

Abstract
The homoprotocatechuate 2,3-dioxygenase from Arthrobacter globiformis (MndD) catalyzes the oxidative ring cleavage reaction of its catechol substrate in an extradiol fashion. While this reactivity is more typically associated with nonheme iron enzymes, MndD exhibits an unusual specificity for manganese(II). MndD is structurally very similar to the iron(II)-dependent homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum (HPCD), and we have previously shown that both MndD and HPCD are equally active towards substrate turnover with either Fe(II) or Mn(II) [J P Emerson, et al. (2008) Proc Natl Acad Sci USA, 105: 7347–7352]. However, expression of MndD in E. coli under aerobic conditions in the presence of excess iron results in the isolation of inactive blue-green Fe-substituted MndD (BG-FeMndD). Spectroscopic studies indicate that this form of Fe-substituted MndD contains an Fe(III) center with a bound catecholate, which is presumably generated by in vivo self-hydroxylation of a second-sphere tyrosine residue, as found for other self-hydroxylated nonheme iron oxygenases,. The absence of this modification in either the native Mn-containing MndD or Fe-containing HPCD suggests that the metal center of Fe-substituted MndD is able to bind and activate O2 in the absence of its substrate, employing a high-valent oxoiron oxidant to carry out the observed self-hydroxylation chemistry. These results demonstrate that the active site metal in MndD can support two dramatically different O2 activation pathways, further highlighting the catalytic flexibility of enzymes containing a 2-His-1-carboxylate facial triad metal binding motif.
Keywords: 2-His-1-carboxylate facial triad, extradiol dioxygenase, homoprotocatechuate 2, 3-dioxygenase, nonheme iron enzymes, self-hydroxylation
Introduction
Mononuclear nonheme iron enzymes that activate O2 have attracted recent interest, primarily as a consequence of the large range of metabolically and environmentally important transformations that they catalyze [1–4]. This superfamily of enzymes almost universally shares a common iron(II) binding site referred to as the 2-His-1-carboxylate facial triad [5]. This binding motif provides three adjacent sites on the metal center to bind substrate, cofactor, and/or O2 in proximity to one another in order to tune and direct the desired oxidative outcome. Mechanistic studies [1, 2, 5, 6] have suggested a common framework for O2 activation by this enzyme superfamily that involves initial formation of a metal-superoxo species upon O2 binding to the iron(II) center that gives rise to one of several possible oxidants, depending on the specific enzyme in question. For instance, the ultimate oxidant for -ketoglutarate(KG)-dependent dioxygenases is an oxoiron(IV) species capable of attacking strong substrate C–H bonds [2, 5, 7–10]. On the other hand, the oxidative cleavage of aromatic rings catalyzed by extradiol cleaving catechol dioxygenases involves lower-valent metal-superoxo and metal-peroxo moieties [4, 6, 11, 12]. Direct support for the extradiol dioxygenase reaction pathway has been obtained by Kovaleva and Lipscomb from X-ray crystallographic studies of the iron(II)-dependent homoprotocatechuate 2,3-dioxygenase (HPCD) from Brevibacterium fuscum. Using an electron-deficient 4-nitrocatechol (4NC) substrate, they found direct evidence for the formation of both the iron(II)-superoxo-semiquinone and iron(II)-alkylperoxo species in crystallo [13, 14], yielding the first structurally characterized intermediates for this family of enzymes.
We have had a long-standing interest in the homoprotocatechuate 2,3-dioxygenase from Arthrobacter globiformis (MndD), an extradiol dioxygenase that exhibits an unusual preference for divalent manganese, rather than the more common iron(II) cofactor found in HPCD [15–17]. Both enzymes catalyze oxidative C–C bond cleavage of homoprotocatechuate (HPCA) to 5-carboxymethyl-2-hydroxymuconic semialdehyde (5–CHMSA) (Scheme 1). Surprisingly, MndD and HPCD share 82% sequence identity [18, 19], and X-ray crystallographic studies established that the two enzymes are isostructural in both their as-isolated forms and in complex with substrate (HPCA) [20]. We later found that substitution of the nonnative metal into both MndD and HPCD, giving Fe(II)-MndD and Mn(II)-HPCD, resulted in enzyme preparations having steady-state activities towards HPCA cleavage identical to those of the native forms, along with spectroscopic properties that suggested that metal exchange did not appreciably alter the active site structure. These striking observations led to a proposed mechanism in which the Mn(II) and Fe(II) centers of MndD and HPCD, respectively, act as conduits for electron flow between O2 and the HPCA substrate [21]. This mechanism is further supported by our recent discovery that Co(II)-substituted HPCD is comparably active in HPCA ring-cleavage chemistry [22]. The accumulated experimental data reported to date for HPCD and MndD collectively suggests that this pair of homologous enzymes represents an attractive platform for obtaining further molecular-level insights into the factors that modulate O2 activation chemistry at transition metal centers.
Scheme 1.
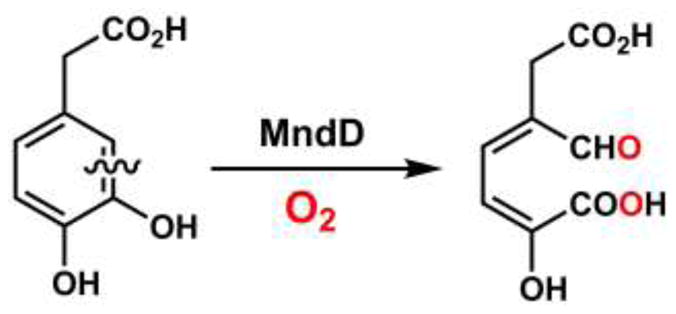
Extradiol catechol cleavage reaction catalyzed by homoprotocatechuate 2,3-dioxygenase from Arthrobacter globiformis (MndD).
