


Abstract
Within the base excision repair (BER) pathway, the DNA N-glycosylases are responsible for locating and removing the majority of oxidative base damages. Endonuclease III (Nth), formamidopyrimidine DNA glycosylase (Fpg) and endonuclease VIII (Nei) are members of two glycosylase families: the helix–hairpin–helix (HhH) superfamily and the Fpg/Nei family. The search mechanisms employed by these two families of glycosylases were examined using a single molecule assay to image quantum dot (Qdot)-labeled glycosylases interacting with YOYO-1 stained λ-DNA molecules suspended between 5 µm silica beads. The HhH and Fpg/Nei families were found to have a similar diffusive search mechanism described as a continuum of motion, in keeping with rotational diffusion along the DNA molecule ranging from slow, sub-diffusive to faster, unrestricted diffusion. The search mechanism for an Fpg variant, F111A, lacking a phenylalanine wedge residue no longer displayed slow, sub-diffusive motion compared to wild type, suggesting that Fpg base interrogation may be accomplished by Phe111 insertion.
INTRODUCTION
Normal cellular metabolism and environmental agents generate large amounts of damage to DNA (1), thus the ability of proteins to locate DNA damages within the vast sea of undamaged DNA is critical to genomic integrity. The two major effects of DNA damage are cytotoxicity resulting from damages that inhibit the progress of DNA polymerase during replication and mutagenesis from insertion of non-cognate bases during replication. Nearly half of all DNA base damages are oxidatively-induced, and if left unrepaired can lead to mutations, malignant transformation, or apoptosis (2). To preserve the integrity of DNA, numerous repair systems have evolved. The major DNA repair system that removes non-bulky DNA lesions, such as oxidative damages, is the base excision repair pathway (BER) (1–3). Since in many cases damaged bases differ only subtly from their original structure, the recognition step in the BER pathway is the most difficult. Moreover, finding these lesions by the DNA N-glycosylases is undertaken without the use of biochemical energy.
In a mammalian cell, over the course of a day, approximately one damage per million bases must be removed by DNA glycosylases (4). Since the glycosylase search process relies on thermal energy, lesions are found through random collisions between the DNA and the glycosylase. However, the rate of lesion recognition through three-dimensional (3D) diffusion alone would be insufficient to maintain genome integrity compared to the generation of DNA damage. Thus, glycosylases are thought to find their target bases via facilitated diffusion, where the glycosylase binds to a non-specific site followed by 1D diffusion along the DNA until the lesion is found or until the enzyme dissociates from the DNA. Therefore, in order to scan the entire genome rapidly, numerous cycles of 1D and 3D diffusion are required for careful base pair interrogation and long-distance translations, respectively (5–7). Initially, this model was inferred from kinetic data [reviewed in ref. (8)] but it recently has been confirmed in single-molecule studies which suggest that glycosylases may slide in close contact while rotating around the DNA helix (9,10).
Glycosylases can be separated into families based on their sequence homology, structural motifs and substrate specificity (4). There are two structurally different families of glycosylases that recognize oxidative damage, the helix–hairpin–helix (HhH) superfamily and the Fpg/Nei family. The HhH superfamily, represented by Escherichia coli endonuclease III (Nth), contains a HhH element followed by a Gly/Pro-rich loop and an essential catalytic aspartate residue (11–13). The principal DNA substrates recognized by Nth and its orthologs are oxidized pyrimidines (14–16). The Fpg/Nei superfamily, represented by E. coli formamidopyrimidine DNA-glycosylase (Fpg) and endonuclease VIII (Nei), contains the helix two turns helix (H2TH) and zinc finger motifs (17–20). Fpg protein is specific for oxidized purines including 8-oxoguanine and formamidopyrimidines (21–24), while Nei, like Nth, prefers oxidized pyrimidines (25,26). All three glycosylases recognize and remove the further oxidation products of 8-oxoguanine, spirodinodihydantoin and guanodinohydantin (27,28). Despite having different structural characteristics and catalytic mechanisms, these two families of glycosylases have overlapping substrate specificities and thus provide an excellent platform to probe unique molecular structure/function relationships.
Although their substrate preferences may differ, glycosylases do share commonalities in their structure and function [for reviews see (29–32)]. Importantly, the glycosylase–DNA binding interface is largely electrostatic, primarily through positively charged amino acids on the protein and negatively charged phosphates on the DNA backbone. Upon lesion binding, glycosylases cause a sharp bend in the DNA that is accompanied by flipping out of the damaged base from the helix into the glycosylase substrate-binding pocket, while amino acid residues of the glycosylase are inserted into the void left by the extruded base (12,17–20). Both kinetics (33) and single molecule (9,10) experiments have shown that glycosylases scan DNA close to diffusion limits indicating that it is thermodynamically impossible for the primary lesion recognition mechanism to involve sequential extrusion of every base. In parallel, crystallographic structural data show that Fpg glycosylases may be able to intrahelically scan for damaged bases by the insertion of a phenyalanine, one of several residues that are inserted into the void left by the extruded 8-oxoguanine (34,35). In Bacillus stearothermophilus Fpg, Phe114 causes a disruption in base stacking at the point of insertion and a buckling of the target base pair at the site of insertion. Thus Phe114 can potentially be a sensor for the deformability of base pairs (34).
To determine whether members (Nth, Nei and Fpg) of two structurally distinct glycosylase families share a similar search mechanism, we imaged single Qdot-labeled glycosylase molecules interacting with elongated λ-DNA with high spatial and temporal resolution. Most glycosylases diffused along DNA with a broad distribution of rates that ranged over two orders of magnitude between 0.001 and 0.1 µm2/s. This diffusive behavior was common to all three glycosylases studied. By mutating a key E. coli Fpg wedge residue, Phe111, to an alanine, we observed a significant increase in the average diffusion constant compared to the wild-type protein. Interestingly, this increase was due to a selective loss of glycosylases diffusing at the slower end of the spectrum (i.e. <0.06 µm/s), which was characteristic of the wild type. We propose that slowly diffusing glycosylases represent molecules that are involved in intrahelical base interrogation, which do so by tracking and diffusing along the DNA helix in a rotational manner. The glycosylases exhibiting faster rates of diffusion also diffuse rotationally but may reflect those that have a lower ‘duty ratio’ in terms of the fraction of the protein's bound lifetime committed to base interrogation versus free diffusion. This bimodal scanning process allows the Nth, Nei and Fpg glycosylases to search efficiently for DNA damage.
