


Abstract
As large-scale re-sequencing of genomes reveals many protein mutations, especially in human cancer tissues, prediction of their likely functional impact becomes important practical goal. Here, we introduce a new functional impact score (FIS) for amino acid residue changes using evolutionary conservation patterns. The information in these patterns is derived from aligned families and sub-families of sequence homologs within and between species using combinatorial entropy formalism. The score performs well on a large set of human protein mutations in separating disease-associated variants (∼19 200), assumed to be strongly functional, from common polymorphisms (∼35 600), assumed to be weakly functional (area under the receiver operating characteristic curve of ∼0.86). In cancer, using recurrence, multiplicity and annotation for ∼10 000 mutations in the COSMIC database, the method does well in assigning higher scores to more likely functional mutations (‘drivers’). To guide experimental prioritization, we report a list of about 1000 top human cancer genes frequently mutated in one or more cancer types ranked by likely functional impact; and, an additional 1000 candidate cancer genes with rare but likely functional mutations. In addition, we estimate that at least 5% of cancer-relevant mutations involve switch of function, rather than simply loss or gain of function.
INTRODUCTION
The importance of amino acid variation and mutations as genetic factors of human diseases has been known for many years. Mutations can affect protein folding and stability (1–6), protein function (7,8) and protein–protein interactions (9–12), as well as protein expression and subcellular localization (13,14). Mutations in proteins have a major role in the onset and development of cancer (15,16). The special role of mutations is determined by the diversity of their impact on molecular function. The mutations observed in cancer cells comprise both apparently random (‘sporadic’) mutations followed by somatic selection, and pre-existing mutations in the germline (17,18). Mutations can contribute to cancer by activating protein function, as in oncogenes (19), or inactivating function, as in tumor suppressors (20). Realizing the central role of mutations in cancer and exploiting recent advances in sequencing technology, the scientific community began systematic massive screening of cancer samples for mutations (21–28). Multiple re-sequencing projects have yielded thousands of cancer-associated protein mutations per year and many thousands more will likely be discovered in the near future. A common, probably simplistic, model view defines two classes of mutations, ‘driver’ mutations, i.e. mutations that give a cancer cell a particular selective advantage, and functionally irrelevant ‘passenger’ mutations. Discovering functionally important mutations, including clear ‘drivers’ is one goal of genome re-sequencing efforts. To understand the functional contribution of molecular alterations to oncogenesis, response to therapy and evolution of resistance to therapy it is important to have tools that predict the functional implications of mutations as early in the discovery process as possible.
Several methods for assessing the effects of mutation on protein function have been developed over the years (29–32). To assess a mutational effect, such methods typically use the physico-chemical properties of amino acids, as well as information about the role of amino acid side chains in protein structure. These methods can be conventionally classified as ‘machine learning’ or ‘direct’. The machine learning methods (33–36) combine all essential properties of both the original and substituted residues (e.g. size, polarity), structural information (e.g. surface accessibility, hydrogen bonding) and evolutionary conservation, and then are trained to distinguish between known functionally deleterious variants (positive set) and presumably neutral variants (negative control set). The direct methods (30,37–43) assess a mutation effect by a phenomenological score computed based on a particular theoretical model. Most of these computational approaches are validated on variants with pronounced phenotypic effects, e.g. functionally deleterious and disease-related variants. Such variants usually involve loss of function of a mutated gene.
However, causative mutations in cancer are not limited to those causing loss of function. There are three particularly important types of mutations that contribute to cancer progression: (i) ‘gain of function’ or activating mutations that convert normal genes into oncogenes (e.g. activating mutations in EGFR, KRAS, BRAF); (ii) ‘loss of function’ mutations that inactivate tumor suppressors (e.g. mutations in TP53, RB1, PTEN); and (iii) ‘drug resistance’ mutations (e.g. mutations in PI3K, EGFR, BCR-ABL) that overcome the (usually) inhibitory effect of a drug on the targeted protein. Here, we also consider a fourth type, ‘switch of function’, intermediate between (i) and (ii), such as the recently much studied mutations in IDH1 in glioblastoma and AML (44).
In general it is not sufficient to assess the functional impact of mutations at the level of protein function alone. Mutations have an effect on cancer as a tissue in an organism in the complicated context of the typically numerous alteration in a given tumor (45) and the host background. It is therefore desirable to be able to complement information about mutations in proteins with information on gene expression and genetic alteration of related genes, e.g. in oncogenic pathways.
The direct prediction of a mutation’s impact on molecular function based on first principles is currently impossible for a number of reasons: e.g. lack of data (3D structures and complexes) and lack of accurate and efficient approaches for de novo modeling of protein structure and function on the molecular level. However, evolutionary analysis does provide a powerful tool, as natural selection of a particular sequence variant by definition reflects the aggregate effect of molecular changes on cell, tissue and organ physiology. We therefore base this method development on phenomenological analysis that extracts information from protein family alignments of large numbers of homologous sequences grouped into aligned sets (families and subfamilies) and exploits 3D structures of sequence homologs. We use this rich evolutionary information for the prediction of the functional impact of mutations in general and in cancer in particular (Figure 1).
Figure 1.

Schematic of the method and validation tests. The functional impact score (FIS) is derived from multiple sequence alignments of sequence homologs. The score is based on the evolutionary conservation of a mutated residue in a protein family and, separately, in each of its subfamilies. Larger scores indicate more likely functional impact of a mutation.
Our use of evolutionary information for this purpose is novel in that it includes a refined class of evolutionarily conserved residues—specificity residues—which are determined by clustering multiple sequence alignments of homologous sequences into subfamilies to analyze functional specificity on the background of conservation of overall function (46). The specificity residues are predominantly located on protein surfaces in known or predicted binding interfaces and often directly linked to protein functional interactions (46). In addition, based on our analysis of mutations that affect predicted specificity residues, we propose a new type of functional impact that results in a ‘switch of function’—a switch from one set to an alternative set of specific interactors of the mutated protein and, consequently, an altered biological function that is not necessarily simply strengthened (gain) or weakened (loss).
We calibrated and tested the scoring function by its ability to separate large sets of disease-associated variants from common polymorphisms presumed to be mostly function-neutral. We applied the approach further to assess the functional impact of amino acid changing (‘missense’) cancer mutations collected in the COSMIC database (28) and ranked mutated genes by their likely significance in cancer. In the context of large-scale cancer genome projects such as The Cancer Genome Atlas (TCGA) project or projects in the International Cancer Genome Consortium (ICGC) ranked lists of mutations reported in various tumor types can facilitate more efficient subsequent computational or experimental investigations or therapeutic development.