In this work, we describe the generation of a unique blue-green form of Fe-substituted MndD (BG-FeMndD) generated in vivo under aerobic expression conditions in E. coli. Characterization of this species using several spectroscopic techniques suggests that the chromophore arises from a post-translationally generated self-hydroxylated form of FeMndD, indicating that the active center of Fe(II)-MndD can support oxygen activation chemistry that is clearly different from that of the typical extradiol dioxygenase chemistry. This unexpected reactivity provides further evidence for an altered, lowered reduction potential for the metal center in Fe(II)-MndD compared to Mn(II)-MndD.
Materials and Methods
Reagents and General Procedures
Reagents and buffers were of the highest grade commercially available and were used as received. All solutions and media were prepared using water purified by a Millipore Ultrapurification system. E. coli DH5 cells harboring the pYB2 plasmid [16] for the A. globiformis MndD were cultured in media containing 100 mg/L ampicillin.
Preparation of Fe-MndD
Fe-substituted MndD was over-expressed and purified as described previously [21], with some modifications. 4 – 5 mL of LB media containing 100 mg/L ampicillin were inoculated with a single colony from a fresh LB/agar plate of E. coli DH5 harboring the pYB2 plasmid for A. globiformis MndD and allowed to grow at 37 °C with shaking for ca. 5 hours. This culture was then transferred to 4 × 75 mL M9 minimal medium (100 mg/L ampicillin) and allowed to grow overnight at 37 °C with shaking for periods of 12 to 16 hours. BG-FeMndD was generated by transferring the overnight cultures to 4 × 1 L of M9 minimal medium (100 mg/L ampicillin) and allowing the cultures to grow aerobically with shaking at 37 °C until OD600 reached 0.5 – 0.7. At this point, 50 mg/L of isopropyl -D-1-thiogalactopyranoside (IPTG) and 30 mg/L of Fe(NH4)2(SO4)2 were added, and the culture was allowed to grow for periods of up to 24 hours prior to harvesting by centrifugation. The colorless form of Fe(II)-MndD (Fe(II)-MndDcolorless) was obtained using a similar approach, except that the large scale growths were carried out anaerobically. Specifically, the overnight culture was transferred to 4 L of M9 minimal medium (100 mg/L ampicillin) in a 5 L fermenter (Bioflow 2000 Fermentor, New Brunswick Scientific) which had been thoroughly sparged with N2 and the entire growth was carried out under a low flow of N2 gas (less than 0.5 L/min) to exclude O2. Anaerobic cell cultures were allowed to grow for 24 hours past induction to ensure a good yield of the colorless enzyme before being harvested via centrifugation. Isolation and purification of both the blue-green and colorless forms of Fe-substituted MndD was carried out aerobically using previously described methods [19].
Metal and Protein Analysis
The metal content of all proteins was determined by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) performed at the Soils Research Analytical Laboratory (University of Minnesota, St. Paul). Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Laboratories, California), with bovine serum albumin (BSA) as a standard. Samples were prepared as described previously [17]. The molar absorptivity at 280 nm of BG-FeMndD preparations was found to be 40000 M−1cm−1, and this value was routinely used to quantify BG-FeMndD concentrations in subsequent studies.
Enzyme Assays and Activation/Inactivation Studies
Activity studies were conducted using spectroscopic changes associated with the rate of formation of 5-CHMSA, the HPCA ring cleavage product, which has a molar extinction coefficient of ε380 ~ 36000 M−1cm−1 in MOPS buffer (pH 7.5). All measurements were carried out at room temperature (23 °C) on a Beckman DU 640 spectrophotometer. All assays employed 100 – 300 nM enzyme in air saturated 50 mM MOPS buffer (pH 7.5) unless stated otherwise. The steady-state kinetic parameters were determined by using non-linear regression techniques to fit a Michaelis-Menten curve to the experimentally derived initial velocities of 5-CHMSA formation plotted versus the concentration of HPCA in the assay mixture.
EPR and Resonance Raman Spectroscopy
EPR samples were prepared by adding 300 L of the as isolated enzyme (200 – 300 M) solution in quartz tubes with an inner diameter of 3 mm, which were subsequently frozen by slow immersion in liquid nitrogen. X-band EPR spectra (9.64 GHz) were measured at liquid helium temperature (4 K) in perpendicular applied magnetic fields on a Bruker E-500 spectrometer equipped with an Oxford ESR-10 cryostat.
Resonance Raman spectra were collected on an Acton AM-506 spectrometer (1200 groove grating) using Kaiser Optical Systems holographic supernotch filters and a Princeton Instruments liquid N2-cooled CCD detector (LN-1100 PB) with 4 cm−1 spectral resolution. Laser excitation lines at 100 mW power were obtained with a Spectra Physics 2060-KR-V krypton ion laser or a Spectra Physics 2030-15 argon ion laser and a 375B continuous wave dye (Rhodamine 6G). Raman spectra were collected at room temperature by 90° scattering from a spinning flat-bottomed NMR tube, and the Raman frequencies were referenced to indene with an accuracy of ± 1 cm−1. For each sample the entire spectral range was obtained by collecting spectra at two different frequency windows, each containing the nonresonance enhanced 1005 cm−1 band corresponding to a phenylalanine ring mode [23]. Laser exposure totaled 60 to 90 min. for each window. In each spectral window, the fluorescent background was subtracted prior to normalizing the intensity of the 1005 cm−1 bands and splicing the spectra together. Baseline corrections (polynomial fits) and curve fits (Gaussian functions) to ascertain peak positions were performed using Grams/32 Spectral Notebook (ThermoGalactic). Excitation profiles were generated by comparing peak intensities to that of the nonresonance enhanced vibration at 1005 cm−1.
