


Abstract
To understand the molecular mechanisms that underlie global transcriptional regulation, it is essential to first identify all the transcriptional regulatory elements in the human genome. The advent of next-generation sequencing has provided a powerful platform for genome-wide analysis of different species and specific cell types; when combined with traditional techniques to identify regions of open chromatin [DNaseI hypersensitivity (DHS)] or specific binding locations of transcription factors [chromatin immunoprecipitation (ChIP)], and expression data from microarrays, we become uniquely poised to uncover the mysteries of the genome and its regulation. To this end, we have performed global meta-analysis of the relationship among data from DNaseI-seq, ChIP-seq and expression arrays, and found that specific correlations exist among regulatory elements and gene expression across different cell types. These correlations revealed four distinct modes of chromatin domain structure reflecting different functions: repressive, active, primed and bivalent. Furthermore, CCCTC-binding factor (CTCF) binding sites were identified based on these integrative data. Our findings uncovered a complex regulatory process involving by DNaseI HS sites and histone modifications, and suggest that these dynamic elements may be responsible for maintaining chromatin structure and integrity of the human genome. Our integrative approach provides an example by which data from diverse technology platforms may be integrated to provide more meaningful insights into global transcriptional regulation.
INTRODUCTION
Consistent proper function of all biologic processes relies on the precise spatial and temporal expression of genes (1–3). Development, differentiation, proliferation, apoptosis, and even aging, are the culmination of cell type-specific and ubiquitous gene expression. Since transcription was first described researchers have sought to define the molecular mechanisms that regulate this phenomenon, driven by the belief that understanding the gene expression profiles of normal and disease states will facilitate discoveries of therapeutic targets to alleviate human and animal suffering. These works have defined several types of cis-acting transcriptional regulators, including promoters, enhancers, insulators and locus control regions (LCR) (1,4,5) and the trans-acting factors that bind to them. Nonetheless, the relative roles of these regulatory DNA elements have yet to be fully elucidated. The introduction of high-throughput sequencing (6) and its massive amounts of data spanning entire genomes of species has provided a platform from which we may begin to examine global patterns of gene expression and compare these patterns among different cell types to gain a clearer understanding of the molecular mechanisms underlying the dynamic and complex processes of life.
Next-generation sequencing (NGS) has become a popular approach to identifying gene regulatory elements and to performing accurate functional analysis (6). NGS of DNA–protein complexes isolated by chromatin immunoprecipitation, a procedure known as (ChIP-seq), has allowed for global localization of regulatory elements associated with a specific protein of interest (7–11). Unfortunately, this combined technique is only applicable to known (previously characterized) trans-acting factors and is limited by its requirement for a high quality ChIP-grade antibody to isolate the transcription factor (TF) to be analyzed (9). By coupling the NGS method with DNaseI hypersensitive (DNaseI HS) site mapping (long considered the gold-standard for comprehensively identifying the location of various classes of transcriptional regulatory elements), a particularly powerful high-resolution procedure, DNase-seq, was developed (12–18). Like the ChIP-seq procedure, though, DNase-seq suffers from some inherent limitations. DNase-seq provides only location data and is unable to directly characterize function or identify the particular TF(s) associated with the region.
The data obtained from each of these combined-NGS procedures may be analyzed in parallel (along with data obtained from gene expression arrays) to facilitate the identification of bona fide transcriptional regulatory elements. First, though, we must obtain a thorough understanding of the different types of cis-regulatory sequence elements and epigenetic modulatory mechanisms in order to accurately investigate their contributions to spatial and temporal gene expression.
The first genome-wide maps of histone methylation (7) and acetylation marks (19) were generated from human resting CD4+ T cells. Histone modifications associated with gene transcription were designated as active, while those associated with repressed transcription were designated as repressive. Intriguingly, some of the ‘active’ were identified in transcriptionally silent genes (7,20–23), suggesting that these modifications may act more as markers of genes primed for transcriptional activity. Not surprisingly, then, histone modification is not the sole mediator of expression level (24). By performing DNase-seq and DNase fragment, hybridization to microarray chip (DNase-chip), Boyle et al. (25) created a comprehensive genome-wide map of the open chromatin regions in CD4+ T cells. Their analysis of the resultant data sets did not identify a clear correlation between DNaseI HS and levels of gene expression. Shortly thereafter, Xi et al. (26) used DNase-chip to comparatively analyze six human cell types in order to identify functional cell type specific and ubiquitous DNaseI HS sites (DHSs). Their examination of 1% of the human genome revealed that cell type-specific DNaseI HS sites co-localized with cell type-specific gene expression. Recently, Stitzel et al. (27) conducted genome-wide analysis of DNaseI hypersensitive sites in human islets. Ling et al. (28) produced a set of detailed, high-quality, genome-wide DNaseI hypersensitivity maps in the mouse liver in vivo. These studies highlight the utility of DNase-seq for systematically uncovering cis-regulatory elements on a genome-wide scale.
In the study presented herein, we performed a genome-wide meta-analysis of DNaseI HS sites identified in 29 different cell types. We sought to determine the relationship between DNaseI HS, histone modifications and gene expression. We found that specific correlations exist between DNaseI HS, gene expression and the amounts of active and repressive histone modifications across different cell types. These correlations displayed four distinct modes (repressive, active, bivalent and primed), reflecting different functions of the chromatin domains. Furthermore, CCCTC binding factor (CTCF) binding sites were newly identified based on these integrative data. Our findings revealed a situation of complex regulation of gene expression mediated by DNaseI hypersensitive chromatin regions and their histone modifications.