MATERIALS AND METHODS
Protein purification
Glycosylase cDNA obtained from Wallace laboratory stocks was cloned into a pET22b vector thereby adding a C-terminal hexahistidine tag, and expressed in BL21 DE3 cells (Novagen, Gibbstown, NJ, USA) and the proteins purified as previously described (36). The active fraction of each enzyme was measured using our previously described fluorescence-based high through put molecular accessibility method (37) (Supplementary Table S1).
Substrates
All single molecule experiments used undamaged lambda DNA (New England Bio Labs, Ipswich, MA, USA). The fluorescence-based protein active fraction assay utilized a 24-bp substrate with 5,6-dihydrouracil (DHU) opposite guanine. For the glycosylase enzyme activity assay, a 35-bp oligonucleotide containing either thymine glycol (Tg) opposite adenine (Nth, Nei) or 8-oxoguanine (8-oxoG) opposite cytosine (Fpg) served as substrates. The 32P labeling of the substrates was previously described (38). Briefly, the damage-containing strand was end labeled with [γ-32P] ATP for 15 min by T4 polynucleotide kinase (New England Biolabs). The reaction was terminated by heat inactivation and by the addition of EDTA. The labeled damage-containing strand was then ethanol precipitated, resuspended and annealed to the complementary strand. Substrates were stored at −20°C in 10 mM, Tris pH 8.0, 50 mM NaCl. The substrates for the fluorescence-based protein active fraction determination (Midland, Midland, TX, USA) were handled differently and are described in detail in a previous publication (37).
Glycosylase assay
All reactions, both gel based and microscope based, were carried out in 50 mM Tris–HCl, pH 8.1, 150 mM KGlu, 10 mM DTT and 1 mg/ml bovine serum albumin (BSA) unless otherwise noted. The glycosylase assay reactions typically contained 20 nM substrate and 2 nM of active Nth, Fpg, or Nei. These reactions were carried out at 37°C and terminated with 98% formamide, 0.2 N NaOH, 10 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanole. Glycosylase treated substrates were loaded onto sequencing acrylamide gels to resolve the amount of product cleaved. Gels were dried, exposed to a radiographic phosphor screen and scanned on a molecular imager FX system (Bio-rad, Hercules, CA, USA). Using spot densitometry allowed for the ratio of product to substrate to be determined, thereby allowing the percent activity to be calculated.
Single molecule assay
To image the glycosylase search mechanism on DNA, we adopted the same experimental approach used previously to monitor the movement of nucleotide excision repair proteins, UvrA and UvrB, on single λ DNA molecules (39). In brief, λ DNA was elongated to ∼90% of its contour length [see Supplementary Data in ref. (39)] using hydrodynamic flow within a specially designed flow through microscope slide chamber that contained poly-l-lysine (Sigma)-coated 5 μm silica beads (Polysciences, Warrington, PA, USA) fixed to a surface (see Figure 1A and B). The beads served as attachment platforms for the extended λ DNA, which resembled ‘tightropes’ measuring ∼6.5 μm between attachment points. DNA tightropes were constructed by flowing 200 µl of DNA elongation buffer [50 mM Tris–HCl (pH 8.0), 10 mM dithiothreitol (DTT) and 150 mM KGlu] containing 31.7 pM λ-DNA into the chamber using a series of infusion withdrawal cycles every 100 µl at a flow-rate of 500 µl/min. Once formed, 5 nM of YOYO-1 dye (Invitrogen) in glycosylase buffer [50 mM Tris–HCl (pH 8.0), 10 mM dithiothreitol (DTT), 1 mg/ml BSA and 0.0–250 mM KGlu] was infused and incubated for 2 min and flushed with more buffer to allow for imaging of the fluorescent DNA. All experiments were performed at room temperature.
Figure 1.

Single molecule glycosylase assay. (A) λ DNA ‘tightropes’ extended between stationary 5 μm silica beads post-hydrodynamic flow-mediated DNA elongation. (B) Image of faint DNA tightrope strung between beads (green) with bound Qdot-labeled glycosylase (red). (C) Conjugation sandwich system used to fluorescently label single glycosylases. Illustration created using crystal structures for Nth (1ORN), IgG (1HZH) and DNA (3BSE) (12,70,71). Biotin conjugated anti-histidine antibody was used to link the his-tagged glycosylase to the streptavidin-coated Qdot. (D) Sample trajectories of two Nth molecules undergoing slow (dotted line) and fast (solid line) diffusion. Trajectories include motion that is both parallel (x) and perpendicular (y) to the DNA longitudinal axis. Glycosylase position was determined as described in the text. (E) The mean square displacement (MSD) plot for slow (square) and fast (circle) trajectories from Figure 1D. The diffusion constant is obtained from the slope of the MSD plot. Error bars represent standard error of the mean (SEM) of a single trace. Inset: The same MSD plots presented on log–log axes with the slope equal to the diffusive exponent, α.
To image single glycosylases, expressed hexahistidine tagged glycosylases (see above) were conjugated to streptavidin-coated Qdots (λ = 655 nm; Invitrogen, Carlsbad, CA, USA) through a biotin-conjugated anti-histidine antibody (Penta-His; Qiagen, Germantown, MD, USA). To maximize the probability that a Qdot has only one active glycosylase, the Qdot to antibody to active glycosylase molar ratio was 1:5:1, obtained through the following conjugation protocol. First, 5 µl of 1 µM antibody was mixed with 1 µl of 1 µM Qdots and incubated on ice for 30 min. Then 2 µl of 0.16 µM active glycosylase was added to 2 µl of the Qdot-antibody mixture and incubated on ice for 15 min. This final mixture was diluted 100-fold in final buffer and infused into the flowcell giving a glycosylase concentration of 0.8 nM. To further characterize this conjugation strategy, we estimated that only one antibody actually bound to the Qdot (see Supplementary Data, Supplementary Figure S1). This most likely resulted from the antibodies being labeled with up to eight biotins (manufacturer's specifications) and thus a single antibody could effectively occupy the 6–8 available biotin sites per Qdot (see Supplementary Data). Therefore, excess non-Qdot bound antibody competed for glycosylase in solution and given the antibody dissociation constant (5 × 10−8 M; manufacturer's specifications), the free glycosylase concentration was reduced by 83% at the mixing concentrations used (see above). Thus, the effective Qdot to glycosylase ratio was 5.7 to 1. Based on Poisson statistics (40), most Qdots (i.e. 84%) will have no bound glycosylases with the majority of the remaining Qdots (i.e. 15%) having only a singly bound glycosylase.