Results
Properties of Aerobically Grown Fe-Substituted MndD
We previously obtained Fe-substituted MndD by growing E. coli DH5 [pYB2] cells in iron-enriched minimal media under anaerobic conditions. Isolation and purification afforded colorless Fe(II)-MndD enzyme preparations that were readily inactivated by air exposure to afford Fe(III)-MndD (Fe(III)-MndDcolorless), which could be fully reactivated by treatment with appropriate reductants such as ascorbate [21]. Conversely, growth of E. coli DH5 [pYB2] cells in iron-enriched minimal media under aerobic conditions leads to isolation of a bluish-green colored protein (BG-FeMndD). BG-FeMndD exhibits a broad visible absorption centered at 675 nm with a molar extinction coefficient of approximately 750 M−1cm−1 on a per monomer basis (Figure 1). Interestingly, while the blue green enzyme can be obtained from cells harvested four hours after induction with IPTG, the yield of this form is maximized for cells grown up to 24 hours following induction, implying that the biochemical process generating the green protein occurs fairly slowly in vivo. We have been unable to identify conditions under which BG-FeMndD can be generated in vitro; extended exposure of fully reduced Fe(II)-MndD to air simply results in oxidation of the iron center to iron(III), but without any apparent color change.
Figure 1.

UV-Vis absorption spectrum of blue-green iron-substituted MndD (BG-FeMndD). The inset shows an expansion of the visible region of the spectrum.
ICP-AES metal analyses indicate that our preparations of BG-FeMndD typically contain ca. 0.6 equiv of iron and trace amounts of manganese. BG-FeMndD exhibits weak steady-state reactivity towards HPCA, with a turnover number (kcat) of ca. 10 min−1 on a per metal per monomer basis, considerably lower than the value of 420 min−1 previously reported for fully reduced Fe(II)-MndD, but comparable to the value of 29 min−1 obtained for Fe(III)-MndDcolorless (the residual activity observed for typical Fe(III)-MndDcolorless preparations can be assigned to residual Fe(II)-MndD that was not air-oxidized) [21]. Unlike Fe(III)-MndDcolorless, treatment of BG-FeMndD with 20-fold excesses of ascorbate or the strong reductant dithionite results in only modest increases in HPCA cleavage activity, affording kcat values of ~ 20 – 30 min−1. Pre-treatment of stock solutions of BG-FeMndD with 20 equiv. of H2O2 resulted in further diminution of as-isolated enzyme activity by about 30%. These observations strongly suggest that the majority of enzyme-bound iron is in the form of iron(III), and we may further conclude that the modest amount of activity observed for BG-FeMndD arises from small amounts of enzyme-bound iron(II), as was the case for Fe(III)-MndDcolorless, as well as contaminating Mn(II)-MndD. However, our inability to reduce the iron center of BG-FeMndD points either to an active site that is no longer accessible to small molecule reductants, or else to an iron(III) site possessing a significantly depressed reduction potential relative to the colorless form, perhaps as a consequence of changes in iron-binding ligands. Treatment of the as-isolated enzyme with exogenous anions such as cyanide, azide, or fluoride did not perturb either the position or intensity of the green chromophore centered at 675 nm, consistent with the notion of a coordinatively saturated or inaccessible iron center.
The green color and depressed reactivity of BG-FeMndD are reminiscent of other mononuclear nonheme iron enzymes that have been shown to oxidatively inactivate themselves via hydroxylation of a protein derived aromatic amino acid in close proximity to the active site [24]. We therefore resorted to EPR and resonance Raman spectroscopies to further characterize the putative Fe(III) center in BG-FeMndD.
Spectroscopic Studies of BG-FeMndD
Electron paramagnetic resonance (EPR) spectroscopy is an obvious technique to test for the presence of iron(III) in our preparations of BG-FeMndD. The X-band EPR spectrum of BG-FeMndD at 4K in perpendicular applied fields exhibits prominent absorption features at g = 8.3, 7.8, and 5.6, a derivative-shaped feature at g = 4.3, and an extremely broad depression centered at g = 2.8 (Figure 2). These EPR features arise from moderately axial high spin Fe(III) (S = 5/2) centers present in the sample with transitions involving both the ground and first excited Kramers doublet. Both the g = 8.3 feature and g = 7.8 feature correspond to ground state transitions involving two different S = 5/2 centers having E/D values of 0.12 and 0.085, respectively, while the asymmetric g = 5.6 feature is associated with transitions in the first excited Kramers doublet for both of the E/D = 0.12 and 0.085 species. We note that the intensity of the g = 4.3 signal is weak, indicating that BG-FeMndD does not contain a significant quantity of a highly rhombic (E/D ~ 0.33) Fe(III) center. Fe(III)-MndDcolorless exhibits a similar EPR spectrum with features at g = 8.4, 8.3, 6.5, 5.6, 4.3, and 2.0 consistent with the presence of multiple S = 5/2 Fe(III) centers in the enzyme, though Fe(III)-MndDcolorless exhibits a much more intense g = 4.3 feature than BG-FeMndD (Figure 2), pointing towards a greater fraction of rhombic (E/D = 0.33) Fe(III) [21]. The EPR properties of BG-FeMndD are reminiscent of those reported for iron-substituted phosphomannose isomerase (blue PMI) [25], which contains a dihydroxyphenylalanine (DOPA) residue chelated to the oxidized Fe(III) center, as well as to those of oxidized tyrosine hydroxylase (TyrH) [26] and oxidized phenylalanine hydroxylase (PheH) [27] in the presence of catecholamines such as dopamine. For example, EPR studies of blue PMI revealed the presence of at least three distinct high-spin Fe(III) centers with E/D values of 0.064, 0.135, and 0.33, similar to our BG-FeMndD observations.