MATERIALS AND METHODS
DNase-seq, ChIP-seq, gene expression and Gencode data
All data used in this study were freely available for download from the University of California, Santa Cruz (UCSC) NCBI36/hg18 Genome Browser (http://genome.ucsc.edu/encode/) (29,30). The Open Chromatin (25) track was used to obtain DNase-seq data from 29 cell lines and ChIP-seq data of CTCF, c-Myc and RNA Pol II. The Broad Histone (22) track provided the ChIP-seq data of histone modifications from nine cell lines. Regions of enriched signal in either DNaseI HS or ChIP experiments were identified using F-Seq (31). The gene annotations presented herein were taken from the Gencode data (32) in the Gencode Genes track (Version 3c, October 2009). The detailed information of these data was presented in Supplementary Table S1. Finally, the 17 751 UCSC known human genes with expression data were obtained from the Gene Expression Omnibus database (series GSE15805 and GSE17793; http://www.ncbi.nlm.nih.gov/projects/geo/). All chromosome Y data were omitted from this study.
Classification of DNaseI HS sites relative to cell types
Considering the lineage specificity observed with DNaseI HS sites (Supplementary Figure S1) (26), we classified DNaseI HS sites according to their occurrence rates in 29 cell lines. A given DNaseI HS site is classified as cell type specific, if it does not overlap (Here, overlap between two binding sites represents that these two regions have at least one common base pair.) with any DNaseI HS site within other 28 cell lines. A given DNaseI HS site is classified ubiquitous, if it overlaps with any DNaseI HS site within all 28 cell lines. The remaining DNaseI HS sites are classified as common, which are present in two to 28 cell lines. The results showed that about one-half of DNaseI HS sites were found in two cell lines (Supplementary Table S2). The cell lines that had the highest DNaseI HS overlap (74.96%) were the two lymphocyte lines, GM12891 and GM12892; the lowest overlap (25.99%) was found between AoSMC (aortic smooth muscle) and Medullo (Medulloblastoma). This agreed with the clustering analysis that we performed on DNaseI HS sites (Supplementary Figure S1).
Tag density profiles
The profiles of tag density at DNaseI HS sites or at transcription start sites (TSSs) were generated as described by Wang et al. (19). Briefly, the DNaseI HS region was examined in 200 bp windows that spanned the 5 kb immediately upstream of the DNaseI HS start site (dxStart) and 5 kb downstream from the end of the DNaseI HS site (dxEnd); each window was evaluated for content of uniquely mapped ChIP-seq data of histone modifications and TFs. The DNaseI HS site itself was divided equally among 10 windows for detailed analysis. All window tag counts were normalized to the total number of bases present in the window and to the total read number of the given library.
To plot the profiles of those DNaseI HSs associated with TSSs, the 17 751 UCSC known genes with expression information were categorized among broad groups according to their reported expression levels: high, median or mainly silent. One thousand genes were selected per group and corresponding DNase-seq data was analyzed after each was aligned by their TSS.
Gene density and TFBSs associated with DNaseI HS sites
The entire genome was scanned by 2 Mb windows. Within each window, the number of genes, DNaseI HS sites, CTCF binding sites, Pol2 and c-Myc binding sites were quantitated. Linear regression was used to determine the correlation between gene density and binding site density.
Enrichment/depletion of histone modifications at DNaseI HS sites
To quantitatively measure histone modification enrichment or depletion at DNaseI HS sites, we evaluated the histone signal based on the profiles of histone modification tag density by using the formula: 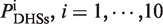 , where
, where 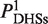 and
and 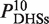 wee tag density at dxStart and dxEnd, respectively. Enrichment or depletion of histone modifications at DNaseI HS sites were defined as
wee tag density at dxStart and dxEnd, respectively. Enrichment or depletion of histone modifications at DNaseI HS sites were defined as  , where ζ > 1.0 indicated enrichment and 0 ≤ ζ < 1.0 indicated depletion.
, where ζ > 1.0 indicated enrichment and 0 ≤ ζ < 1.0 indicated depletion.
DNaseI HS sites associated with histone modifications and expression levels
Cell type specific, common and ubiquitous DNaseI HS sites were divided among 100 groups according to DNaseI hypersensitivity. The average tag density of DNaseI HS and of histone modifications was calculated for each group.
The 17 751 UCSC known genes with expression information were divided into 100 groups, based on expression. The average tag densities of each modification and the DNaseI HS within the gene body were calculated for each group, respectively.
Motif identification
To carry out motif identification, we examined data of DNaseI HS sites that encompassed defined ChIP-enriched regions in each cell line. CUDA-MEME (Version 3.0) (33), which was programmed using hybrid CUDA (Compute Unified Device Architecture) based on MEME (Version 4.4.0) (34) was used to discover consensus motifs with default parameters.
To determine the number of peaks that could be explained by statistically significant motifs, the MEME tool MAST (35) was used to estimate the maximal difference between the total number of peaks containing a motif and the number that could be explained by chance within a range of stringencies (E-values). See Methods in Supplementary Data for generation of MAST curves.
RESULTS
Global properties of DNaseI HS sites
Classification of DNaseI HS sites
We classified DNaseI HS sites according to their occurrence rates: cell type specific, found in one out of 29 cell lines; common, found in 2 to 28 cell lines or ubiquitous, found in all 29 cell lines (Supplementary Table S3).
In the erythroleukemia cell type K562, 9% of the DNaseI HS sites were found to be cell type specific, while the remaining 77% were common and 14% were ubiquitous (Figure 1A). In the strongest DNaseI HS sites (top 20%), 58% were found to be ubiquitous and only 1% of them to be cell type-specific (Figure 1A). In contrast, in the weakest DNaseI HS sites (bottom 20%), we found that 19% were cell type-specific and almost none were ubiquitous (Figure 1A). Similar observations were obtained from other cell lines (Supplementary Figure S2). Taken together, these findings indicated that ubiquitous DNaseI HS sites are extremely hypersensitive to DNaseI digestion, while cell type-specific DNaseI HS sites are less susceptible to digestion, although still significantly more susceptible than the genome average. This is expected since ubiquitous DNaseI HS sites are accessible in all 29 cell types analyzed, and cell type-specific DNaseI HS sites are only accessible in one cell type.
Figure 1.