MATERIALS AND METHODS
A protein sequence is subject to mutations in natural evolution as well as in somatic development, especially in cancer tissues. The direct effect of a mutation on a protein can be an effect on protein function by a number of different mechanisms. These include (i) changes in protein stability, e.g. destabilization leading to higher degradation rates, and, in the steady state, altered protein concentration and (ii) change in the interaction of the protein with other biomolecules, such as other proteins or DNA or RNA or lipids, or change in the interaction with ligands, such as enzyme substrates. Changes in the molecular function of a protein can affect the phenotype cells, tissues and the organism. Mutations that decrease replicative fitness below a certain threshold are eliminated from a population (of organism or of cells). Conversely, mutations can be fixed in a population, if they significantly increase replication rates. Thus, variability of protein sequences is restricted affected by natural selection, whether germ line or somatic. These restrictions are apparent in residue conservation in certain positions of aligned proteins in a protein family. One can use the analysis of sequence conservation to derive numerical estimates of the functional impact of mutation.
We make numerical estimates of the functional impact of a mutation assuming that protein family sequences reflect continuity of functional constraints and can be treated as a statistical ensemble, i.e. can be represented by a statistical model that expresses the likelihood that any particular sequence belongs to the family. In other words, we assume that many mutations were tried in evolution in each sequence position sufficiently often such that the observed distributions of residues in aligned positions of homologous sequences reflect the functional constraints on these residues. Thus, evolutionarily unfavorable (for whatever reason) residues are not observed or observed less frequently than neutral or critically important residues, while critically important residues are conserved in diverse evolutionary settings (paralogs or orthologs). These assumptions provide the basis for converting the observed frequencies into a numerical estimate of the functional impact of a mutation. We also assume that the distribution of residues in any (aligned) sequence position of a protein family can be treated independently of other positions (Supplementary Data S6).
With these assumptions, we use the entropy of the residue distribution in an alignment column as a measure of residue conservation (47) and estimate the mutation impact using the difference of the entropy caused by the mutation.
The entropy of an alignment column i is defined as:
| (1) |
with  and, by definition,
and, by definition,  ; α is a residue type (α = 1,2, … ,21 indexing 20 residues types and gaps);
; α is a residue type (α = 1,2, … ,21 indexing 20 residues types and gaps); 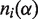 is the number of residues of type α in an alignment column i; N is the total number of residues in a column (
is the number of residues of type α in an alignment column i; N is the total number of residues in a column (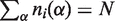 ), i.e. the number of proteins in the family alignment.
), i.e. the number of proteins in the family alignment.
The entropy difference caused by a mutation of a residue of type α to a residue of type β then becomes:
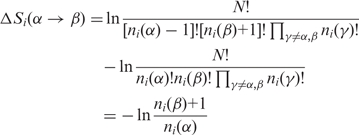 |
(2) |
We interpret this entropy difference as a measure of the impact of a mutation (conservation score).
| (3) |
The value of the entropy difference in Equation (3) is large when 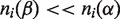 i.e. when the mutated residue of type α in position i is conserved across many sequences in the protein family and residues of type β rarely or never occurs in this position. We therefore call the mutation impact term of Equation (3) ‘the conservation score’ (superscript ‘c’) to underline that it takes into account conservation across the entire family.
i.e. when the mutated residue of type α in position i is conserved across many sequences in the protein family and residues of type β rarely or never occurs in this position. We therefore call the mutation impact term of Equation (3) ‘the conservation score’ (superscript ‘c’) to underline that it takes into account conservation across the entire family.
The conservation score of Equation (3) depends both on the types of the original and the mutated residues, and on the position of the altered residue in the protein family alignment. To what extent does this entropy term reflect physical effects, such as the change in residue–residue interactions in a protein? Assuming that physical constraints dictate the nature of residues at particular positions and that functional constraints require the approximate satisfaction of these physical constraints, we argue that evaluation of the entropy change also takes into account the change in the physico-chemical nature of the amino acid as a result of the mutation α to β, albeit indirectly.
The actual numerical value of the impact term of Equation (3) is determined by the particular frequencies of residues in an alignment column. Equation (3) is also defined for cases in which ni(β) is equal to 0, i.e. the mutated residue has never been observed in this column. However, columns for which no family sequence alignment is available are outside the scope of this formulation and they are not scored in the current implementation of the method.
The impact term of Equation (3) is the same for all residues of the same type aligned in the same sequence position (Supplementary Data S6). More precisely, the mutation impact is the same for all sequences of a protein family, for which a residue of type α in a position i is mutated to a residue of type β.
To refine the assessment of conservation patterns, we proceed to consider patterns of a subtler type, in which the evolutionary constraint on a residue type in a particular position is not constant in the entire family, but only appears to operate in a protein subfamily, e.g. because of different interaction partners or substrates on the background of similar, conserved biochemical or cellular function.
Among available approaches to quantify subfamily conservation patterns (48–54), we use our own combinatorial entropy approach, which simultaneously determines protein subfamilies, by clustering, and residues, called specificity residues, which characteristically differ between these subfamilies (46). The clustering algorithm groups the sequences of a protein family alignment into distinct subfamilies, so as to minimize the sequence diversity within subfamilies and to maximize the overall difference between subfamilies at a select number of ‘specificity’ positions. Evolutionary constraints can then be inferred from the patterns of residue conservation in the protein subfamilies (46). The objective function minimized in the process of determining optimally distinct subfamilies quantifies the extent of order in the M subfamilies compared to an even residue distribution over subfamilies (46):
| (4) |
where L `is a number of columns in a protein family alignment, M is a number of subfamilies and
| (5) |
is the difference of the entropy of the residue distribution in column i of subfamily m and the entropy of the reference (uniform) distribution of residues in column i (46). Note that m is and index, not an exponent. Here,  and
and  are, respectively, the actual (observed) and the expected (uniform distribution) number of residues of type α in position i of subfamily m; Nm is the number of sequences in subfamily m;
are, respectively, the actual (observed) and the expected (uniform distribution) number of residues of type α in position i of subfamily m; Nm is the number of sequences in subfamily m;  is the number of residues of type α in a column i.
is the number of residues of type α in a column i.
The larger the absolute value of the (negative) entropy difference 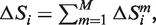 the bigger is the difference between the observed and the uniform distribution of residues in a column i. Larger absolute values of
the bigger is the difference between the observed and the uniform distribution of residues in a column i. Larger absolute values of 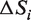 correspond to evolutionarily selected specificity residues, i.e. residue distributions constrained at the level of one or more subfamilies.
correspond to evolutionarily selected specificity residues, i.e. residue distributions constrained at the level of one or more subfamilies.