Figure 2.

Perpendicular-mode X-band EPR spectra of as-isolated BG-FeMndD (top) and air-oxidized Fe(III)-MndDcolorless (bottom) (data for air-oxidized Fe(III)-MndDcolorless is from reference [21]). Conditions: temperature, 4 K; microwave frequency, 9.64 GHz; microwave power, 0.2 mW (BG-FeMndD) or 1.0 mW (Fe(III)-MndDcolorless).
Resonance Raman studies of BG-FeMndD provided compelling evidence for self-hydroxylation of an aromatic amino acid in the Fe-substituted MndD active site. Laser excitation into the 675 nm chromophore afforded a set of resonance enhanced vibrations at 530, 569, 586, 646, 666, 1161, 1271, 1318, and 1423 cm−1 (Figure 3a) that gain intensity as the excitation wavelength approaches the absorption maximum (Figure 3b) The pattern of vibrations is evocative of those observed previously for the F208Y variant of RNR R2 [28], blue PMI [25], (S)-2-hydroxypropylphosphonic acid epoxidase (HppE) [29], and taurine/-ketoglutarate dioxygenase (TauD) [30–32], all of which were shown to contain DOPA moieties chelated to Fe(III) centers, as well as ferric TyrH in complex with dopamine and other catecholamines [33, 34] (Table 1). The vibrations in the 500 – 670 cm−1 region can be assigned to metal-ligand vibrations associated with the iron(III)-catecholate chelate, while features between 1100 and 1500 cm−1 reflect catecholate ring deformation modes. The pattern of vibrations is distinct from those observed for uteroferrin [35, 36] or the W48F/D84E mutant of RNR R2 [37], both of which contain iron(III)-phenolate moieties (derived from self-hydroxylation of a Phe residue in the case of W48F/D84E RNR R2), as well as that of the 2,4-dichlorophenoxyacetate/KG dioxygenase (TfdA), which contains a self-hydroxylated Trp residue (Table 1) [38]. This distinction can be made from the presence of multiple vibrations in the 500 – 670 cm−1 region, whereas iron(III) adducts with phenolates and hydroxylated Trp residues will give only a single (Fe–O) mode. Similarly, the feature at 1318 cm−1 has been shown to be uniquely associated with a catechol ring vibration [34], and is not observed in resonance Raman studies of chromophores containing phenolates or hydroxylated Trp. The excitation profiles obtained for BG-FeMndD also closely resemble those reported earlier for other examples of iron(III)-catecholate moieties in enzymes, including the TyrH-dopamine complex [34] and self-hydroxylated HppE [29]. Unfortunately, our inability to identify conditions for the preparation of BG-FeMndD in vitro precluded the use of oxygen isotope (H218O, 18O2) labeling methods to gain further insight into the vibrational properties of the bound DOPA moiety. Nonetheless, our resonance Raman data provide strong evidence for self-hydroxylation of a tyrosine residue in MndD to yield an Fe(III)-DOPA adduct.
Figure 3.

(a) Resonance Raman spectrum of BG-FeMndD obtained with 632.8 nm laser excitation. (b) Excitation profiles for selected bands as a function of excitation wavelength. The intensities are relative to the normalized intensity of the non-resonance enhanced 1005 cm−1 band corresponding to a phenylalanine ring mode [23].
Table 1.
Resonance Raman vibrations of nonheme iron proteins with Fe(III)-catecholate and related chromophores.a
| Enzyme Complex | Resonance Raman Vibration (cm−1) | Reference | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fe(III)-catecholate | ||||||||||
| Fe(III)-MndD-DOPA | 530 | 569 586 |
646 | 666 | 1273 | 1318 | 1423 | 1464 | this work | |
| Fe(II)-TauD-KG + O2 | 544 | 580 | 623 | 644 | 1261 | 1314 | 1425 | 1475 | [30] | |
| Fe(II)-TauD-succinate + O2 | 534 | 582 | 623 | 646 | 1263 | 1314 | 1421 | 1474 | [32] | |
| Fe(II)-HppE + O2 | 531 | 591 | 636 | 1264 | 1315 | 1425 | 1478 | [29] | ||
| Fe(II)-[F208Y]RNR R2 + O2 | 512 | 592 | 619 | 1263 | 1319 | 1420 | 1475 | [28] | ||
| Fe(III)-TyrH-dopamine | 528 | 592 | 631 | 1275 | 1320 | 1425 | 1475 | [34] | ||
| blue PMIb | 591 | 631 | 1266 | 1330 | 1428 | 1482 | [25] | |||
| Fe(III)-phenolate and hydroxylated Trp | ||||||||||
| uteroferrin (O-Tyr) | 575 | 805 872 |
1168 1285 |
1503 | 1603 | [36] | ||||
| W48F/D84E RNR R2 (m-O-Phe) | 615 | 1281 | 1484 | 1580 | [37] | |||||
| Fe(II)-TfdA-KG + O2 (O-Trp) | 564 | 750 | 898 970 |
1240 1274 |
1344 | 1622 | [38] | |||
MndD, Mn(II)-dependent homoprotocatechuate 2,3-dioxygenase; DOPA, dihydroxyphenylalanine; KG, -ketoglutarate; TauD, taurine/KG dioxygenase; HppE, (S)-2-hydroxypropylphosphonic acid epoxidase; RNR R2, R2 subunit of ribonucleotide reductase; TyrH, tyrosine hydroxylase; PMI, phosphomannose isomerase; TfdA, 2,4-dichlorophenoxyacetate/KG dioxygenase.