Identification and characterization of DNaseI HS sites. (A) Genome-wide distribution of DHSs relative to cell type. Total numbers of cell type specific, common and ubiquitous DHSs are indicated. The proportions of DHSs in the K562 cell line among the strongest scoring (top 20%) and weakest scoring DHSs (bottom 20%) are shown. (B) Cumulative percentage of the genome and cumulative numbers covered by DNaseI HS sites from increasing numbers of cell lines (x-axis). Cumulative percentage of the genome (solid lines) and numbers (dashed lines) covered by all (red), cell type specific (green) and ubiquitous (blue) DNaseI HS sites from any cell line. Each point represents an averaged value of all possible cell line combinations. (C) The genome-wide distribution of DNaseI HS sites in proximal promoters (defined as 1 kb upstream and downstream of TSS), exons, introns and intergenic regions. Total numbers of DHSs relative to gene annotations are shown for the representative K562 cell line. (D) The densities of DNaseI HS sites and transcription factor binding sites on each chromosome. The points plotted on the x- and y-axes represent the number of binding sites/2 Mbp and the number of DNaseI HS sites/2 Mb, respectively.
Genome-wide coverage of DNaseI HS sites represented
To determine whether the majority of DNaseI HS sites that exist in the human genome were represented in the data sets under examination, we computed the cumulative percentage of the genome covered by DNaseI HS sites and the cumulative numbers of DNaseI HS sites with respect to the number of cell lines tested [Methods in Supplementary Data; described in (26)]. Therefore, as additional cell lines were included in the analysis, the total number of DNaseI HS sites being investigated increased; ultimately, we examined approximately 900 000 DNaseI HS sites from 29 cell lines (Figure 1B). Accordingly, the total percentage of base pairs (bps) in the human genome covered by DNaseI HS sites was calculated at 8.28%. A total of 18 943 ubiquitous and 10 100 cell type-specific DNaseI HS sites were identified from the 29 cell types, which covered 0.67 and 0.07% of bp in the human genome, respectively. However, even after addition of the 29th cell line we were unable to reach a significant saturation level of total DNaseI HS sites, which would have been represented by equal levels of cell type specific, common and ubiquitous DNaseI HS sites. This finding was consistent with previously published results using only six cell types and examining 1% of the human genome (26), and supported the suggestion by those authors that substantially more cell lines/types must be included in future analysis to identify the majority of the DNaseI HS sites that exist in the entire human genome.
Genome-wide localization of DNaseI HS sites
To obtain an overall picture of the DNaseI HS site distribution pattern relative to genes, we divided the human genome into four regions: proximal promoter, 1 kb upstream and downstream of the TSS; exon; intron and intergenic regions. Groups were assigned based on the ‘GENCODE’ annotation published in the UCSC Genome Browser.
In the K562 cell line, only 14 and 9% of all DNaseI HS sites mapped to proximal promoters and exons, respectively, while 24% mapped to introns (Figure 1C). The majority (53%) mapped within intergenic regions. However, we discovered that over 20% of DNaseI HS sites located within intergenic region, on average, are associated with strong Pol II signals; this correlation indicated these DNaseI HS sites may in fact represent a promoter, exon or intron of an unannotated gene (Supplementary Data and Supplementary Figure S3). The genome-wide location results were well agreed with previously published results in human islets (27) and CD4+ T cells (25). Of note, the DNaseI HS sites found to be located within promoters and exons were over 2-fold larger in size and twice as hypersensitive than those located in the introns or intragenic regions (Supplementary Table S4). When we considered only the cell type-specific DNaseI HS sites, we found that ∼9% were located in proximal promoters or exons. In stark contrast, nearly one-half of the ubiquitous DNaseI HS sites were located in proximal promoters or exons (Supplementary Table S5). Similar distribution patterns were observed in other cell lines (Supplementary Tables S4 and S5). These findings, when considered along with the results from our analysis of overlapping CpG islands and sequence conservation within those sites (Supplementary Figures S4 and S5; Methods in Supplementary Data), indicate that ubiquitous DNaseI HS sites are generally associated with promoters of so-called ‘housekeeping’ genes.
Gene density and binding sites associated with DNaseI HS sites
To examine the correlation of DNaseI HS sites with gene density in the surrounding regions and presence of TFBSs, we segmented each chromosome into 2 Mb windows and counted the numbers of DNaseI HS sites and genes and binding sites within each. In general, the DNaseI HS sites were found to strongly correlate with genes in all of the cell lines examined (R2 = 0.9122 in K562; Figure 1D). Additionally, the DNaseI HS sites were highly correlated with Pol II in each cell line (R2 = 0.9553 in K562; Figure 1D). These results were consistent with observations by others that the open chromatin state is affiliated with gene dense regions in the genome (36). Interestingly, we found that DNaseI HS sites correlated with TFBSs, as evidenced by analysis of the CTCF and c-Myc binding sites (R2 = 0.9052 and R2 = 0.9383, respectively; Figure 1D). This finding confirmed the Ling et al. (28) study for a larger set of TFs that binding sites show up to 90% overlap with DNaseI HS sites. The property of DNaseI HS site distribution is consistent with its role at identification of TFs and suggests a widespread function of DNaseI HS sites in the genome.
Genome-wide correlation of DNaseI HS sites and histone modifications
Histone modifications near and in DNaseI HS sites
To characterize the histone modification patterns at DNaseI HS sites, we aligned the DNaseI HS sites of each group (cell type specific, common, ubiquitous) and compared each with the histone modifications of that region. Methylation and acetylation were examined, as distinct forms of each have been associated with activation, repression or both according to context. For example, the H3K27me3 methylation modification has been reported as being present at sites of repressed a gene expression (19).