To quantify the entropy difference resulting from a mutation that affects conserved residue patterns in protein subfamilies, we (i) determine distinct sequence subfamilies from protein family alignments using Equation (4) (46) and (ii) compute a specificity conservation score in analogy to the family conservation score of Equation (3). We define the specificity score (superscript s) as:
| (6) |
where the index p refers to the particular subfamily to which the mutated sequence is assigned as the result of clustering and 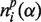 and
and 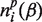 are, respectively, the numbers of residues of types α and β in sequence position i of subfamily p.
are, respectively, the numbers of residues of types α and β in sequence position i of subfamily p.
The conservation [Equation (3)] and the specificity [Equation (6)] scores are complementary measures of evolutionary conservation; therefore, a combination of these scores should provide more information about the potential functional impact of a mutation. In general, a good mathematical model for combining two scores with effective relative weights would be derived from a comprehensive statistical model, e.g. derived from an optimization procedure (as in machine learning) involving cross-validation tests of predictive power. Here, for simplicity, we tested two simple forms of combining the two scores: (i) the maximal value of the conservation and the specificity scores; and (ii) the average of both scores (details in Supplementary Table S1). The simple averaging (or a sum) of the two scores gave the higher prediction accuracy in the validation tests.
The combined score for the functional impact of changing an amino acid residue of type α to one of type β in sequence position i, as assessed in the context of evolutionary patterns in a multiple sequence family alignment, as used in this study, is therefore defined as follows:
| (7) |
In the current implementation, multiple sequence alignments were derived using the BLAST program (60) retrieving up to 700 homologous sequences at an E-value threshold of 0.05 from the Uniprot sequence database (61); the resulting hits were then aligned using the MUSCLE program (62). In place of  we also use the notation FIS (functional impact score based on evolutionary information) or simply the word ‘score’ in tables and computer output. (Details of derivation of the score are in Supplementary Data S6).
we also use the notation FIS (functional impact score based on evolutionary information) or simply the word ‘score’ in tables and computer output. (Details of derivation of the score are in Supplementary Data S6).
RESULTS
Validation test
To test the ability of the FIS score to predict the functional impact of a mutation, we applied the computational protocol to known ‘disease-associated’ and ‘common polymorphism’ variants and mutations as annotated in UniProt (HUMSAVAR, release 2010_08). Comparing the FIS scores for disease-associated and common polymorphic variants, we tested the hypothesis that disease-associated variants and mutations typically have a negative effect on protein function and are therefore more likely to be observed in positions with low rates of mutation fixation, i.e. in conserved positions, while functionally neutral or weakly deleterious polymorphic variants are more likely to be observed in evolutionarily promiscuous positions. We therefore require that the evolutionary conservation score distinguishes between disease-associated and polymorphic variants and can be used as a measure of functional impact of mutations. To explore how to best satisfy this requirement, we tested 1000 discrete thresholds in FIS and counted the number of disease-associated and polymorphic variants on either side of the threshold. The recognition accuracy, defined as the percentage of correctly assigned variants is 79% when false positives and false negatives are balanced (equal fractions) (Figure 2). The summary results of the validation tests and a calibration curve of the functional score are in Figures 2 and 3 and in the Supplementary Data (Supplementary Table S1).
Figure 2.

Separation of disease-associated and polymorphic variants by functional impact score. (A) Normalized smoothed distributions of the values of the functional score as computed for 19 179 known ‘disease-associated’ and 35 608 ‘common polymorphism’ variants and mutations annotated in UniProt (HUMSAVAR, release 2010_08; http://www.uniprot.org/docs/humsavar). (B) The cumulative distributions of the score values computed for disease-associated and polymorphic variants, same data as in (A). An equally balanced separation (79%) between the two variant classes is achieved at a score threshold of FIS∼1.9. At this threshold, ∼79% of all disease associated variants are scored higher than this threshold and ∼79% of all polymorphic variants are scored lower. The maximal separation (∼80.3%) between the two classes is achieved at the threshold value of 2.26; at this threshold, ∼70% of disease-associated variants are scored higher and 86% of polymorphic variants are scored lower.
Figure 3.

ROC analysis of classification between disease-associated and polymorphic variants. The observed score range (−6, 6) was divided into 1000 discrete thresholds, and for each of the thresholds, percentages of disease-associated and polymorphic variants above and below the score threshold were determined. The percentage of disease associated variants above the score threshold is defined as ‘true positives’, while the percentage of polymorphic variants above the score threshold is defined as ‘false positives’. The ROC curves are built for two test sets: in the first set, all available ∼55.7 K variants (∼19.2 K disease-associated and ∼36.5 K polymorphic) were used; the scores of the variants that fall on regions with no sequence homology were taken equal to zero; in the second set, the scores for a reduced set of ∼27.4 K variants (∼13.7 K disease-associated and ∼13.6 K polymorphic) were computed using alignments of 75 or more sequences.
Robustness analysis
A significant fraction of polymorphic variants falls in regions with low coverage of sequence homologs (Figure 2). Out of the total ∼55 K variants, ∼10.5 K variants fall into regions of low homology coverage (MSA has <10 sequences). Among these variants, ∼90% were polymorphic variants and only ∼10% were disease-associated variants. By definition, variants with low homology coverage get low score values. How does the uneven distribution of variants with low homology coverage affect the overall accuracy of separation between disease-associated and polymorphic variants? How does the accuracy of separation between disease-associated and polymorphic variants depend on the size of a multiple sequence alignment? To answer these questions, we compared the score distributions for disease-associated and polymorphic variants that have homology coverage from 1 to up to 600 or more sequences in a family alignment. We found that the enrichment of low-homology polymorphic variants disappears, when the minimal alignment size exceeds 75 sequences per family. With coverage of 75 or more sequences, ∼14 K disease-associated and 14 K polymorphic variants (∼1/2 of all tested mutations) are separated with an accuracy of better than 76% and an AUC in ROC analysis 0.83 (Figure 3). We repeated this test taking larger sizes of the minimal alignment. The accuracy of separation between disease-associated and polymorphic variants remained the same, when the minimal alignment size varied from 30 to up to 350 sequences. Thus, the observed enrichment of polymorphic variants in regions of low homology coverage in practice does not affect the discrimination of disease-associated and polymorphic variants. Additional details of the recognition tests at different alignment sizes are given in at Supplementary Table S1.