PMI is a zinc(II)-dependent metalloprotein, but up to 0.5 equiv Fe/PMI monomer are present in the as-isolated enzyme.
Consideration of the X-ray crystal structures of nonheme iron oxygenases in which self-hydroxylation has been shown to occur shows that the modified residue is typically within 10 Å of the iron center, presumably reflecting the importance of proximity to the incipient iron-based oxidant [24, 39]. The crystal structure of as-isolated Mn(II)-MndD (1F1U.pdb) reveals three tyrosine residues, Tyr255 (8.3 Å distant), Tyr257 (5.2 Å), and Tyr269 (7.2 Å) within 10 Å of the Mn(II) center [20]. We have attempted LC-ESI-MS/MS mass spectrometry studies of trypsin digests of BG-FeMndD to determine whether one of these three residues, or another tyrosine elsewhere in the protein, is the target for self-hydroxylation chemistry. Unfortunately, we have been unable to obtain convincing evidence for the site of modification, as the digested fragment of interest is either not observed or else its signal intensity is extremely weak.
Discussion
In this work, we have shown that expression of the normally manganese(II)-dependent extradiol dioxygenase MndD under aerobic conditions in the presence of excess iron(II) in E. coli leads to isolation of an inactive greenish blue protein, BG-FeMndD. Based on our spectroscopic results, this chromophore can be attributed to self-hydroxylation of one of the tyrosine residues near the active site, affording an Fe(III)-DOPA adduct. Fe-substituted MndD thus represents a new addition to a growing collection of nonheme iron enzymes that can self-hydroxylate an endogenous aromatic amino acid close to the active site in the absence of their prime substrate [24].
Our results suggest that only a portion of the protein contains hydroxylated Tyr residues. The strongest evidence for this comes from the extinction coefficient associated with Fe(III)-bound DOPA, which we found to be approximately 750 M−1cm−1 at the 675 nm absorption maximum on a per monomer basis. Given that this feature is a LMCT transition specifically associated with the Fe(III) center, correcting for our typical metal occupancy of 0.6 equiv iron per monomer would afford an 675 value of 1250 M−1cm−1 on a per iron, per monomer basis, assuming that all iron is present as iron(III) with bound DOPA. This value is approximately half that of the values of 2000 – 2500 M−1cm−1 that have been reported for iron(III) model complexes containing a single catecholate ligand [40]. The incomplete modification suggested by our UV/Vis absorption data is consistent with the presence of multiple S = 5/2 Fe(III) centers as shown by EPR spectroscopy. Indeed, the majority of self-hydroxylated nonheme iron enzymes reported to date show only partial modification, based primarily on the lower extinction coefficients associated with their LMCT transitions and mass spectrometry results [24, 41]. For example, in vitro self-hydroxylation of Tyr73 of TauD affords chromophores with extinction coefficients ranging from 300 – 700 M−1cm−1 depending upon the reaction conditions employed [30–32], and a significant fraction of enzyme activity can be restored simply by treating self-hydroxylated TauD with dithionite [31]. The only examples where the iron(III)-catecholate absorptivity approaches the expected values are those where the transformation occurs in vivo, including F208Y RNR R2 ( 720 = 2500 M−1cm−1) and iron-substituted PMI ( 680 = 2100 M−1cm−1). It is, however, simplistic to attribute this to a distinction between in vivo and in vitro conditions, as illustrated by the observation that the in vivo generated chromophore of HppE exhibited a molar absorptivity at 680 nm of 680 = 450 M−1cm−1 [29].
An alternate, and perhaps more plausible, explanation is that the extent of modification will be related to the distance of the target residue from the iron center and the dynamic flexibility of the protein. This is nicely illustrated by a structural analysis of unmodified forms of F208Y RNR R2 and HppE. The crystal structure of diiron(II) R208Y RNR R2 shows direct ligation of the mutant Tyr208 residue to Fe1 of the active site [42], assuring that the target residue is close to the diiron cluster during the entire chemical cycle leading to formation of DOPA208 [28, 43]. Studies of HppE showed that both Tyr103 and Tyr105 could be modified [29], and X-ray crystallographic analysis of the unmodified HppE indicated that Tyr105 is 8.7 Å away from the iron(II) center, while Tyr103 is even more distant at 10.8 Å [44], thus suggesting that HppE must rearrange itself more extensively than F208Y RNR R2 to self-hydroxylate the target amino acids. Although a crystal structure of Fe-substituted MndD has not been reported to date, the near identity of the reported Fe(II)-HPCD, Mn(II)-HPCD, and Mn(II)-MndD structures to one another [20–22] suggests that the Fe(II)-MndD structure should be quite similar to that of Mn(II)-MndD. Although we were unsuccessful in identifying the site of self-hydroxylation in BG-FeMndD via mass spectrometry methods, there are at least three tyrosine residues, Tyr255, Tyr257, and Tyr269, within 10 Å of the metal center of MndD that could potentially be self-hydroxylated without requiring a substantial protein backbone rearrangement. Indeed, self-hydroxylation of more than one of these residues is an attractive rationale to account for the multiplicity of S = 5/2 Fe(III) centers observed by EPR, as well as the fact that the intensity of the green chromophore in BG-FeMndD is greater than those observed for other self-hydroxylated enzymes aside from F208Y RNR R2 and PMI. Also, the resonance Raman spectrum of BG-FeMndD is considerably more complex in the 500 – 700 cm−1 region than expected (Figure 3), as there are at least five distinct resonance-enhanced vibrations as opposed to the three vibrations expected for a typical Fe(III)-DOPA adduct (Table 1) [34]. This is reminiscent of earlier observations for self-hydroxylated TauD generated by reaction of Fe(II)-TauD-succinate with H2O2, in which the addition of bicarbonate anion was shown to result in the appearance of a new feature in the low-frequency region of the resonance Raman spectrum [32]. This was attributed to a downshift of the Fe–O4 vibration of the bound DOPA residue caused by bicarbonate modulation of the Lewis acidity of the Fe(III) center in modified TauD [31, 32]. A similar effect may also be operant in BG-FeMndD, such that there are two (or more) Fe(III)-DOPA adducts in slightly different environments giving rise to different Raman vibrations.