As shown in Figure 2, all three states of H3K4 methylation and acetylation of both H3K9 and H3K27 were sharply elevated in different types of DNaseI HS sites. H3K9me1 and H4K20me1 were modestly elevated in cell type specific and common DNaseI HS sites, and the elevated levels of these two marks were diminished in ubiquitous DNaseI HS sites (Figure 2). We did not observe elevated levels of trimethylation of either H3K27 or H3K36 in areas surrounding the DNaseI HS sites (Figure 2). The tag densities of these histone modifications in ubiquitous DNaseI HS sites were much higher than those in cell type-specific DNaseI HS sites, except for H3K4me1 and H3K27me3. The percentages of various histone modifications overlapping with DNaseI HS sites from each of the different types are shown in the inset of Figure 2. Only a small fraction of the cell type-specific DNaseI HS sites overlapped with H3K4me2, H3K4me3, H3K9ac and H3K27ac; in contrast, about one-half of cell type-specific DNaseI HS sites overlapped with H3K4me1, H3K9me1, H3K27me3 and H4K20me1 (Figure 2A). Nearly 70% of the ubiquitous DNaseI HS sites overlapped with all of the histone modifications (Figure 2C), and of these, about one-fourth were located in proximal promoters.
Figure 2.


Histone modifications proximal to the DNaseI HS sites in K562 cell type. Histone modification profiles of cell type specific (A), common (B) and ubiquitous (C) DNaseI HS sites. The tag density for modifications is shown across the DNaseI HS sites and extending 5 kb upstream of the dxStart and downstream of the dxEnd. Inset shows the percentage of DNaseI HS sites of each different type overlapping with histone modifications. The numbers on the x-axis correspond to: 1, H3K4me1; 2, H3K4me2; 3, H3K4me3; 4, H3K9ac; 5, H3K9me1; 6, H3K27ac; 7, H3K27me3; 8, H3K36me3; 9, H4K20me1. The vertical dashed line indicates the peak of DNaseI HS signal within each DNaseI HS site.
We next examined the enrichment/depletion of histone modification profiles at each type of the DNaseI HS sites. We were intrigued to discover that both active and repressive histone modifications corresponded to the depletion at the peak signal of ubiquitous DNaseI HS sites, albeit to differing extents (Figure 2 and Supplementary Table S6). However, we did not observe similar modified histone troughs for the cell type-specific DNaseI HS sites, except in the cases of H3K9 and H3K27 acetylation (Figure 2; Supplementary Table S6). This analysis provided supportive evidence that the DNaseI HS peaks within ubiquitous DNaseI HS sites are nucleosome depleted. Noticeably, we found that the enrichment/depletion at DNaseI HS sites was strongly correlated with the level of DNaseI HS (Supplementary Figure S6 and Supplementary Table S7). Moreover, the correlation of the enrichment/depletion at the ubiquitous DNaseI HS sites was much stronger than that found at the cell type-specific DNaseI HS sites. Trimethylation of H3K27 was the only outlier to this trend, suggesting that the degree of nucleosome depletion is related to the level of DNaseI HS.
Correlation between DNaseI HS and histone modifications
To examine the correlation between DNaseI HS sites and the different histone modifications across the entire human genome, we separated cell type specific, common and ubiquitous DNaseI HS sites into 100 groups for each, based on level of DNaseI hypersensitivity. These groups were then plotted against their average modification levels in DNaseI HS sites (Figure 3; see ‘Materials and Methods’ section).
Figure 3.

Correlation between DNaseI HS and histone modifications in K562 cell type. Cell type specific (A), common (B) and ubiquitous (C) DNaseI HS sites were grouped among 100 sets (dot) based on their hypersensitive levels (from high to low, left to right on the x-axis). The average tag density of DNaseI hypersensitive and of histone modifications was calculated for each group and plotted according to the average tag density of DNaseI hypersensitive (right y-axis) and the histone methylation (left y-axis).
For the cell type-specific DNaseI HS sites, we observed a strongly negative correlation between H3K27me3 and DNaseI hypersensitivity, but a strongly positive correlation between DNaseI hypersensitivity and all other histone modifications (Figure 3A and Supplementary Table S8). Similar correlations between histone modifications and DNaseI hypersensitivity were found for the common DNaseI HS sites, with the exception of a weak correlation with H3K36me3 (Figure 3B and Supplementary Table S8). In ubiquitous DNaseI HS sites, however, modest positive correlations between DNaseI hypersensitivity and H3K4me2, H3K4me3 and H3K9ac were detected and a weak correlation was found with H3K27ac (Figure 3C and Supplementary Table S8). In this type of DNaseI HS sites, DNaseI hypersensitivity was negatively correlated with H3K4me1, H3K9me1, H3K27me3, H3K36me3 and H4K20me1; the H3K36me3 was most negatively correlated, followed in descending order by H4K20me1, H3K9me1, H3K27me3 and H3K4me1 (Figure 3C and Supplementary Table S8).
Genome-wide correlation of DNaseI HS sites and gene expression
DNaseI HS proximal to TSSs
To identify the general distribution pattern of DNaseI HS sites near and around TSSs, we generated composite profiles (n = 1000 each) of the 1000 most active, 1000 median and 1000 least active genes. The genomic region that was analyzed encompassed the entire defined gene body (exons and introns) and extended 5 kb upstream and 5 kb downstream of the 5′- and 3′-boundaries (Figure 4A). Numbers of tags in the gene body were quantitated in windows representing 10 equal parts, and in the 5′- and 3′-proximal regions in 0.2 kb windows; total numbers for each window were then summed to obtain to the overall methylation level for each gene. These numbers were then normalized by the total number of base pairs in each region. It was notable that the DNaseI hypersensitivity signal peaked near both the 5′- and 3′-ends. As such, this signature may represent a useful method by which to confirm annotated TSSs, to identify novel TSSs or to determine alternative TSSs functioning in particular cell types (37). In addition, the DNaseI hypersensitivity level of active genes was consistently much higher than that of silent genes, suggesting that DNaseI hypersensitivity is associated with transcriptional activation.
Figure 4.