Validation of the FI score on experimentally tested TP53 mutations
Cases in which the predicted functional impact of mutations can be compared to direct measurements of functional activity are of special interest for validation of the method. Therefore we tested the FI score on data obtained from experimental studies of the functional impact of TP53 mutations as collected in the IARC TP53 database (55). TP53 mutants in cancer can result in both ‘loss of function’ and, in some cases, ‘gain of function’ (56). Although the biological effects of TP53 mutations in cancer are affected by a various post-transcriptional factors (56), direct measurements of transcriptional activity of mutant TP53 are very useful for assessment of the impact of TP53 mutations in cancer.
For each of the 2314 mutations, the ‘TP53MUTfunction2R15’ table of IARC TP53 database gives eight promoter-specific transcriptional activities measured in yeast functional assays and expressed as percent of wild-type activity. Although these eight normalized activities are correlated across all studied mutants, the individual activities measured for a particular TP53 mutant can vary significantly. In particular, the average value of the standard deviation of eight activities is ∼25%. To reduce the mutation impact divergences and experimental error, we computed average values of transcriptional activities and compared the FIS distributions for mutations, of which the average activities fell into eight distinct bins: [0–20], [20–40], [40–60], [60–80], [80–100], [100–120], [120–140], [140–250] (Figure 4). The activity of the normal TP53 is equal to 100.
Figure 4.

FIS distributions of mutations in TP53 binned into eight classes based on mutational impact. The normalized transcriptional activities of 2314 TP53 mutants were averaged and, depending on the average activity value, the mutations were binned into eight classes; the ranges of the average transcriptional activity are given below the bin marks. The FIS distributions are presented by the box plots; thick black lines show the medians of the distributions; each of the boxes is drawn between the lower and upper quartiles of the distributions; the dotted lines extend to the minimum and maximum values of the distributions. The mutations with larger functional impact, i.e. higher or lower than normal transcriptional activity (‘loss of function’ or ‘gain of function’) tend to have the higher values of the FIS score.
Obviously, mutations in bins [0–20] and [80–100] differ significantly by their functional impact. Mutations in bin [0–20] strikingly reduce transcriptional activity of TP53, while mutations in bin [80–100] are close to normal. The difference between two mutation classes is clearly depicted by the corresponding FIS distributions. The mutations of bin [0–20] are scoring significantly higher than mutations of bin [80–100]. The area under the receiver–operating–characteristic curve for two–class distinction between mutations of bin [0–20] and mutations of bin [80–100] is close to 0.93. The FIS distributions of bins [20–40], [40–60] and [60–80] are shifted from the higher to the lower values, which is in agreement with increase of transcriptional activity of TP53. The FIS distributions in bins [100–120], [120–140], [140–250] are shifted from lower values to higher values, also in agreement with increase of the transcriptional activity of TP53. Thus, the functional impact score is correlated with experimentally measured functional impact of mutations: the score is higher for mutations that result in ‘loss of function’ and in ‘gain of function’ of TP53. More details on the FIS distributions of TP53 mutations are given in Supplementary Data S4.
Functional mutations in the COSMIC database
There are currently ∼10.7 K non-synonymous point mutations in various tumors listed in the Catalog of Somatic Mutations in Cancer (COSMIC, v49). Many of these mutations have been studied experimentally and their functional impact and role in cancer are fairly well characterized. However, for the majority of mutations, their functional impact and role in cancer remains unknown.
Cancer mutations can be ranked by the number of occurrences of particular mutations; similarly, the genes implicated in cancer can be ranked by the total number of mutations detected for a particular gene. Obviously, particular numbers of mutations depend on sampling. However, in general, mutations that promote cancer will be selected more frequently than neutral mutations, and therefore, recurrent mutations and recurrently mutated genes are likely to play a key role in cancer. Thus, mutations of frequently mutated genes are likely to be functional.
In this study, we ranked mutations and mutated genes by their potential role in cancer by combining several factors: the predicted impact of a mutation on protein function, the occurrence of an individual mutation in different tumors, the total numbers of mutations detected for a particular gene and the gene’s role in cancer (tumor suppressor or oncogene) provided by a Cancer Gene resource at MSKCC (57).
To substantiate ranking of mutations and genes, we conducted three computational tests. In the first test, we tested the hypothesis that recurrently observed cancer mutations are significantly enriched by mutations of predicted high functional impact and therefore can be differentiated from single mutations, many of which are passenger mutations with low functional impact. In the second test, we tested a similar hypothesis that is mutations of frequently mutated genes are enriched by predicted functional mutations as compared to mutations of solitary mutated genes. In the third test, the scores of mutations in tumor suppressors (TS) or oncogenes (OG) were compared to the scores of mutations in the genes non-annotated as TS or OG. We tested the hypothesis that mutations in primary cancer genes (TS and OG) are enriched by high scoring functional mutations as compared to the mutations of non-cancer genes and therefore can be differentiated from all other mutations.
The ‘case and control’ sets of mutations used in these tests are not completely independent because many of recurrent mutations affect multiply mutated genes with key roles in cancer (TS or OG). However, conducted together, these tests give a more complete presentation of the distribution of functional mutations in cancer than each of the tests conducted individually.
The results of the tests are in Figures 5 and 6.
Figure 5.

Cumulative score distributions computed for recurrent cancer mutations in the COSMIC database (release 49, September, 2010), the scores were computed for 10 005 unique non-synonymous point mutations affecting 3630 genes. Recurrent cancer mutations observed two or more times (1828) and highly recurrent mutations observed five or more times (712) are scoring significantly higher compared to mutations observed only once (8177); the ROC analysis (not shown) of separation of recurrent mutations from one-time-observed mutations gives AUC = 0.75; the accuracy of separation is ∼69%, when a percentage of false positives is equal to a percentage of false negatives.
Figure 6.

Cumulative score distributions computed for mutations of multiply mutated genes in the COSMIC database; mutations in COSMIC are distributed non-uniformly across genes: one mutation per gene is detected in 1349 genes; two or more mutations are detected in 620 genes, three or more—in 265 genes, five or more in 96 genes, 10 or more—in 51 genes, 19 or more—in 37 genes. Multiply mutated genes (mutated two or more times) are enriched in high score mutations compared to single mutated genes and polymorphisms.
The FIS score distributions of Figure 5 show that cancer mutations collected in COSMIC are more significantly enriched in high score mutations than are polymorphic variants. Interestingly, recurrent mutations (observed in two or more samples) have a score distribution very close to the score distribution of disease-associated variants, while highly recurrent mutations (observed in five or more samples) are even more significantly enriched in high-score mutations than disease-associated variants (Figure 5). We also found that mutations of singly mutated genes in COSMIC are two times more enriched in high scoring mutations than are polymorphic mutations.