With the notable exception of iron-substituted PMI, ordinarily a zinc(II)-dependent isomerase, all reported instances of aromatic self-hydroxylation have involved nonheme iron enzymes known to employ a high-valent iron oxidant in their catalytic cycles. Although no such intermediate has been directly detected during enzyme self-hydroxylation, the oxygen-isotope incorporation patterns observed by resonance Raman spectroscopy [28–32, 37, 38] and the EPR characterization of a tyrosyl radical + Fe(III) combination in TauD in the course of self-hydroxylation [31] strongly imply the involvement of high-valent iron-oxo centers as the oxidant in these reactions [24]. How then might this chemistry occur in Fe-substituted MndD, which employs a very different O2 activation pathway for extradiol cleavage of the catechol ring [11, 12, 21]? We have found that self-hydroxylation does not occur in the native Mn(II) form of MndD, nor has it been observed to date in Fe(II)-HPCD, which exhibits a highly similar sequence and active site structure [19, 20]. The metal-substitution studies of HPCD and MndD showed that the as-isolated forms of Fe(II)-HPCD and Fe(II)-MndD were susceptible to inactivation by oxidants such as H2O2 and ferricyanide, while their Mn-substituted congeners were not oxidized. These observations logically imply that the FeIII/II reduction potentials of Fe(II)-HPCD and Fe(II)-MndD are lower than the MnIII/II potentials of the Mn(II)-HPCD and Mn(II)-MndD enzymes. This can be expected, given that the structural congruence of Fe(II)-HPCD and Mn(II)-HPCD suggests that the protein environment should alter the intrinsic aqueous FeIII/II (Eo = +0.77 V) and MnIII/II (Eo = +1.56 V) reduction potentials [45] to a comparable degree for both metal centers; this notion presumably applies to Mn(II)-MndD and Fe(II)-MndD as well. Fe(II)-MndD may also have a lower FeIII/II reduction potential relative to Fe(II)-HPCD, based on its increased susceptibility to air oxidation [21] and the generation in vivo of the self-hydroxylated BG-FeMndD described herein. The fact that BG-FeMndD is only generated during aerobic growths implies that an O2-derived oxidant is likely to be involved in effecting self-hydroxylation of Fe-substituted MndD. Similarly, as anaerobically-grown Fe(II)-MndD does not convert to the blue-green form during aerobic purification or incubation, a simple combination of iron and O2 is insufficient to catalyze self-hydroxylation. We therefore propose that intracellular biological reducing cofactors play a role in the in vivo O2 activation process that leads to BG-FeMndD. Iron-substituted PMI [25], HppE [29], and the DNA repair enzyme AlkB [46] provide precedent for this proposal, as all three have been found to self-hydroxylate in vivo, and only AlkB utilizes a well-defined biologically available cofactor ( KG). We favor the involvement of a high-valent iron-oxo intermediate in the self-hydroxylation of Fe-substituted MndD, by analogy to past examples of this transformation that suggest a potent oxidant is required to carry out this modification (Scheme 2). The lack of self-hydroxylation in Mn(II)-MndD implies that the 2-His-1-carboxylate facial triad may not be able to support analogous O2 activation chemistry at a Mn(II) center that would lead to a putative high-valent manganese-oxo oxidant. This is consistent with observations on several KG-dependent dioxygenases, in which Mn(II) was found to be unable to support substrate turnover [47, 48]. Indeed, Mn(II) substitution has frequently been used as a means to obtain crystal structures of several mononuclear nonheme oxygenase enzyme-substrate complexes such as isopenicillin N-synthase and AlkB under aerobic conditions, further highlighting the lack of O2 reactivity of Mn(II)-substituted Fe(II)-dependent oxygenases [48–50].
Scheme 2.