Correlation between DNaseI HS and gene expression in K562 cell type. (A) Profiles of DNaseI HS sites across the gene bodies of the most active, median or most silent genes (n = 1000 each group). Inset shows the percentage of DNaseI HS sites of each different type that composed the three gene groups. The numbers on the x-axis correspond to: 1, cell type specific; 2, common; 3, ubiquitous. (B) DNaseI HS sites were grouped into 100-gene sets (dot) based on their expression levels (high to low, left to right on the x-axis). The average tag densities of the DNaseI HS within the gene body and the average gene expression level were calculated for each group and plotted according to the average gene expression level (left y-axis) and the average tag density of DNaseI hypersensitive (right y-axis).
We then examined the distribution of DNaseI HS sites that overlapped these genes. As shown in the inset of Figure 4A, 849 of the most active genes were found to be associated with DNaseI HS sites, whereas only 421 of the least active genes were similarly associated. Of those most active genes, 258 (>30%) associated with ubiquitous DNaseI HS sites, while only 22 (∼5%) of the most silent genes associated with ubiquitous DNaseI HS sites. Association with cell type-specific DNaseI HS sites was found with 22% (189) of the most active genes and >30% (148) of the most silent genes, respectively. These results suggested that ubiquitous DNaseI HS sites are associated with gene activation, whereas cell type-specific DNaseI HS sites are associated with gene repression.
DNaseI HS and gene expression
To investigate whether the DNaseI hypersensitivity level was correlated with gene expression level, we grouped the genes into 100 gene sets according to expression level. The sets were then plotted against the DNaseI hypersensitivity levels in the transcribed regions (Figure 4B). This analysis indicated that a strong positive correlation exists between the level of DNaseI hypersensitivity and gene expression (R = 0.8713, P = 4.73E-32). We then used the same method to assess the correlation of gene expression with the level of DNaseI hypersensitivity within the different types of DNaseI HS sites (Supplementary Figure S7), and found that the positive correlation remained for each. Cell type-specific DNaseI HS sites were more robustly correlated with gene expression than were the ubiquitous DNaseI HS sites, suggesting that cell type-specific DNaseI HS sites are involved in cell type-specific gene regulation (Supplementary Figure S7).
Four distinct modes of chromatin domains
DNaseI HS related to both histone modifications and gene expression
To further clarify whether the relationship between the DNaseI HS sites and histone modifications correlates with gene expression, we compared the DNaseI HS sites and histone modifications with gene expression levels of 17 751 genes. We generated an image plot to determine the average signals of active and repressive histone modifications relative to the tag density of DNaseI hypersensitivity and gene expression levels (Materials in Supplementary Data). Trimethylation of H3K4 and H3K27 were examined as representatives of active and repressive histone modifications, respectively. The results further confirmed previous observations that DNaseI hypersensitive signal is strongly positively correlated with active histone modifications and inversely correlated with repressive histone marks (Supplementary Figure S8). The genes with higher levels of the H3K4me3 signal tended to be expressed at higher levels, while the presence of H3K27me3 signals tended to correlate with decreased levels of expression. This was also consistent with previously published findings by others in which gene expression was significantly associated with presence of histone modifications (7,19). The interrelatedness of DNaseI hypersensitivity, gene expression and histone modifications indicated that active histone modifications generally correlate with the open chromatin state and active gene expression, whereas repressive histone marks indicate closed chromatin and gene silencing (Supplementary Figure S8).
H3K4me3 and H3K27me3 are referred to as a pair of ‘active-repressive’ modifications with regard to their effects on gene activity (7). The overlapping islands of H3K4me3 (‘active’) and H3K27me3 (‘repressive’) histone modifications are defined as ‘bivalent domains’, which have been implicated in the development and differentiation of mammalian embryonic stem cells and differentiated cells (7,21,22,38–41). By counting the total numbers of H3K4me3 and H3K27me3 modifications within each of the different types of DNaseI HS sites from the K562 cell line, we determined that, 15 and 45% of all DNaseI HS sites were associated with H3K4me3 and H3K27me3, respectively (Figure 5A). These numbers were consistent with other cell lines examined (Supplementary Figure S9). Intriguingly, >10% of all DNaseI HS sites were associated with both H3K4me3 and H3K27me3 modifications in multiple cell lines of different origins (Figure 5A and Supplementary Figure S9), indicating that bivalent domains are a widespread phenomenon in mammalian cells and suggesting that the ‘active-repressive’ switch is functionally relevant. However, recent studies suggested that bivalent marks, as described for mammalian embryonic stem cells, do not exist in Xenopus embryos (42,43). Future studies in different model organisms at various developmental stages are essential to elucidate the curious case of the occurrence or absence of bivalent marks (43).
Figure 5.

Four distinct modes of function for chromatin domains. (A) The percentage of DNaseI HS sites associated with H3K4me3 alone, both H3K4me3 and H3K27me3 or H3K27me3 alone, or not associated with H3K4me3 or H3K27me3 in K562 cells. The bar indicates the composition of those DNaseI HS sites that overlapped with both H3K4me3 and H3K27me3. (B) Distribution of DNaseI HS, gene expression, H3K4me3 and H3K27me3 signal at DNaseI HS sites associated with H3K4me3 alone, both H3K4me3 and H3K27me3 or H3K27me3 alone, or not associated with H3K4me3 or H3K27me3 in K 562 cells. (C) Four distinct modes of chromatin domains. Region I, active chromatin domains; Region II, silent chromatin domains; Region III, bivalent chromatin domains; Region IV, primed chromatin domains.