Image acquisition and data analysis
Flowcells were placed on a custom built, through-the-objective (PlanApo 100×, 1.49 n.a.), total internal reflectance fluorescence (TIRF), microscope (Nikon TE2000 inverted). To image the DNA attached to the 5 µm beads, the excitation light from a 488 nm, 50 mW Argon-ion laser (Spectra-Physics, Santa Clara, CA, USA) was adjusted to a sub-critical angle, yielding an obliquely angled illumination ray. This was accomplished by defocusing the beam at the edge of the objective's back aperture with the use of a movable lens that could easily switch between TIRF and oblique modes of operation. Emitted light was passed through a beam splitter (Optical Insights, Pleasanton, CA, USA) to provide simultaneous dual-color imaging of the YOYO-1 stained λ-DNA and Qdot-labeled glycosylase.
Image stacks of 1000 frames were captured at 16–66 frames/s with 2 × 2 pixel binning (58.5-nm pixel resolution) using an intensified CCD camera (XR Mega-S30 running Piper Control v2.3.14 software, Standard Photonics, Stanford, CA, USA). Two-dimensional trajectories were generated using ImageJ v1.37 (National Institutes of Health, Bethesda, MD, USA) and SpotTracker 2D (41). The spatial resolution of the system was calculated by tracking immobile Qdots on a glass surface for 1000 frames. The 15 nm positional error for the x- and y-axes was determined by the standard deviation of each axis. The glycosylase movement was analyzed using mean squared displacement (MSD) analysis (39). The general expression for the MSD is given by:
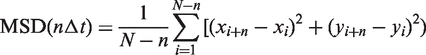 |
(1) |
where N is the total number of frames in the trajectory, n is the number of frames for different time intervals, Δt is the time (typically 66.6 ms) between frames, and xi and yi are the positions of the Qdot in frame i (42). The diffusion constant (D) was estimated from the slope of the first 10% of the MSD versus Δt when plotted on linear axes. Furthermore, the MSD when plotted on a log-log axis provides insight as to the type of motion being observed (43). For various types of motion the relationship between the MSD and the time interval (nΔt) can be generalized to MSD ∼ (nΔt)α, where α is the ‘diffusive exponent’ (43). For immobile particles, α = 0, randomly diffusing particles have an α = 1 and for directed motion at constant velocity, α = 2. Thus, by plotting the MSD versus Δt on log–log axis, α is the slope of the MSD.
RESULTS
The DNA tightrope assay and glycosylase–DNA interactions
To study glycosylase–DNA interactions at the single molecule level, we used DNA tightropes which are YOYO-1 dyed λ DNA molecules elongated to ∼90% of their contour length [see Supplementary Data in ref. (39)] by hydrodynamic flow, allowing for its attachment to poly-lysine-coated microspheres in a custom flowcell (39) (Figure 1A and ‘Materials and Methods’ section). Qdot-labeled glycosylases were then added to the flowcell and both the DNA and glycosylases simultaneously imaged in the absence of flow using oblique angle fluorescence microscopy (Figure 1B and C) at 15 nm spatial and 67 ms temporal resolution. Image fields typically contained >20 fluorescent single λ DNA molecules with on average ∼2 bound glycosylases per DNA tightrope (Movie S1 in Supplementary Data and Figure 1B). Only those glycosylases that were observed to bind to, diffuse along and then dissociate from the DNA molecule within the ≥16 s imaging period were characterized in terms of their bound lifetime and motion characteristics (see below). Before analyzing these data, a series of controls were performed to confirm that: (i) the DNA and glycosylase labeling strategies did not affect glycosylase function and (ii) the assay reports on the behavior of a single glycosylase interacting with a single λ DNA molecule.
Since the intercalating dye, YOYO-1, perturbs DNA structure (44,45), we assessed whether YOYO-1 affects the ability of the glycosylase to cleave out damaged bases. Using a standard gel-based glycosylase assay (see ‘Materials and Methods’ section), we observed no significant differences in the activities of Nth, Nei, and Fpg using YOYO-1 stained DNA substrates compared to the unstained DNA control (Figure 2). Equally important, there was no effect of Qdot-labeling on the cleavage activity of the three glycosylases (Figure 2).
Figure 2.

Gel-based glycosylase assays under single molecule conditions. Time course reactions contained 20 nM substrate and 2 nM of Nth, Nei or Fpg enzyme. Black curve/filled circles represent the control oligodeoxyribonucleotide substrates containing either a thymine glycol lesion opposite A for Nth and Nei or an 8-oxoguanine lesion opposite C for Fpg. Gray curve/filled triangles depict reactions where the oligodeoxyribonucleotide substrates had been pretreated with 10 pM YOYO-1. Dashed black curve/filled squares represent reactions where the glycosylase was conjugated to a quantum dot. Error bars represent SEM. (A) Nth; (B) Nei; (C) Fpg.
To observe individual His-tagged glycosylases, we relied on biotinylated, anti-histidine antibodies coupled to a single streptavidin-functionalized Qdot (Figure 1C). First, we confirmed that in the absence of glycosylase, neither the Qdot nor the histidine antibody-conjugated Qdot bound nonspecifically to the elongated λ DNA (data not shown). In contrast, only Qdots conjugated to glycosylases interacted with DNA. In addition, to ensure glycosylase binding events to elongated λ DNA molecules did not represent the glycosylase non-specifically interacting with multiply elongated λ DNA molecules in close proximity, ensemble averaged axial and longitudinal intensity profiles (46) allowed us to decipher single from multiple elongated λ DNA molecules (data not shown). When conjugated at an effective Qdot:glycosylase molar ratio of 6:1, the majority of Qdots will be glycosylase-free (84%) with the remaining Qdots having a 94% probability of being associated with only a single glycosylase, based on Poisson statistics (see ‘Materials and Methods’ section).