These results confirm the hypothesis that recurrent mutations are likely to be functional mutations and can be differentiated from single mutations by the evolutionary derived functional impact score.
The score distributions of Figures 6 and 7 also confirm that mutations of multiply mutated gene and mutations in annotated tumor suppressors and oncogenes are enriched in functional mutations: multiply mutated genes (mutated two or more times) are more enriched in high score mutations than singly mutated genes and polymorphisms.
Figure 7.

Cumulative score distributions computed for mutations in genes annotated as tumor suppressors and oncogenes in the COSMIC database; 4413 mutations in tumor suppressors and oncogenes are enriched in high-scoring mutations compared to 5592 mutations in genes non-annotated as TS and OG. The ROC analysis (not shown) of separation of recurrent mutations from one-time-observed mutations gives AUC = 0.6745; accuracy of separation is 64%, when the percentage of false positives is equal to the percentage of false negatives.
We found that the more mutations are observed in a gene, the bigger the fraction of high scoring mutations in this gene (Figure 6). However, the portion of high-scoring mutations in multiply mutated genes is smaller than in disease-associated variants or in recurrent individual mutations. We found similar results for mutations detected in key cancer genes—tumor suppressors and oncogenes (Figure 7). Mutations in TSs and OGs are scoring significantly higher than mutations in genes non-annotated as TSs or OGs. However a fraction of high-scoring mutations in TSs and OGs is less, than in a reference set of disease-associated mutations. Taking into account that the score generally correctly distinguish functional and non-functional mutations (Figures 2–5), this difference emphasizes the fact that not all mutations in multiply mutated genes or in known cancer genes are automatically functional and, hence, not all of them play role in cancer. Thus, a functional analysis of mutations is necessary to narrow down a list of potential driver mutations.
Ranked list of cancer mutations and cancer genes
Ranking mutations by a functional impact score makes possible the determination of mutation sets that are enriched by either functional or non-functional mutations. Obviously, there is no strict value of the score that can definitely separate functional and non-functional mutations. However there is a score threshold that separates sets of likely functional and likely non-functional mutations. Using this threshold, one can assess a number of functional mutations in a given set of mutations.
Using available sequence data, the automated procedure could assess a functional impact of ∼10 K unique mutations of the total ∼10.7 K unique mutations of COSMIC database (release 49). Based on the computed scores and the optimal separation threshold (Figure 4), a portion of mutations of high and medium impact can be estimated as ∼51%. A summary of functional analysis of missense mutations from COSMIC database is given in Table 1 and Figure 8. Note significant enrichment of predicted functional mutations in recurrent mutations, cancer genes (TS or OG) and in genes with multiple mutations.
Table 1.
Prediction of the functional impact of mutations observed in cancer (COSMIC databasea)
| Mutations/Genes | All scored (taken as 100%) n | Scored (FIS ≤ 0.8) neutral impactb, n (%) | Scored (0.8 < FIS ≤ 1.9) low impact, n (%) | Scored (1.9 < FIS ≤ 3.5) medium impact, n (%) | Scored (FIS > 3.5) high impact, n (%) |
|---|---|---|---|---|---|
| Mutations total | 10 005 | 2049 (20) | 2814 (28) | 3748 (37.5) | 1349 (13.5) |
| Mutations observed ≥2 times | 1828 | 89 (5) | 254 (14) | 862 (47.2) | 623 (34.1) |
| Mutations observed 1 time | 8177 | 1960 (24) | 2560 (31) | 2886 (35.3) | 771 (9.4) |
| Mutations in one-time-mutated genes not annotated as TSc or OGc | 2324 | 699 (30) | 777 (33) | 693 (29.8) | 155 (6.7) |
| Mutations in genes mutated ≥5 times | 5174 | 616 (12) | 1198 (23) | 2314 (44.7) | 1046 (20.2) |
| Mutations in TS or OG | 4413 | 477 (11) | 996 (23) | 2000 (45.3) | 940 (21.3) |
| Mutations in genes not annotated as TS or OG | 5592 | 1572 (28) | 1818 (33) | 1748 (31.3) | 454 (8.1) |
| Genes totald | 3629 | 841 (23) | 1115 (31) | 1268 (34.9) | 405 (11) |
| Genes annotated as TS or OG | 338 | 50 (15) | 124 (37) | 92 (27.2) | 72 (21) |
| Genes mutated ≥5 times | 188 | 2 (1) | 9 (5) | 96 (51.1) | 81 (43) |
aOf the total 10716 missense mutations in COSMIC 45, 10 005 mutations were mapped on sequences of UniProt, and determined as unique non-synonymous.
bApproximately 50% of polymorphic variants and ∼7% of disease-associated variants got FIS score <0.8; ∼27% of polymorphic variants and ∼14% of disease-associated variants got FIS score between 0.8 and 1.9; ∼17% of polymorphic variants and ∼50% of disease-associated variants got FIS score between 1.9 and 3.5; ∼21% of polymorphic variants and ∼79% of disease-associated variants got FIS score >1.9; ∼3% of polymorphic variants and ∼30% of disease-associated variants got FIS score >3.5.
cTS and OG stand, respectively, for tumor suppressor and oncogene.
dA gene is scored according to the highest FIS bin of any of its mutations.
Figure 8.

Ranking mutated genes by significance for cancer. The cancer gene ranking score (Rs), derived from information reported in the COSMIC database, is defined as Rs = log2(Nm*Nc), where Nm is a number of unique cancer-associated mutations reported in the gene, and Nc is a number of different cancer types with mutations in this gene. All analyzed 3629 genes were divided into four categories depending on presence or absence of predicted functional mutations and known association to cancer (gene is considered as cancer associated, if it is annotated as TS or OG, or it interacts with one or more of TS or OG). Cancer associated genes are enriched with predicted functional mutations (P < 10−20 in two-tail Fisher test) compared to genes with unknown cancer association. Using a reasonable cutoff, one nominates a list of 957 genes with significance for cancer (arrow). A gene is above the cut either because it is observed to be multiply mutated (Rs > 1, three or more mutations) or, for Rs = 1 (two mutations), if at least one of the mutations in the gene is predicted as functional. Detailed statistical information on mutated genes is in Supplementary Table SM2. The higher proportion of genes with at least one predicted functional mutation (orange or brown) in frequently mutated genes (peak at left) is not surprising—in fact, a fair number of these mutations have been functionally validated in the literature. A particularly interesting set of genes (998, bottom left) are those that (so far) have been observed just once (Rs = 0) but contain a mutation predicted to be functional. Such genes may be rare, but functionally significant, contributors to oncogenesis and are good candidates for experimental follow-up.