Examining the composition of these DNaseI HS sites showed that over one-third of those with H3K4me3 alone or with both 3K4me3 and H3K27me3 were of the ubiquitous type (Figure 5A and Supplementary Figure S10). However, only 7% of the DNaseI HS sites overlapping with either H3K4me3 alone, both 3K4me3 and H3K27me3 or H3K27me3 alone, were of the cell type-specific type (Figure 5A and Supplementary Figure S10). We then sought to determine whether the signals of DNaseI hypersensitivity, gene expression and H3K4me3 and H3K27me3 significantly differed among the DNaseI HS sites that were associated with H3K4me3 alone, both H3K4me3 and H3K27me3 or H3K27me3 alone, or not associated with H3K4me3 or H3K27me3 at all. As shown in Figure 5B, the highest levels of gene expression, DNaseI hypersensitivity and H3K4me3, and the lowest levels of H3K27me3 were associated with DNaseI HS sites that overlapped with H3K4me3 alone. The lowest levels of gene expression, DNaseI hypersensitivity and H3K4me3, and the highest levels of H3K27me3 were associated with DNaseI HS sites that overlapped with H3K27me3 alone. The DNaseI HS sites that overlapped with both H3K4me3 and H3K27me3 were characterized by high levels of gene expression, DNaseI hypersensitivity, and H3K4me3 and H3K27me3.
Four distinct functions of chromatin structure
Based on the relationship of DNaseI hypersensitivity with active and repressive histone modifications and gene expression, we theorized that at least four major functional modes existed for the different chromatin domain structures observed in the human genome across different cell types (Figure 5C). (i) Active chromatin domains were characterized by relatively higher levels of active histone modifications, DNaseI hypersensitivity and gene expression, and lower levels of repressive histone modifications (Figure 5B; Region I in Figure 5C), (ii) Silent chromatin domains were characterized by lower levels of active histone modifications, DNaseI hypersensitivity and gene expression and higher levels of repressive histone modifications (Figure 5B; Region II in Figure 5C), (iii) The bivalent chromatin domains were characterized by high levels of both active and repressive histone modifications, and high levels of DNaseI HS and gene expression (Figure 5B; Region III in Figure 5C) and (iv) The primed chromatin domains are characterized by patterns of active histone modifications and gene expression similar to the active chromatin domains, but also have similar patterns to the repressive chromatin domains of repressive histone modifications and DNaseI hypersensitivity (Figure 5B; Region IV in Figure 5C).
Identification of CTCF binding sites
Next, we investigated whether the specific correlation pattern between the data from DNase-seq and ChIP-seq, in combination with gene expression from microarray analysis, could be used to predict transcription factor binding sites (TFBSs) in the human genome.
In vertebrates, the CTCF, a ubiquitously expressed 11 zinc finger (ZF) protein (44,45), is necessary for insulator element function (46–48). To characterize how the CTCF binding sites are distributed along the human genome, we performed computational meta-analysis of the DNase-seq and ChIP-seq data to identify potential CTCF binding sites. We examined the DNaseI HS sites associated with CTCF ChIP-enriched regions across cell lines (Supplementary Table S9). Of the 35 307 DNaseI HS sites that overlapped with CTCF enriched regions in cell type K562, 13 007 were distal (>1 kb) to an annotated TSS or RNA Pol II signal. Then, we applied the de novo motif finder MEME to identify CTCF binding sites within these 13 007 distal DNaseI HS sites.
Our MEME analysis revealed that the CTCF consensus DNA binding motif was enriched in these DNaseI HS sites (Figure 6A). Furthermore, the consensus motif was identical in each of the cell types studied (Supplementary Figure S11) and was consistent with previous findings from other cell lines (human islets, CD4+ T cells, HeLa cells and Jurkat cells) (27,49). Most of the distal DNaseI HS sites (10 397 out of 13 007, 79.9%) contained at least one consensus motif (Figure 6B). Of those, 149 (1.4%) were cell type specific, whereas 7969 (76.7%) and 2279 (21.9%) were common and ubiquitous, respectively. This agreed well with previous observations that CTCF binding in insulator regions is similar across diverse cell types (50). In a statistical context, the consensus motif explains 65% (8456 out of 13 007) of the distal DNaseI HS sites after accounting for motifs that are expected to occur by chance. Compared to using DNaseI-Seq data (29%, 39 951 out of 139 121; Supplementary Figure S12A) or ChIP-seq data (57%, 46 328 out of 81 688; Supplementary Figure S12B) alone, our result illustrated the high accuracy of CTCF binding sites discovery based on the integrative data (Figure 6B). Most often, these distal DNaseI HS sites contained only a single motif (Figure 6C).
Figure 6.

Motif analysis of DNaseI HS sites. (A) Significantly enriched CTCF consensus motifs are graphically depicted using Weblogos. (B) MAST curves. Horizontal axis represents the E-value, the number of peaks expected to contain a given motif by chance. The vertical axis represents the number of peaks identified. The red curve (peaks with motifs) represents the number of peaks containing a given motif at the corresponding E-value. The blue curve (peaks explained) represents the number of peaks that contained a motif not by chance. The maximum of the peaks explained curve is displayed. (C) The histograms of the distribution of the motif number within the distal DNaseI HS sites that overlapped with CTCF binding sites.
The canonical motif was highly located on the peak of the DNaseI HS signal within each DNaseI HS site (Supplementary Figure S13A), which was consist with previous finding (27). This indicated that these peaks serve as the point of contact by the protein in vivo. Not surprisingly, we also found that the identified motif is located far distally from the nearest upstream or downstream associated genes (Supplementary Figure S13B), a finding that would explain the widespread and fundamental role of CTCF. Although the identified motif appeared to represent the major CTCF binding sequence, a significant number of the sites lacked the consensus sequence. Another study recently found that CTCF can bind to genomic regions that apparently lack the defined motif (51); this may be a result of DNA–CTCF interactions mediated by contacts with distinct arrangements of CTCF's 11-ZFs (44,48,52–54).