Once bound to DNA, the position of the Qdot-labeled glycosylase was tracked both parallel and perpendicular to the DNA's long axis (Figure 1D). The extent of perpendicular motion that could be attributed to oscillations of the taut DNA molecule was characterized by tracking an immobile Qdot-labeled glycosylase located at the center of an extended DNA molecule. The y-axis positional error of 21 nm was calculated from the standard deviation of twenty 1000 frame trajectories, where the mean λ DNA length was ∼6.5 µm between attachment points. These perpendicular displacements were extremely small and comparable to those of motile glycosylases. In contrast, the glycosylase motion parallel to and thus along the DNA was random in appearance with excursions as large as 350 nm. These random movements were characterized using mean square displacement (MSD) analysis (Figure 1E). When plotted on linear axes, a linear MSD relationship is indicative of Brownian motion, which was observed for all glycosylases over a 1 s time scale (Figure 1E). Diffusion constants were determined from the slope of the linear regression to the MSD (Figure 1E, see ‘Materials and Methods’ section) (9,10,47–52). Further insight as to the type of diffusive motion is obtained when the MSDs are plotted on log–log axes, where the slope of the relationship, α, defines the diffusive exponent (43). Specifically, α = 1 is characteristic of unrestricted diffusion, α = 2 is directed, while α < 1 is indicative of sub-diffusive behavior (Figure 1E, inset) (42,53). Although sub-diffusive motion has been reported previously in various biological systems (54), the mechanistic basis for such sub-diffusive behavior is far from certain. Our data suggest that glycosylase sub-diffusive motion defines specific interactions between the protein and DNA that prolong the residency time at a given location.
Nth-, Fpg- and Nei-DNA interactions are similarly diffusive and ionic strength-dependent
The two structurally different families of glycosylases, E. coli Nth from the HhH superfamily and E. coli Fpg and Nei from the Fpg/Nei family were characterized in terms of their motion on DNA and the effect of ionic strength on these interactions. As salt concentration was increased from 0 to 250 mM potassium glutamate (KGlu), the mean bound lifetime of the glycosylase on DNA decreased significantly (P < 0.002) for all glycosylases (Figure 3 and Supplementary Figure S2), although, enzyme activity at 250 mM was also reduced (data not shown). Moreover, these results are consistent with structural data that show the glycosylase–DNA interactions to be electrostatic in nature (11,12,17–20,55).
Figure 3.

Salt dependence of the mean binding lifetime for Nth, Nei and Fpg. The open square represents FpgFIIIA. The mean binding lifetime represents the period of time from initial glycosylase binding to dissociation from the DNA. Values were generated from single-exponential fits to binding lifetime histograms at 0.00, 0.05, 0.15 and 0.25 M KGlu (Supplementary Figure S2). Error bars represent SEM.
Based on MSD analysis of glycosylase trajectories along DNA at physiological salt concentration (150 mM KGlu), there was no apparent difference in the diffusion constants for Nth, Nei and Fpg (Figure 4). The distribution of diffusion constants covered a broad range of values with the majority below 0.06 μm2/s (Movies S2 and S3 in Supplementary Data). The observed diffusion is characteristic of glycosylase motion and not merely an artifact of DNA thermal fluctuations, given that the apparent diffusion constant for a ‘stationary’ Qdot-labeled glycosylase measured parallel to the longitudinal DNA axis was 0.0006 ± 0.0002 µm2/s (n = 19), two orders of magnitude lower than the mean diffusion constant for all ‘diffusing’ glycosylases. Therefore, the diffusion constant associated with the longitudinal DNA motion was set as the threshold below which a glycosylase was considered stationary, accounting for 12% of the glycosylases analyzed.
Figure 4.

Distribution of diffusion constants (D) for Nth, Nei and Fpg at 0.15 M KGlu. (A) Nth: D = 0.058 ± 0.015 µm2/s, n = 95. (B) Nei: D = 0.034 ± 0.010 µm2/s, n = 81. (C) Fpg: D = 0.023 ± 0.005 µm2/s, n = 88.
For the 88% of glycosylases that were motile, their motion was further characterized by the diffusive exponent, α, which ranged between 0.1 and 1.4. Interestingly, when the diffusive exponent is plotted against the diffusion constant, D, at physiological salt conditions (Figure 5A), the range of diffusion constants and diffusive exponents define a continuum from relatively slow (D < 0.06 μm2/s), sub-diffusive (α < 0.5) motion to faster (D > 0.06 μm2/s), unrestricted (α ∼ 1.0) diffusion. This continuum was characteristic of Nth, Nei and Fpg and was unaffected by changes in salt concentration (Figure 5A–C).
Figure 5.

Effect of salt concentration on the diffusive behavior of Nth, Nei and Fpg. The diffusive exponent, α, is plotted versus the diffusion constant, D, at 150 mM KGlu (A) for Nth: n = 95, Nei: n = 81 and Fpg: n = 88; at 50 mM KGlu (B) for Nth: n = 54, Nei: n = 55 and Fpg: n = 66; at 0 mM KGlu (C) for Nth: n = 53, Nei: n = 31 and Fpg: n = 38.
Phenylalanine111, an Escherichia coli Fpg wedge residue, is responsible for base interrogation
The mechanism(s) by which glycosylases find specific DNA damages is still unknown. However, structural studies suggested that for Fpg, intrahelical base interrogation may occur by insertion of a Phe residue into the DNA helix (34,35,55). To test this hypothesis we generated an E. coli Fpg mutant, F111A, using site-directed mutagenesis (see ‘Materials and Methods’ section). Similar to a previous report which claimed that Fpg F111A had no enzymatic activity on 8-oxoG opposite cytosine (8-oxoG:C) (56), we found substantially reduced activity of Fpg F111A on 8-oxoG:C compared to the wild-type protein (Supplementary Figure S3). Furthermore, Fpg F111A showed little or no ability to form a Schiff base (37) with 8-oxoG:C or 5,6-dihydrouracil opposite guanine (DHU:G) compared to wild-type; however, Schiff base formation with AP:C was still observed (Supplementary Table S1). At 150 mM KGlu, Fpg F111A interacted with DNA in the single molecule assay with a bound lifetime (1.7 ± 0.2 s) that was not significantly different (P > 0.05) from wild type (1.2 ± 0.1 s) (Figure 3). However, the variant's diffusive motion was dramatically different from wild type (Movie S4 in Supplementary Data and Supplementary Figure S7), as evidenced by the distribution of diffusive exponents versus diffusion constants. As can be seen in Figure 6 there was a noticeable loss of Fpg F111A diffusing at the slower rates (i.e. D < 0.01 µm2/s) and the mean diffusion constant for Fpg F111A (0.062 ± 0.019 µm2/s) was 3-fold higher compared to wild-type Fpg (0.023 ± 0.010 µm2/s). Moreover, the Fpg F111A diffusive motion was characterized by an α = 0.9 ± 0.2, indicative of unrestricted motion over all time scales, compared to the sub-diffusive motion of the wild type (α = 0.5 ± 0.3).
Figure 6.