In Supplementary Table SM1, we present a list of COSMIC mutations (10 005) for which the functional impact score was computed. For each of the mutations, the table provides its sequence and genomic coordinates, the functional impact characteristics of the mutation, the characteristics of the protein domain family, the statistics of cancer mutations in a gene and the basic oncogenic annotations, the URL links presenting mutation in context of MSA and homologous 3D structures of PDB.
We used our assessments of mutation impact to rank genes by their significance for cancer. To that end, we divided all genes into four categories taking into account the presence or absence of predicted functional mutations in a gene and gene’s known associations with cancer. Genes annotated as TS or OG and genes interacting with TS or OG genes were defined as associated with cancer; gene interactions were taken from the PIANA database (58); cancer annotations were taken from the Cancer Gene resource at MSKCC (57).
Genes were classified into the following categories: (i) genes with functional mutations and known cancer association; (ii) genes with functional mutations and no available associations with cancer; (iii) genes with no functional mutations and with known cancer association; and (iv) genes with no functional mutations and no available associations with cancer. It is reasonable to assume that the more unique mutations are detected in a gene, and the more cancer types are affected by these mutations, the more important this gene is for development of cancer. Therefore we used as a gene ranking score the product of the ‘number of unique mutations’ and the ‘number of different cancer types’ affected by these mutations. Note that truncating mutations, i.e. premature stop codons (so-called non-sense mutations) are not taken into account.
The ranked list of 3629 genes is given in Supplementary Table SM2. Distributions of the gene ranking scores are in Figure 8. Genes with multiple mutations and genes with cancer associations are at the top of the list. Based on this ranking, we nominated ∼957 genes as genes with very likely cancer implications (Figure 8). These genes are of primary interest for experimental cancer genomics projects. The specific oncogenic roles of many of the top-scoring mutated genes are known or being studied. However, there are many genes of ‘moderate significance’, i.e. mutated approximately two times in approximately two cancers, for which the specific oncogenic roles in cancer is not yet well determined. A particularly interesting set of genes (Figure 8, bottom left) are those that have been observed just once, as reported in the COSMIC database, but contain a mutation predicted to be functional. Such genes may be rare, but functionally significant, contributors to oncogenesis and are good candidates for experimental follow-up.
Switch-of-function: a new type of functional impact?
The effect of a functional mutation can be described as a change of the specificity (selectivity) of interactions between a mutated protein and its specific interactors—proteins, nucleic acids or small molecules. One can imagine a set of free energies of interactions between a given protein and all other proteins and ligands. As a result of a mutation, the native spectrum of the binding free energies will change. An extreme example of a strong functional impact of a mutation is a destabilization of a protein globule resulting in the complete loss of the specificity, i.e. a ‘loss of function’ (LOF). The opposite example of a functional impact is a ‘gain of function’ (GOF), which can result from a change in the specificity of particular protein-substrate interactions or a change in the specificity of interactions with regulatory proteins. Both LOF and GOF mutations assume changes of free energies of interaction with native binders.
However, a mutation impact can also result in a ‘switch of function’ (SOF), which is an acquisition of new specific interactors and, consequently, a new biological function. A mutation in a protein-binding site can result in new specific interactions. One mutation in a binding site of isocitrate dehydrogenase 1 (IDH1) that resulted in a switch of molecular function was recently discovered in glioma (44). Mutations of R132 to C, H, L or S alter the activity of IDH1, such that isocitrate is no longer converted to alpha-ketoglutarate, but, instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate, which elevates the risk of brain tumors (44). The affected position, R132 is highly conserved in the protein family alignment and all the above mutations get a high FIS score.
In cells, there are many families of homologous proteins (and protein domains); each protein in such a family has its own specific function and specific interactors. Mutations can switch the specific interaction between a protein family members resulting in a drastic impact on the phenotype. Mutations in evolutionarily selected specificity residues are likely candidates for SOF. Switch of the specific signaling of Rho GTPases caused by mutations was studied experimentally Heo and Meyer (59). All of the experimentally studied functional mutations reported in (59) have a high specificity component of the FIS score. In Figure 9, we show the multiple sequence alignment and the 3D position of one the key mutations that switch the Rac1-signaling phenotype (lamellipodia) to the Cdc42-signaling phenotype (filopodia) (59). This mutation affects one of the key predicted specificity positions of Rho family.
Figure 9.

Functional mutation in a predicted specificity position of RAC1 (Ras-related C3 botulinum toxin substrate 1). (A) The mutation affects a residue that is conserved as A (Ala) in subfamily #1 (top sequences, close homologues of RAC1) and as E (Glu) in subfamily #2 (bottom sequences, close homologues of CDC42); Uniprot name, species identifier, residues number range and subfamily number are in left columns. The sequence subfamilies and specificity scores (vertical bars at top) were computed from a non-redundant MSA (multiple sequence alignment) of 274 sequences using CEO clustering. The mutation A95E of RAC1 has a high specificity score in RAC1. (B) The position affected by the mutation is in the binding interface of RAC1 in contact with the T-lymphoma invasion and metastasis factor 1 (Tiam1); (PDB code 1foe).
The mutation-caused rewiring of protein interaction network is of the high interest in cancer, where missense mutations are one of the common factors of oncogenesis. Currently, it is impossible to determine such mutations by direct de novo modeling. However, one can narrow down a list of potential SOF mutations by determining mutations in binding sites that change the identity of an amino acid residue located in one of the functional evolutionarily selected (predicted specificity) positions, and, especially by determining mutations that change amino acid identities between already existing groups (classes) of residue specificity. We estimated a number of such mutations by identifying mutations that fall into binding sites and that have high specificity score and low conservation score. Among ∼10 K mutations of COSMIC, 3631 affect annotated functional regions and binding sites, and, among those, 554 mutations (∼5%) have a specificity score >2.5 (top 25%) and a conservation score less than the specificity score. This set of mutations is enriched in potential SOF mutations. A few examples of putative SOF mutation are given in Supplementary Data S5.