DISCUSSION AND CONCLUSION
Global properties of DNaseI HS sites
The dynamic and complex regulatory elements of gene transcription that underlie every biological process, from cell type-specific functions to systemic response to the environment, remain to be completely defined or understood. Whole-genome mapping of DNaseI HS sites has provided crucial clues to regions of transcriptional regulation. By combining these data with data from ChIP-seq and gene expression microarray experiments, we may gain a better understanding of this process. To this end, we performed a meta-analysis using each of these datasets that are publically available. By classifying the DNaseI HS sites according to their characteristic of cell type specific, common or ubiquitous, we were able to identify approximately 900 000 DNaseI HS sites from 29 diverse cell lines. These sites of presumed transcriptional regulatory function encompassed 8.28% of the human genome, which was in agreement with the proportion reported in a related study (26). Detailed examination of all the DNaseI HS sites in each of the 29 cell lines revealed that only approximately 10 000 (∼7%) are cell type specific and approximately 19 000 (∼14%) were ubiquitous, covering 0.67 and 0.07% of the human genome, respectively. The percentage of cell type-specific DNaseI HS sites was much smaller than that obtained previously by Xi et al. (26). This discrepancy may be due to the particular cell types examined in our study or the fact that we examined more different cell types in total. Nonetheless, even though 29 cell types were considered in our study, we were also unable to reach the statistical saturation point for the DNaseI HS sites that are likely to exist throughout the human genome. This finding indicated that functional TFBSs make up a more significant percentage of the genome.
By investigating the ubiquitous DNaseI HS sites, we hoped to gain insight into the function of housekeeping genes and overall chromatin structures. Our location analysis of DNaseI HS sites relative to annotated genes indicated that nearly 50% of the ubiquitous DNaseI HS sites in these data sets were located in proximal promoters or exons. This analysis, when considered in tandem with the overlapping with CpG islands between DNaseI HS sites and levels of sequence conservation among different cell lines (Supplementary Figures S4 and S5), indicated that ubiquitous DNaseI HS sites are generally associated with housekeeping promoters of genes. The strong correlation of DNaseI HS sites with TFBSs revealed that DNaseI HS sites are highly restricted to gene regions and their transcriptional regulatory elements. This provides further proof that DNaseI HS sites in the genome are valuable markers of TF binding regions.
Correlations among DNaseI HS, histone modifications and gene expression
Other groups have noticed sharp declines in the presence of active histone marks near TSSs, termed as nucleosome depleted regions (7,25,55). Interestingly, we observed that both active and repressive histone modifications are characterized by modified histone troughs, which are centered at DNaseI HS peaks of ubiquitous DNaseI HS sites, suggesting that these regions are nucleosome depleted. The degree of nucleosome depletion is undoubtedly related to chromatin accessibility and level of DNaseI hypersensitivity (Supplementary Figure S6; Supplementary Table S7). Our finding extends previous observations made in yeast, flies and mammalian systems, particularly the human, that nucleosome depletion is a general characteristic of active promoters. Furthermore, our finding provides supportive evidence that nucleosome depletion is associated with ubiquitous DNaseI HS sites and the ubiquitous DNaseI HS sites are nucleosome depleted.
Both cell type specific and ubiquitous DNaseI HS sites are generally positively correlated with active histone modifications H3K4me2/3, H3K9ac, H3K27ac and negatively correlated with the repressive modification H3K27me3. Interestingly, monomethylations of H3K4, H3K9, H3K36 and H4K20 display a more complex functional relationship with chromatin, as they are positively correlated with cell type-specific DNaseI HS sites and negatively correlated with ubiquitous sites. Correlation of DNaseI hypersensitivity with gene expression suggests that DNaseI hypersensitivity is highly correlated with transcriptional activation. Ubiquitous DNaseI HS sites were associated with active genes, while cell type-specific DNaseI HS sites were correlated with silent genes. Correlations of DNaseI hypersensitivity, histone modifications and gene expression indicated that active histone modifications generally correlated with active gene expression and the open (accessible) chromatin state, whereas repressive histone marks correlated with gene silencing and tightly packed (inaccessible) chromatin state (Figure 5C). These specific correlations were summarized to reveal four distinct modes of chromatin domain function: repressive, active, primed and bivalent. These modes of association, which are much more finely described than the traditional classification of heterochromatin (open) or euchromatin (closed), provide insights into the complex structure and function of chromatin.
Identification of regulatory elements via an integrative approach
After systematic exploration of the relationships between data emanating from DNase-seq, ChIP-seq and gene expression microarrays, we were able to identify transcriptional regulatory elements by using the de novo motif finder MEME. Our limited MEME analysis of the CTCF binding sites served to illustrate the high specificity and accuracy in identification of transcriptional regulatory elements using the integrated data sets (Figure 6B and Supplementary Figure S12). The advent of high-throughput methods, including DNase-chip/DNase-seq, ChIP-chip/ChIP-seq and gene expression arrays, has encouraged large storage databases of related data and public availability for more significant analysis. Combining distinct genome scale data in meta-analysis approaches represents the next era of genomic research.
Our integrative analysis presented herein can be considered as first-order data integration. The simplicity of the overlapping approach to determine regions and characteristics of enriched domains from DNase-seq and ChIP-seq data, along with the motif discovery and Gene Ontology strategies, represent an efficient means by which to perform integrative analysis. The next higher order, comprehensive, integrative analysis should integrate diverse high-throughput sequence tags directly. Additionally, interactome data (56) can be applied to facilitate identification of the target genes of DNaseI HS sites, since TFs and their three-dimensional interactions are crucial to gene regulation (57,58). Technologies based on chromosome conformation capture (3C) (59), circularized chromosome confirmation capture (4C) (60), carboncopy chromosome confirmation capture (5C) (61) and the recently developed methods of Hi-C (62) and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) (63,64), can be used to provide a high-resolution genome-wide map of long-range genomic interactions. Integrating a wide variety of genomic data sets, including genomes, epigenomes (55), transcriptomes (65) and interactomes (56) will significantly enhance our understanding of the complex genomic systems of all life forms and enhance our efforts to understand and modulate health and optimize well-being (66).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National High Technology Research and Development Program of China (No. 2007AA02Z311 to W.S.); National Nature Science Foundation of China (No. 30700139 and No. 31070639 to W.S.). Funding for open access charge: National Nature Science Foundation of China (No. 31070639 to W.S.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank the ENCODE Project Consortium for making their data publicly available and the ENCODE Open Chromatin and Broad Histone groups for providing the DNase-seq, ChIP-seq data and gene expression data. We thank Yongchao Liu (School of Computer Engineering, Nanyang Technological University, Singapore) for sharing CUDA-MEME source codes for us. The authors would like to thank the anonymous reviewers for their constructive comments, which contributed to an improved presentation.