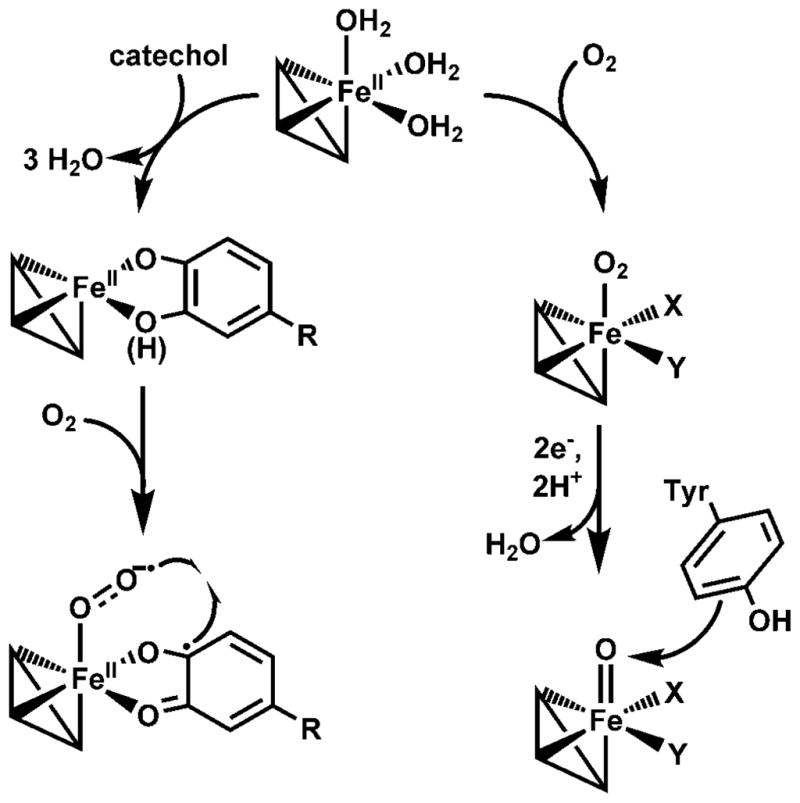
Two distinct O2 activation pathways in Fe-substituted MndD. The iron coordination sites X and Y in the structures on the right may be vacant or consist of ligands derived from solvent or some other unidentified source.
In closing, we have shown that substitution of iron into the manganese(II)-dependent extradiol dioxygenase MndD can result in the self-hydroxylation of one or more endogenous Tyr residues under in vivo conditions. This unexpected reactivity requires O2 activation to occur in the absence of its catecholic substrate, leading to the formation of a high-valent iron-oxo species capable of Tyr hydroxylation. This O2 activation mechanism differs from that associated with extradiol ring cleavage of catechols [4, 13], a reaction that the iron(II) form of FeMndD can nonetheless catalyze. The two modes of reactivity observed for Fe(II)-MndD (Scheme 2) provide a striking illustration of the diverse O2 activation processes that can be supported by the 2-His-1-carboxylate facial triad metal binding motif in a single enzyme.
Acknowledgments
This work was supported by the National Institutes of Health (grant GM 33162 to L.Q., a postdoctoral fellowship GM 072287 to J.P.E., and a pre-doctoral traineeship GM 08700 to E.R.F.). We thank Andrew Fielding for his contributions to the attempted mass spectrometry studies and insightful discussions.
Abbreviations
- KG
-ketoglutarate
- BG-FeMndD
blue-green iron-substituted MndD
- 5-CHMSA
5-carboxymethyl-2-hydroxymuconic semialdehyde
- DOPA
dihydroxyphenylalanine
- EPR
electron paramagnetic resonance
- HPCA
homoprotocatechuate or 3,4-dihydroxyphenylacetate
- HPCD
Fe(II)-dependent homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum
- HppE
(S)-2-hydroxypropylphosphonic acid epoxidase
- LC-ESI-MS/MS
liquid chromatography-electrospray ionization-mass spectrometry/mass spectrometry
- MndD
Mn(II)-dependent homoprotocatechuate 2,3-dioxygenase from Arthrobacter globiformis
- 4NC
4-nitrocatechol
- PheH
phenylalanine hydroxylase
- PMI
phosphomannose isomerase
- RNR R2
R2 subunit of ribonucleotide reductase
- TauD
taurine/KG dioxygenase
- TfdA
2,4-dichlorophenoxyacetate/KG dioxygenase
- TyrH
tyrosine hydroxylase
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 3.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 4.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koehntop KD, Emerson JP, Que L., Jr J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 6.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 8.Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM, Jr, Krebs C, Fitzpatrick PF. J Am Chem Soc. 2007;129:11334–11335. doi: 10.1021/ja074446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaillancourt FH, Bolin JT, Eltis LD. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 12.Lipscomb JD. Curr Op Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovaleva EG, Lipscomb JD. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emerson JP, Farquhar ER, Que L., Jr Angew Chem Int Ed. 2007;46:8553–8556. doi: 10.1002/anie.200703057. [DOI] [PubMed] [Google Scholar]
- 15.Olson PE, Qi B, Que L, Jr, Wackett LP. Appl Environ Microbiol. 1992;58:2820–2826. doi: 10.1128/aem.58.9.2820-2826.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boldt YR, Sadowsky MJ, Ellis LB, Que L, Jr, Wackett LP. J Bacteriol. 1995;177:1225–1232. doi: 10.1128/jb.177.5.1225-1232.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiting AK, Boldt YR, Hendrich MP, Wackett LP, Que L., Jr Biochemistry. 1996;35:160–170. doi: 10.1021/bi951979h. [DOI] [PubMed] [Google Scholar]
- 18.Miller MA, Lipscomb JD. J Biol Chem. 1996;271:5524–5535. doi: 10.1074/jbc.271.10.5524. [DOI] [PubMed] [Google Scholar]
- 19.Wang YZ, Lipscomb JD. Prot Expr Purif. 1997;10:1–9. doi: 10.1006/prep.1996.0703. [DOI] [PubMed] [Google Scholar]