The diffusive behavior of Fpg F111A compared to wild-type Fpg. The diffusive exponent, α, is plotted versus the diffusion constant, D, for wild-type Fpg (gray triangles) and Fpg F111A (black circles). Lines represent the median values for D and α for Fpg wild-type (gray lines) and Fpg mutant F111A (black lines).
DISCUSSION
The BER pathway requires that glycosylases search and identify damaged DNA bases in a rapid and efficient manner to maintain genomic integrity. Single molecule biophysical techniques have allowed investigators to observe individual glycosylases during this process as they move along DNA in vitro (9,10). Here we report that structurally distinct glycosylases from the HhH (Nth) and Fpg/Nei glycosylase families search for damage by diffusing along DNA and when doing so probe the DNA for base damage by insertion of a wedge amino acid. This conclusion is based on the dramatically different diffusive behavior observed for an Fpg F111A variant, where this putative wedge phenylalanine has been changed to an alanine. Specifically, the Fpg F111A mutant no longer displays the slow, sub-diffusive motion that we associate with interrogation of the DNA molecule, but rather faster, unrestricted diffusion because the mutant glycosylase no longer probes for damage. Finally, given both the similarities in their binding pockets (11,12,17–20,55) and their observed scanning behavior reported here, the HhH and the Fpg/Nei families have adopted a similar search mechanism for DNA base damage.
Nth, Nei and Fpg glycosylases slide along DNA to find their targets
Glycosylases must search for DNA base damage in vivo through a combination of displacement events that are slow enough to identify damage and fast enough to cover long distances along the DNA (3). With thermal energy being the driving force for these displacements, the glycosylase must rely on Brownian motion to accomplish its task. By directly imaging three glycosylases from two structural families using a newly developed in vitro DNA tightrope assay (39), we confirm that all three, Nth, Nei and Fpg, diffuse along DNA in a similar manner. The diffusive properties for each glycosylase were characterized by the mean squared displacement versus time relationship (MSD). This yields characteristic diffusion constants (D) and diffusive exponents (α); the latter offers a statistical description of whether the diffusive behavior is sub-diffusive (α < 1), directed (α = 2) or unbiased (α = 1). Based on this analysis, glycosylases appear extremely variable in their diffusion constants and mode of diffusive behavior (Figure 5). This variability is best described as a continuum where at one extreme, glycosylases diffuse slowly (D < 0.01 μm2/s) in a sub-diffusive mode (α ∼ 0.5) and at the other extreme, in an unrestricted (α ∼ 1.0), fast diffusive mode (D > 0.1 μm2/s) (Movie S3 in Supplementary Data and Supplementary Figure S6). In addition, glycosylases can be observed interconverting between the sub-diffusive and fast diffusive search modes (Movie S2 in Supplementary Data and Supplementary Figure S5) and the relative contribution of these search modes determines the value of the diffusion constant and diffusive exponent, which may account for the continuum observed in the diffusive behavior of the glycosylases. Nevertheless, how these modes of DNA interaction relate to the capacity of the glycosylase to rapidly target DNA damage and which of the described modes reflects the actual base pair interrogation process was inferred from the diffusive behavior of an Fpg amino acid wedge variant, as discussed below.
Glycosylase search is accomplished through intrahelical base interrogation
At least three theories exist for how glycosylases find single damaged DNA bases (3,17). Glycosylases may (i) extrude each base out of the helix for interrogation (57), (ii) capture those damaged bases that have spontaneously flipped out of the helix as has been shown for uracil DNA glycosylase (58), or (iii) may search by sampling the relative strength and flexibility of DNA base pairs by inserting a single residue into the DNA duplex (34,35). Given that the first two models are difficult to reconcile thermodynamically and may have to be modified (9), for the DNA glycosylases that recognize oxidative damages, intrahelical residue insertion seems the most likely mechanism for locating damage while scanning DNA at diffusion limits. A crystal structure of a Bacillus stearothermophilus Fpg–DNA complex (34) suggests that Phe114 can insert into the helix of an undamaged DNA molecule and test for damage by buckling the base pair and disrupting base stacking without extrusion. If an 8-oxoG is present, an alternative sugar pucker may be recognized and the damaged base is extruded into the substrate binding pocket. When we mutated this residue in the E. coli Fpg gene to an alanine (F111A), there was a dramatic increase in the overall diffusion rate of the variant compared to the wild-type protein (Figure 6 and Movie S4 in Supplementary Data). Even more striking was the apparent loss of a specific population of Fpg molecules that would have been characterized as slow and sub-diffusive (i.e. D < 0.01 µm2/s, α ∼ 0.5) (Figure 6). Although, the gel-based assay showed that Fpg F111A maintained ∼15% of the wild-type capacity to cleave an oligodeoxynucleotide containing 8-oxoguanine (Supplementary Figure S3), it may be doing so in part by capturing spontaneously extruded damages. Furthermore, the gel-based assay uses a relatively short substrate where lesion location and removal may occur via random collisions with the glycosylase rather than by 1D diffusion. Therefore, based on these data, together with structural (34) and kinetic (56,59) data in the literature, the Fpg search and interrogation mechanism is most likely mediated through intrahelical insertion of the Phe111 residue.
Glycosylases rotationally diffuse while interrogating DNA for damage
It is clear from observing how glycosylases interact with the DNA that their motion is achieved through 1D diffusion. However, the observed movement could occur either by the glycosylase tracking the contour of the DNA minor groove (i.e. rotational diffusion) or by translating along the longitudinal surface of the elongated DNA molecule. This is consistent with observations of uracil DNA glycosylase (UNG), which is thought to slide short distances along one strand of the DNA molecule and use longer range translations in order to trap the uracils in their spontaneously extruded state (33). Furthermore, human alkyladenine DNA glycosylase (AAG) has been shown to locate damages that occur on both strands of the DNA by intramolecular hopping, which may also permit glycosylases searching on opposite strands to bypass one another (60). Our data do not allow us to decipher between intramolecular hopping and sliding events. However, theoretical models have allowed for a description of whether glycosylases rotate while undergoing 1D diffusion (61,62). Briefly, by ensemble averaging diffusion constants of three different glycosylases conjugated to molecules with increasing Stokes radii, Blainey et al. (10) were able to show that glycosylases search by rotating around the DNA helix with a diffusion constant of 5.87 × 106 bp2/s (i.e. 0.68 µm2/s) for hOgg1-Cy3B. Using a similar theoretical calculation (Supplementary Figure S4), we estimate that the motion of Qdot-labeled glycosylases having diffusion constants of ∼0.05 µm2/s is consistent with rotational diffusion along DNA. This value, which is an order of magnitude lower than that of Blainey et al. (10), is due to the Stokes drag associated with the ∼20 nm Qdot label (see Supplementary Data, Supplementary Figure S4).