DISCUSSION AND CONCLUSION
Here, we introduced and tested a new computational approach for predicting the functional impact of protein mutation on protein function and, by implication, for providing a rough estimate of the probability that the mutation has a phenotypic consequence at the level of the organism. The principal strength of the approach is that it uses information based on the analysis of evolutionary conservation patterns in protein family multiple sequence alignments which are subject to selective forces at the level of the ability of the organism to survive and reproduce. Strong selection patterns across an entire protein family or within protein subfamilies are very likely the result of strong selection that disfavors amino acid residues not consistent with the conservation pattern, no matter what the precise mechanism of de-selection of unfavorable variants might be. In other words, evolutionary conservation patterns effectively integrate information from any effect of residue changes, without the need to dissect them into separate contributing factors, such as effects on protein stability or protein–protein interactions.
The computational protocol performs the analysis as follows: given a mutated protein name and a mutated residue position, it searches for sequence homologs, builds a multiple sequence alignment, clusters sequences into subfamilies and scores a mutation by global and sub-family specific conservation patterns. Mutations affecting either type of conserved residue are likely to be functional.
The functional impact score was tested on a large set of disease-associated and polymorphic variants, with the adjustment of only a single threshold parameter (optimal sensitivity ∼79% at a score threshold ∼2).
We applied the approach to evaluate the functional impact of amino-acid changing cancer mutations in the curated catalog of somatic mutations in cancer, the COSMIC database. Mutations in this database are distributed non-uniformly across genes. In spite of potential investigator bias, this non-uniform distribution plausibly reflects clonal selection of cancer mutations, i.e. functional mutations in key proteins give selective advantage to cancer cells and are thus observed more frequently than others in cancer genome re-sequencing. Therefore, the COSMIC list of cancer mutations is a useful positive test set for testing the predictive power of methods to assess the functional impact of protein mutations. We conducted three comparisons of the functional impact score in pairs of sets of mutations in which one would expect higher functional impact in the first set compared to the second set: (i) mutations recurrent in many samples versus mutations observed only in one sample; (ii) mutations in multiply mutated genes versus mutations in genes reported only once as mutated; and (iii) mutations of genes annotated as cancer genes versus mutations of genes without such annotation. The tests indeed showed that (i) recurrent mutations; (ii) mutations of multiply mutated genes; and (iii) known cancer genes on average have higher functional impact scores, confirming that the score reflects likely functional impact to a non-trivial extent.
Based on this analysis of COSMIC mutations, we are making three experimentally testable predictions: we predict a non-negligible fraction of non-functional mutations in well known cancer genes. If such mutations are in fact non-functional, then a simple detection of a non-synonymous mutation in an oncogene cannot be automatically interpreted as a cancer-causative event. One would need to assess the functional impact of a mutation experimentally to confidently take it into account in molecular diagnostics or choice of therapy. We also predict that a non-negligible fraction of mutations in one-time mutated genes are functional, in spite of their (currently observed) low recurrence. What is a role of these mutations? Should these mutations be ignored as ‘not significant’ as is commonly done as the result of applying simple statistical models of recurrence in a set of samples? And, we introduce a new type of functional impact of mutations in cancer—involving a ‘switch of specificity’—that may account for ∼5% of predicted functional mutations.
We conclude that the functional impact score based on evolutionary information (FIS) is useful for ranking mutations by likely functional impact, especially for understanding the role of mutated proteins in cancer, for nominating newly discovered mutations contributing to cancer and for prioritizing mutations for further analyses and experiments involving these alterations. Our computational protocol is fully automated and implemented as a publicly available server (http://mutationassessor.org) for use in cancer research.
SUPPLEMENTARY DATA
Supplementary Data is available at NAR Online.
FUNDING
Funding for open access charge: National Institutes of Health (grant R01 CA132744-02).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Alex Lash and Caitlin Byrne from MSKCC Bioinformatics Core for protein-to-genomic position mapping tables (cbio.mskcc.org/Public/products/ human_mapped/), to Joanne Edington for systems support; and to Robert Klein for commenting on the manuscript; to Niki Schultz and Barry Taylor for constructive discussions. All contributed to the design of the project; BR derived the entropy formulation; BR and YA developed the software and performed data analyses; all wrote the article.
REFERENCES
- 1.Ode H, Matsuyama S, Hata M, Neya S, Kakizawa J, Sugiura W, Hoshino T. Computational characterization of structural role of the non-active site mutation M36I of human immunodeficiency virus type 1 protease. J. Mol. Biol. 2007;370:598–607. doi: 10.1016/j.jmb.2007.04.081. [DOI] [PubMed] [Google Scholar]
- 2.Lorch M, Mason JM, Sessions RB, Clarke AR. Effects of mutations on the thermodynamics of a protein folding reaction: implications for the mechanism of formation of the intermediate and transition states. Biochemistry. 2000;39:3480–3485. doi: 10.1021/bi9923510. [DOI] [PubMed] [Google Scholar]
- 3.Lorch M, Mason JM, Clarke AR, Parker MJ. Effects of core mutations on the folding of a beta-sheet protein: implications for backbone organization in the I-state. Biochemistry. 1999;38:1377–1385. doi: 10.1021/bi9817820. [DOI] [PubMed] [Google Scholar]
- 4.Alfalah M, Keiser M, Leeb T, Zimmer KP, Naim HY. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology. 2009;136:883–892. doi: 10.1053/j.gastro.2008.11.038. [DOI] [PubMed] [Google Scholar]
- 5.Koukouritaki SB, Poch MT, Henderson MC, Siddens LK, Krueger SK, VanDyke JE, Williams DE, Pajewski NM, Wang T, Hines RN. Identification and functional analysis of common human flavin-containing monooxygenase 3 genetic variants. J. Pharmacol. Exp. Ther. 2007;320:266–273. doi: 10.1124/jpet.106.112268. [DOI] [PubMed] [Google Scholar]
- 6.De Cristofaro R, Carotti A, Akhavan S, Palla R, Peyvandi F, Altomare C, Mannucci PM. The natural mutation by deletion of Lys9 in the thrombin A-chain affects the pKa value of catalytic residues, the overall enzyme's stability and conformational transitions linked to Na+ binding. FEBS J. 2006;273:159–169. doi: 10.1111/j.1742-4658.2005.05052.x. [DOI] [PubMed] [Google Scholar]