REFERENCES
- 1.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 2.Lemon B, Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;14:2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- 3.Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- 4.Heintzman ND, Ren B. The gateway to transcription: identifying, characterizing and understanding promoters in the eukaryotic genome. Cell Mol. Life Sci. 2007;64:386–400. doi: 10.1007/s00018-006-6295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nightingale KP, O'Neill LP, Turner BM. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 2006;16:125–136. doi: 10.1016/j.gde.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Metzker ML. Sequencing technologies - the next generation. Nat Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 7.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 9.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mardis ER. ChIP-seq: welcome to the new frontier. Nat. Methods. 2007;4:613–614. doi: 10.1038/nmeth0807-613. [DOI] [PubMed] [Google Scholar]
- 11.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 12.Crawford GE, Holt IE, Mullikin JC, Tai D, Blakesley R, Bouffard G, Young A, Masiello C, Green ED, Wolfsberg TG, et al. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc. Natl Acad. Sci. USA. 2004;101:992–997. doi: 10.1073/pnas.0307540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16:123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, Green R, Meltzer PS, Wolfsberg TG, Collins FS. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods. 2006;3:503–509. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabo PJ, Hawrylycz M, Wallace JC, Humbert R, Yu M, Shafer A, Kawamoto J, Hall R, Mack J, Dorschner MO, et al. Discovery of functional noncoding elements by digital analysis of chromatin structure. Proc. Natl Acad. Sci. USA. 2004;101:16837–16842. doi: 10.1073/pnas.0407387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabo PJ, Humbert R, Hawrylycz M, Wallace JC, Dorschner MO, McArthur M, Stamatoyannopoulos JA. Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries. Proc. Natl Acad. Sci. USA. 2004;101:4537–4542. doi: 10.1073/pnas.0400678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat. Methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005;123:233–248. doi: 10.1016/j.cell.2005.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc. Natl Acad. Sci. USA. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr. Opin. Genet. Dev. 2009;19:127–134. doi: 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xi H, Shulha HP, Lin JM, Vales TR, Fu Y, Bodine DM, McKay RD, Chenoweth JG, Tesar PJ, Furey TS, et al. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS. Genet. 2007;3:e136. doi: 10.1371/journal.pgen.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stitzel ML, Sethupathy P, Pearson DS, Chines PS, Song L, Erdos MR, Welch R, Parker SC, Boyle AP, Scott LJ, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12:443–455. doi: 10.1016/j.cmet.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling G, Sugathan A, Mazor T, Fraenkel E, Waxman DJ. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol. Cell. Biol. 2010;30:5531–5544. doi: 10.1128/MCB.00601-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr. Protoc. Bioinformatics. 2007 doi: 10.1002/0471250953.bi0104s17. Chapter 1: Unit 1.4. [DOI] [PubMed] [Google Scholar]
- 31.Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrow J, Denoeud F, Frankish A, Reymond A, Chen CK, Chrast J, Lagarde J, Gilbert JG, Storey R, Swarbreck D, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl. 1):S4–S9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Schmidt B, Liu W, Maskell DL. CUDA-MEME: accelerating motif discovery in biological sequences using CUDA-enabled graphics processing units. Pattern Recogn. Lett. 2010;31:2170–2177. [Google Scholar]
- 34.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 35.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 38.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 39.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 42.Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, Francoijs KJ, Stunnenberg HG, Veenstra GJ. A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev. Cell. 2009;17:425–434. doi: 10.1016/j.devcel.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herz HM, Nakanishi S, Shilatifard A. The curious case of bivalent marks. Dev. Cell. 2009;17:301–303. doi: 10.1016/j.devcel.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 44.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 1996;16:2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell Biol. 1993;13:7612–7624. doi: 10.1128/mcb.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunn KL, Davie JR. The many roles of the transcriptional regulator CTCF. Biochem. Cell Biol. 2003;81:161–167. doi: 10.1139/o03-052. [DOI] [PubMed] [Google Scholar]
- 47.Klenova EM, Morse HC, III, Ohlsson R, Lobanenkov VV. The novel BORIS + CTCF gene family is uniquely involved in the epigenetics of normal biology and cancer. Semin. Cancer Biol. 2002;12:399–414. doi: 10.1016/s1044-579x(02)00060-3. [DOI] [PubMed] [Google Scholar]
- 48.Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520–527. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 49.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burcin M, Arnold R, Lutz M, Kaiser B, Runge D, Lottspeich F, Filippova GN, Lobanenkov VV, Renkawitz R. Negative protein 1, which is required for function of the chicken lysozyme gene silencer in conjunction with hormone receptors, is identical to the multivalent zinc finger repressor CTCF. Mol. Cell. Biol. 1997;17:1281–1288. doi: 10.1128/mcb.17.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filippova GN. Genetics and epigenetics of the multifunctional protein CTCF. Curr. Top. Dev. Biol. 2008;80:337–360. doi: 10.1016/S0070-2153(07)80009-3. [DOI] [PubMed] [Google Scholar]
- 54.Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006;7:703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- 55.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van CS, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 56.Collins SR. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 57.Farnham PJ. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007;8:104–115. doi: 10.1038/nrg2041. [DOI] [PubMed] [Google Scholar]
- 59.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 60.Simonis M. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 61.Dostie J. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11:R22. doi: 10.1186/gb-2010-11-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guttman M. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat. Rev. Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.