Interestingly, the population of Fpg proteins within the continuum of diffusive modes of DNA interactions that is lost by the F111A variant are glycosylases that were slow diffusers with D < 0.01 µm2/s (Figure 6). These data also suggest that the Qdot label itself, although responsible for the lower diffusion constant, does not interfere with the glycosylase's normal interaction with DNA. If the Qdot, in some manner, is the sole determinant of the observed D versus α relationship, the F111A mutation would have had no effect on diffusion (Figure 6). Therefore, we propose that the very slow diffusing (D < 0.01µm2/s) population are those that rotationally diffuse along the DNA, sampling for damage using a wedge amino acid residue (Figure 7). As further support for this idea, the diffusive exponent (α) for the wild-type glycosylase population containing the wedge residue is much <1, suggestive of sub-diffusive behavior even at the shortest time scales (Figure 1E, inset). Although, it may be possible that sub-diffusive motion could arise from the glycosylase occasionally being restricted between proximal intercalated YOYO-1 dye molecules, it has been shown in other single molecule experiments that the motion of the mismatch repair complex Msh2–Msh6 was not affected by YOYO-1 intercalation (52). Therefore, we favor a model where the glycosylase diffuses freely by rotation until it encounters sites along the DNA where the F111 insertion confines the glycosylase during the base interrogation phase. A similar effective ‘trap’ has been previously proposed to explain a particle's sub-diffusive behavior (63,64). Specifically, Barbi et al. (63) propose that DNA binding proteins locate their target site in a sequence-dependent manner by diffusion that is punctuated by the protein pausing at discrete sites along the DNA due to variations in hydrogen bonding between the protein and DNA, thus exhibiting motion that is sub-diffusive at short time scales with a diffusive exponent of ∼0.5 (63), comparable to the Fpg population lost by the F111A mutation. Therefore, the continuum in diffusive behavior (Figure 5), described by variations in α and D may reflect the glycosylase's ‘duty ratio’, i.e. the proportion of its bound lifetime that the glycosylase probes the DNA helix for base damage. Thus, frequent interrogation may be characterized by low α and D, whereas infrequent interrogation may result in faster, freely diffusing (α ∼ 1) glycosylases.
Figure 7.

The glycosylase search model. (A) Glycosylases rotate around the DNA molecule interrogating bases (red) or by non-specific rotation when interrogation does not occur (blue). (B) A structural model of Fpg glycosylase undergoing slow diffusion, sampling for damage using a wedge amino acid (red) or fast diffusion without intrahelical interrogation (blue) (PDB 2F5O).
Rapid target location through facilitated diffusion has been inferred from kinetics data (33,56,65–67) as well as through direct visualization (9,10,39,47–49,52,68). It has been proposed that a combination of 3D ‘jumping’ or ‘hopping’ and 1D ‘sliding’ non-specifically over DNA dramatically increases the rate of target location. If hopping was the predominate mode of search then the glycosylase's diffusion constant would increase with increasing salt concentrations as it would spend more time in an unbound state, free to move greater distances along the DNA. This was not observed for hOGG1 glycosylase (9) nor did we observe a salt effect on the diffusion constants of Nth, Nei and Fpg (Figure 5). However, increasing salt concentration did reduce the bound lifetime of a glycosylase encounter with DNA (Figure 3). The Nth, Nei and Fpg bound lifetimes reported here are an order of magnitude longer than previously reported for hOGG1 glycosylase (9). The use of constant hydrodynamic flow to maintain elongated DNA molecules in the hOGG1 studies may have shortened the bound lifetimes (9,10), as was demonstrated with restriction endonuclease EcoRV (47). With ionic strength affecting only the glycosylase's bound lifetime and not its diffusion constant, underlying structural interactions that govern diffusion and dissociation of the glycosylase from DNA may be different, with dissociation more dependent on electrostatic interactions.
CONCLUSIONS
Efficient target location by two families of DNA glycosylases appears to be accomplished through rotational diffusion, as previously suggested (9,10). During this scanning process, glycosylases may intrahelically examine DNA, which in E. coli Fpg may be accomplished primarily through Phe111 insertion. The Phe111 insertion may act as a ‘phonograph needle’ to sense the topography of the minor groove, pausing either to check for damage at random locations or in response to subtle deformations of the DNA helix. This search mechanism appears to answer the question of how glycosylases find single damages amongst an overwhelming number of undamaged DNA bases.
We have found on average that the activation energy barrier for Nth, Nei and Fpg is ∼1.5 kBT at physiological salt concentrations (see Supplementary Data). This activation energy barrier is below the theoretical maximum of ∼2 kBT for efficient search (67), however ample time to search the E. coli genome can be achieved with much higher energy barriers (39). Previous studies have estimated the 1D diffusion maximum for a glycosylase to be 0.98 µm2/s or 8.5 × 106 bp2/s (61). With the addition of our estimated activation energy barrier, we calculate the mean diffusion constant for the slow sub-diffusive population of Nth, Nei and Fpg to be Dslow = 1.3 × 106 bp2/s. Given this diffusion constant and the observed mean binding lifetime of 1.3 s, we predict that a slow diffusing glycosylase, which may actually be interrogating the DNA for damage, to have a mean scan distance of ∼1600 bp [see Supplementary Data in ref. (9)]. Given that there are ∼400 molecules of each glycosylase per E. coli cell (69) and both strands of the DNA need to be scanned, it would take ∼16 s for each population of Nth, Nei and Fpg molecules to interrogate the E. coli genome searching for damages they recognize. This does not take into account the many other complexities that may contribute to damage location, such as the presence of other proteins bound to the DNA. A future challenge will be to develop and implement a method for observing glycosylases removing engineered damaged bases in known sites along the DNA. Through such studies, visualizing the targeting, damage identification and removal of a single base at a time will provide critical insight to the molecular mechanism of glycosylase activity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (ES007122 to A.R.D.; HL059408 to D.M.W.; CA098993 to S.S.W.). Funding for open access charge: National Institutes of Health P01 CA098993.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank April Averill for protein preparation and purification; Sam Walcott and Jeffrey Bond for helpful discussions and theoretical modeling; Stephanie Duclos, Scott Kathe, Jake Gockley and Samantha Beck for superb technical assistance; Guy Kennedy for his microscope expertise; and Jeffrey Blasidell for MSD analysis software Lindifplot (software available upon request).
