virus early genes. Proc. Natl. Acad. Sci. USA. 1987;84:6417–6421. doi: 10.1073/pnas.84.18.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]