- 7.Yamada Y, Banno Y, Yoshida H, Kikuchi R, Akao Y, Murate T, Nozawa Y. Catalytic inactivation of human phospholipase D2 by a naturally occurring Gly901Asp mutation. Arch. Med. Res. 2006;37:696–699. doi: 10.1016/j.arcmed.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Takamiya O, Seta M, Tanaka K, Ishida F. Human factor VII deficiency caused by S339C mutation located adjacent to the specificity pocket of the catalytic domain. Clin. Lab. Haematol. 2002;24:233–238. doi: 10.1046/j.1365-2257.2002.00449.x. [DOI] [PubMed] [Google Scholar]
- 9.Jones R, Ruas M, Gregory F, Moulin S, Delia D, Manoukian S, Rowe J, Brookes S, Peters G. A CDKN2A mutation in familial melanoma that abrogates binding of p16INK4a to CDK4 but not CDK6. Cancer Res. 2007;67:9134–9141. doi: 10.1158/0008-5472.CAN-07-1528. [DOI] [PubMed] [Google Scholar]
- 10.Ung MU, Lu B, McCammon JA. E230Q mutation of the catalytic subunit of cAMP-dependent protein kinase affects local structure and the binding of peptide inhibitor. Biopolymers. 2006;81:428–439. doi: 10.1002/bip.20434. [DOI] [PubMed] [Google Scholar]
- 11.Rignall TR, Baker JO, McCarter SL, Adney WS, Vinzant TB, Decker SR, Himmel ME. Effect of single active-site cleft mutation on product specificity in a thermostable bacterial cellulase. Appl. Biochem. Biotechnol. 2002;98–100:383–394. doi: 10.1385/abab:98-100:1-9:383. [DOI] [PubMed] [Google Scholar]
- 12.Hardt M, Laine RA. Mutation of active site residues in the chitin-binding domain ChBDChiA1 from chitinase A1 of Bacillus circulans alters substrate specificity: use of a green fluorescent protein binding assay. Arch. Biochem. Biophys. 2004;426:286–297. doi: 10.1016/j.abb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Tiede S, Cantz M, Spranger J, Braulke T. Missense mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTA) in a patient with mucolipidosis II induces changes in the size and cellular distribution of GNPTG. Hum. Mutat. 2006;27:830–831. doi: 10.1002/humu.9443. [DOI] [PubMed] [Google Scholar]
- 14.Krumbholz M, Koehler K, Huebner A. Cellular localization of 17 natural mutant variants of ALADIN protein in triple A syndrome - shedding light on an unexpected splice mutation. Biochem. Cell. Biol. 2006;84:243–249. doi: 10.1139/o05-198. [DOI] [PubMed] [Google Scholar]
- 15.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 17.Weir B, Zhao X, Meyerson M. Somatic alterations in the human cancer genome. Cancer Cell. 2004;6:433–438. doi: 10.1016/j.ccr.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croce CM. Oncogenes and cancer. N. Engl. J. Med. 2008;358:502–511. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- 20.Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–246. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- 21.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 23.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr. Protoc. Hum.Genet. 2008 doi: 10.1002/0471142905.hg1011s57. Chapter 10, Unit 10 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mooney S. Bioinformatics approaches and resources for single nucleotide polymorphism functional analysis. Brief. Bioinform. 2005;6:44–56. doi: 10.1093/bib/6.1.44. [DOI] [PubMed] [Google Scholar]
- 30.Ng PC, Henikoff S. Predicting the effects of amino Acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 31.Lee W, Yue P, Zhang Z. Analytical methods for inferring functional effects of single base pair substitutions in human cancers. Hum.Genet. 2009;126:481–498. doi: 10.1007/s00439-009-0677-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teng S, Michonova-Alexova E, Alexov E. Approaches and resources for prediction of the effects of non-synonymous single nucleotide polymorphism on protein function and interactions. Curr. Pharm. Biotechnol. 2008;9:123–133. doi: 10.2174/138920108783955164. [DOI] [PubMed] [Google Scholar]
- 33.Karchin R, Diekhans M, Kelly L, Thomas DJ, Pieper U, Eswar N, Haussler D, Sali A. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- 34.Kaminker JS, Zhang Y, Waugh A, Haverty PM, Peters B, Sebisanovic D, Stinson J, Forrest WF, Bazan JF, Seshagiri S, et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007;67:465–473. doi: 10.1158/0008-5472.CAN-06-1736. [DOI] [PubMed] [Google Scholar]
- 35.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc. Natl Acad. Sci. USA. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee W, Zhang Y, Mukhyala K, Lazarus RA, Zhang Z. Bi-directional SIFT predicts a subset of activating mutations. PLoS One. 2009;4:e8311. doi: 10.1371/journal.pone.0008311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue P, Moult J. Identification and analysis of deleterious human SNPs. J. Mol. Biol. 2006;356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 44.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reva BA, Antipin YA, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- 48.Oliveira L, Paiva AC, Vriend G. Correlated mutation analyses on very large sequence families. Chembiochem. 2002;3:1010–1017. doi: 10.1002/1439-7633(20021004)3:10<1010::AID-CBIC1010>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 49.Casari G, Sander C, Valencia A. A method to predict functional residues in proteins. Nat. Struct. Biol. 1995;2:171–178. doi: 10.1038/nsb0295-171. [DOI] [PubMed] [Google Scholar]
- 50.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 51.Mirny LA, Shakhnovich EI. Universally conserved positions in protein folds: reading evolutionary signals about stability, folding kinetics and function. J. Mol. Biol. 1999;291:177–196. doi: 10.1006/jmbi.1999.2911. [DOI] [PubMed] [Google Scholar]
- 52.Kalinina OV, Novichkov PS, Mironov AA, Gelfand MS, Rakhmaninova AB. SDPpred: a tool for prediction of amino acid residues that determine differences in functional specificity of homologous proteins. Nucleic Acids Res. 2004;32:W424–W428. doi: 10.1093/nar/gkh391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rausell A, Juan D, Pazos F, Valencia A. Protein interactions and ligand binding: from protein subfamilies to functional specificity. Proc. Natl Acad. Sci. USA. 107:1995–2000. doi: 10.1073/pnas.0908044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 55.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 56.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Higgins ME, Claremont M, Major JE, Sander C, Lash AE. CancerGenes: a gene selection resource for cancer genome projects. Nucleic Acids Res. 2007;35:D721–D726. doi: 10.1093/nar/gkl811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aragues R, Jaeggi D, Oliva B. PIANA: protein interactions and network analysis. Bioinformatics. 2006;22:1015–1017. doi: 10.1093/bioinformatics/btl072. [DOI] [PubMed] [Google Scholar]
- 59.Heo WD, Meyer T. Switch-of-function mutants based on morphology classification of Ras superfamily small GTPases. Cell. 2003;113:315–328. doi: 10.1016/s0092-8674(03)00315-5. [DOI] [PubMed] [Google Scholar]
- 60.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 61.The UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2010;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.