REFERENCES
- 1.Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 3.Fromme JC, Verdine GL. Base excision repair. Adv. Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T, editors. DNA Repair and Mutagenesis. 2nd edn. Washington, DC: American Society for Microbiology; 2006. [Google Scholar]
- 5.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 6.von Hippel PH, Berg OG. Facilitated target location in biological systems. J. Biol. Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 7.Riggs AD, Bourgeois S, Cohn M. The lac repressor-operator interaction. 3. Kinetic studies. J. Mol. Biol. 1970;53:401–417. doi: 10.1016/0022-2836(70)90074-4. [DOI] [PubMed] [Google Scholar]
- 8.Zharkov DO, Grollman AP. The DNA trackwalkers: principles of lesion search and recognition by DNA glycosylases. Mutat Res. 2005;577:24–54. doi: 10.1016/j.mrfmmm.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl Acad. Sci. USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blainey PC, Luo G, Kou SC, Mangel WF, Verdine GL, Bagchi B, Xie XS. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009;16:1224–1229. doi: 10.1038/nsmb.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo CF, McRee DE, Fisher CL, O'Handley SF, Cunningham RP, Tainer JA. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- 12.Fromme JC, Verdine GL. Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J. 2003;22:3461–3471. doi: 10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armel PR, Strniste GF, Wallace SS. Studies on Escherichia coli x-ray endonuclease specificity. Roles of hydroxyl and reducing radicals in the production of DNA lesions. Radiat Res. 1977;69:328–338. [PubMed] [Google Scholar]
- 15.Gates FT, Linn S. Endonuclease from Escherichia coli that acts specifically upon duplex DNA damaged by ultraviolet light, osmium tetroxide, acid, or x-rays. J. Biol. Chem. 1977;252:2802–2807. [PubMed] [Google Scholar]
- 16.Katcher HL, Wallace SS. Characterization of the Escherichia coli X-ray endonuclease, endonuclease III. Biochemistry. 1983;22:4071–4081. doi: 10.1021/bi00286a013. [DOI] [PubMed] [Google Scholar]
- 17.Gilboa R, Zharkov DO, Golan G, Fernandes AS, Gerchman SE, Matz E, Kycia JH, Grollman AP, Shoham G. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. J. Biol. Chem. 2002;277:19811–19816. doi: 10.1074/jbc.M202058200. [DOI] [PubMed] [Google Scholar]
- 18.Serre L, Pereira de Jesus K, Boiteux S, Zelwer C, Castaing B. Crystal structure of the Lactococcus lactis formamidopyrimidine-DNA glycosylase bound to an abasic site analogue-containing DNA. EMBO J. 2002;21:2854–2865. doi: 10.1093/emboj/cdf304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugahara M, Mikawa T, Kumasaka T, Yamamoto M, Kato R, Fukuyama K, Inoue Y, Kuramitsu S. Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8. EMBO J. 2000;19:3857–3869. doi: 10.1093/emboj/19.15.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zharkov DO, Golan G, Gilboa R, Fernandes AS, Gerchman SE, Kycia JH, Rieger RA, Grollman AP, Shoham G. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 2002;21:789–800. doi: 10.1093/emboj/21.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breimer LH. Enzymatic excision from gamma-irradiated polydeoxyribonucleotides of adenine residues whose imidazole rings have been ruptured. Nucleic Acids Res. 1984;12:6359–6367. doi: 10.1093/nar/12.16.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chetsanga CJ, Lindahl T. Release of 7-methylguanine residues whose imidazole rings have been opened from damaged DNA by a DNA glycosylase from Escherichia coli. Nucleic Acids Res. 1979;6:3673–3684. doi: 10.1093/nar/6.11.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung MH, Kasai H, Jones DS, Inoue H, Ishikawa H, Ohtsuka E, Nishimura S. An endonuclease activity of Escherichia coli that specifically removes 8-hydroxyguanine residues from DNA. Mutat Res. 1991;254:1–12. doi: 10.1016/0921-8777(91)90035-n. [DOI] [PubMed] [Google Scholar]
- 24.Tchou J, Kasai H, Shibutani S, Chung MH, Laval J, Grollman AP, Nishimura S. 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl Acad. Sci. USA. 1991;88:4690–4694. doi: 10.1073/pnas.88.11.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melamede RJ, Hatahet Z, Kow YW, Ide H, Wallace SS. Isolation and characterization of endonuclease VIII from Escherichia coli. Biochemistry. 1994;33:1255–1264. doi: 10.1021/bi00171a028. [DOI] [PubMed] [Google Scholar]
- 26.Jiang D, Hatahet Z, Melamede RJ, Kow YW, Wallace SS. Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem. 1997;272:32230–32239. doi: 10.1074/jbc.272.51.32230. [DOI] [PubMed] [Google Scholar]
- 27.Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, Mitra S. Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res. 2001;29:1967–1974. doi: 10.1093/nar/29.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leipold MD, Muller JG, Burrows CJ, David SS. Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- 29.Perry JJ, Cotner-Gohara E, Ellenberger T, Tainer JA. Structural dynamics in DNA damage signaling and repair. Curr. Opin. Struct. Biol. 2010;20:283–294. doi: 10.1016/j.sbi.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair. 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Yang W. Structure and mechanism for DNA lesion recognition. Cell Res. 2008;18:184–197. doi: 10.1038/cr.2007.116. [DOI] [PubMed] [Google Scholar]
- 32.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc. Natl Acad. Sci. USA. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerjee A, Santos WL, Verdine GL. Structure of a DNA glycosylase searching for lesions. Science. 2006;311:1153–1157. doi: 10.1126/science.1120288. [DOI] [PubMed] [Google Scholar]
- 35.Qi Y, Spong MC, Nam K, Karplus M, Verdine GL. Entrapment and structure of an extrahelical guanine attempting to enter the active site of a bacterial DNA glycosylase, MutM. J. Biol. Chem. 2010;285:1468–1478. doi: 10.1074/jbc.M109.069799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bandaru V, Blaisdell JO, Wallace SS. Oxidative DNA glycosylases: recipes from cloning to characterization. Methods Enzymol. 2006;408:15–33. doi: 10.1016/S0076-6879(06)08002-5. [DOI] [PubMed] [Google Scholar]
- 37.Blaisdell JO, Wallace SS. Rapid determination of the active fraction of DNA repair glycosylases: a novel fluorescence assay for trapped intermediates. Nucleic Acids Res. 2007;35:1601–1611. doi: 10.1093/nar/gkm021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kathe SD, Barrantes-Reynolds R, Jaruga P, Newton MR, Burrows CJ, Bandaru V, Dizdaroglu M, Bond JP, Wallace SS. Plant and fungal Fpg homologs are formamidopyrimidine DNA glycosylases but not 8-oxoguanine DNA glycosylases. DNA Repair. 2009;8:643–653. doi: 10.1016/j.dnarep.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol. Cell. 2010;37:702–713. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 41.Sage D, Neumann FR, Hediger F, Gasser SM, Unser M. Automatic tracking of individual fluorescence particles: application to the study of chromosome dynamics. IEEE Trans. Image Process. 2005;14:1372–1383. doi: 10.1109/tip.2005.852787. [DOI] [PubMed] [Google Scholar]
- 42.Weihs D, Teitell MA, Mason TG. Simulations of complex particle transport in heterogeneous active liquids. Microfluid Nanofluidics. 2007;3:227–237. [Google Scholar]
- 43.Saxton MJ, Jacobson K. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- 44.Larsson A, Carlsson C, Jonsson M. Characterization of the binding of YO to [poly(dA-dT)]2 and [poly(dG-dC)]2, and of the fluorescent properties of YO and YOYO complexed with the polynucleotides and double-stranded DNA. Biopolymers. 1995;36:153–167. doi: 10.1002/bip.360360205. [DOI] [PubMed] [Google Scholar]
- 45.Shimizu M, Sasaki S, Kinjo M. Triplet fraction buildup effect of the DNA-YOYO complex studied with fluorescence correlation spectroscopy. Anal. Biochem. 2007;366:87–92. doi: 10.1016/j.ab.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 46.Muramatsu H, Homma K, Yamamoto N, Wang J, Sakata-Sogawa K, Shimamoto N. Imaging of DNA molecules by scanning near-field microscope. Mater. Sci. Eng. 2000;12:29–32. [Google Scholar]
- 47.Bonnet I, Biebricher A, Porte PL, Loverdo C, Benichou O, Voituriez R, Escude C, Wende W, Pingoud A, Desbiolles P. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008;36:4118–4127. doi: 10.1093/nar/gkn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang YM, Austin RH, Cox EC. Single molecule measurements of repressor protein 1D diffusion on DNA. Phys. Rev. Lett. 2006;97:048302. doi: 10.1103/PhysRevLett.97.048302. [DOI] [PubMed] [Google Scholar]
- 49.Kim JH, Larson RG. Single-molecule analysis of 1D diffusion and transcription elongation of T7 RNA polymerase along individual stretched DNA molecules. Nucleic Acids Res. 2007;35:3848–3858. doi: 10.1093/nar/gkm332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabata H, Kurosawa O, Arai I, Washizu M, Margarson SA, Glass RE, Shimamoto N. Visualization of single molecules of RNA polymerase sliding along DNA. Science. 1993;262:1561–1563. doi: 10.1126/science.8248804. [DOI] [PubMed] [Google Scholar]
- 51.Graneli A, Yeykal CC, Robertson RB, Greene EC. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl Acad. Sci. USA. 2006;103:1221–1226. doi: 10.1073/pnas.0508366103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banks DS, Fradin C. Anomalous diffusion of proteins due to molecular crowding. Biophys. J. 2005;89:2960–2971. doi: 10.1529/biophysj.104.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saxton MJ. Anomalous diffusion due to obstacles: a Monte Carlo study. Biophys. J. 1994;66:394–401. doi: 10.1016/s0006-3495(94)80789-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fromme JC, Verdine GL. DNA lesion recognition by the bacterial repair enzyme MutM. J. Biol. Chem. 2003;278:51543–51548. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- 56.Sidorenko VS, Zharkov DO. Correlated cleavage of damaged DNA by bacterial and human 8-oxoguanine-DNA glycosylases. Biochemistry. 2008;47:8970–8976. doi: 10.1021/bi800569e. [DOI] [PubMed] [Google Scholar]
- 57.Verdine GL, Bruner SD. How do DNA repair proteins locate damaged bases in the genome? Chem. Biol. 1997;4:329–334. doi: 10.1016/s1074-5521(97)90123-x. [DOI] [PubMed] [Google Scholar]
- 58.Cao C, Jiang YL, Stivers JT, Song F. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 59.Koval VV, Kuznetsov NA, Ishchenko AA, Saparbaev MK, Fedorova OS. Real-time studies of conformational dynamics of the repair enzyme E. coli formamidopyrimidine-DNA glycosylase and its DNA complexes during catalytic cycle. Mutat. Res. 2010;685:3–10. doi: 10.1016/j.mrfmmm.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 60.Hedglin M, O'Brien PJ. Hopping enables a DNA repair glycosylase to search both strands and bypass a bound protein. ACS Chem. Biol. 2010;5:427–436. doi: 10.1021/cb1000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bagchi B, Blainey PC, Xie XS. Diffusion constant of a nonspecifically bound protein undergoing curvilinear motion along DNA. J. Phys. Chem. B. 2008;112:6282–6284. doi: 10.1021/jp077568f. [DOI] [PubMed] [Google Scholar]
- 62.Schurr JM. The one-dimensional diffusion coefficient of proteins absorbed on DNA. Hydrodynamic considerations. Biophys. Chem. 1979;9:413–414. [PubMed] [Google Scholar]
- 63.Barbi M, Place C, Popkov V, Salerno M. A model of sequence-dependent protein diffusion along DNA. J. Biol. Phys. 2004;30:203–226. doi: 10.1023/B:JOBP.0000046728.51620.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saxton MJ. Anomalous diffusion due to binding: a Monte Carlo study. Biophys. J. 1996;70:1250–1262. doi: 10.1016/S0006-3495(96)79682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Berg,H. (1993) Random walks in biology. Princeton University Press, Princeton.
- 66.Hughes BD. Random Walks and Random Environments. New York: Clarendon Press; 1995. [Google Scholar]
- 67.Slutsky M, Mirny LA. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Oijen AM, Blainey PC, Crampton DJ, Richardson CC, Ellenberger T, Xie XS. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–1238. doi: 10.1126/science.1084387. [DOI] [PubMed] [Google Scholar]
- 69.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 70.Narayana N, Weiss MA. Crystallographic analysis of a sex-specific enhancer element: sequence-dependent DNA structure, hydration, and dynamics. J. Mol. Biol. 2009;385:469–490. doi: 10.1016/j.jmb.2008.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.