- 20.Vetting MW, Wackett LP, Que L, Jr, Lipscomb JD, Ohlendorf DH. J Bacteriol. 2004;186:1945–1958. doi: 10.1128/JB.186.7.1945-1958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emerson JP, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L., Jr Proc Natl Acad Sci USA. 2008;105:7347–7352. doi: 10.1073/pnas.0711179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fielding AJ, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L., Jr J Biol Inorg Chem. 2011 doi: 10.1007/s00775-010-0732-0. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lord RC, Yu N-t. J Mol Biol. 1970;50:509–524. doi: 10.1016/0022-2836(70)90208-1. [DOI] [PubMed] [Google Scholar]
- 24.Farquhar ER, Koehntop KD, Emerson JP, Que L., Jr Biochem Biophys Res Commun. 2005;338:230–239. doi: 10.1016/j.bbrc.2005.08.191. [DOI] [PubMed] [Google Scholar]
- 25.Smith JJ, Thomson AJ, Proudfoot AE, Wells TN. Eur J Biochem. 1997;244:325–333. doi: 10.1111/j.1432-1033.1997.00325.x. [DOI] [PubMed] [Google Scholar]
- 26.Haavik J, Martinez A, Olafsdottir S, Mallet J, Flatmark T. Eur J Biochem. 1992;210:23–31. doi: 10.1111/j.1432-1033.1992.tb17386.x. [DOI] [PubMed] [Google Scholar]
- 27.Martinez A, Andersson KK, Haavik J, Flatmark T. Eur J Biochem. 1991;198:675–682. doi: 10.1111/j.1432-1033.1991.tb16066.x. [DOI] [PubMed] [Google Scholar]
- 28.Ling J, Sahlin M, Sjöberg B-M, Loehr TM, Sanders-Loehr J. J Biol Chem. 1994;269:5595–5601. [PubMed] [Google Scholar]
- 29.Liu P, Mehn MP, Yan F, Zhao Z, Que L, Jr, Liu H-w. J Am Chem Soc. 2004;126:10306–10312. doi: 10.1021/ja0475050. [DOI] [PubMed] [Google Scholar]
- 30.Ryle MJ, Liu A, Muthukumaran RB, Ho RYN, Koehntop KD, McCracken J, Que L, Jr, Hausinger RP. Biochemistry. 2003;42:1854–1862. doi: 10.1021/bi026832m. [DOI] [PubMed] [Google Scholar]
- 31.Ryle MJ, Koehntop KD, Liu A, Que L, Jr, Hausinger RP. Proc Natl Acad Sci USA. 2003;100:3790–3795. doi: 10.1073/pnas.0636740100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koehntop KD, Marimanikkuppam S, Ryle MJ, Hausinger RP, Que L., Jr J Biol Inorg Chem. 2006;11:63–72. doi: 10.1007/s00775-005-0059-4. [DOI] [PubMed] [Google Scholar]
- 33.Andersson KK, Cox DD, Que L, Jr, Flatmark T, Haavik J. J Biol Chem. 1988;263:18621–18626. [PubMed] [Google Scholar]
- 34.Michaud-Soret I, Andersson KK, Que L, Jr, Haavik J. Biochemistry. 1995;34:5504–5510. doi: 10.1021/bi00016a022. [DOI] [PubMed] [Google Scholar]
- 35.Gaber BP, Sheridan JP, Bazer FW, Roberts RM. J Biol Chem. 1979;254:8340–8342. [PubMed] [Google Scholar]
- 36.Antanaitis BC, Strekas T, Aisen P. J Biol Chem. 1982;257:3766–3770. [PubMed] [Google Scholar]
- 37.Baldwin J, Voegtli WC, Khidekel N, Moënne-Loccoz P, Krebs C, Pereira AS, Ley BA, Huynh BH, Loehr TM, Riggs-Gelasco PJ, Rosenzweig AC, Bollinger JM., Jr J Am Chem Soc. 2001;123:7017–7030. doi: 10.1021/ja002114g. [DOI] [PubMed] [Google Scholar]
- 38.Liu A, Ho RYN, Que L, Jr, Ryle MJ, Phinney BS, Hausinger RP. J Am Chem Soc. 2001;123:5126–5127. doi: 10.1021/ja005879x. [DOI] [PubMed] [Google Scholar]
- 39.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM., Jr Proc Natl Acad Sci USA. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox DD, Benkovic SJ, Bloom LM, Bradley FC, Nelson MJ, Que L, Jr, Wallick DE. J Am Chem Soc. 1988;110:2026–2032. [Google Scholar]
- 41.Chen Y-H, Comeaux LM, Eyles SJ, Knapp MJ. Chem Commun. 2008:4768–4770. doi: 10.1039/b809099h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Logan DT, deMaré F, Persson BO, Slaby A, Sjöberg B-M, Nordlund P. Biochemistry. 1998;37:10798–10807. doi: 10.1021/bi9806403. [DOI] [PubMed] [Google Scholar]
- 43.Åberg A, Ormö M, Nordlund P, Sjöberg B-M. Biochemistry. 1993;32:9845–9850. doi: 10.1021/bi00088a040. [DOI] [PubMed] [Google Scholar]
- 44.Higgins LJ, Yan F, Liu P, Liu H-w, Drennan CL. Nature. 2005;437:838–844. doi: 10.1038/nature03924. [DOI] [PubMed] [Google Scholar]
- 45.Bratsch SG. J Phys Chem Ref Data. 1989;18:1–21. [Google Scholar]
- 46.Henshaw TF, Feig M, Hausinger RP. J Inorg Biochem. 2004;98:856–861. doi: 10.1016/j.jinorgbio.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 47.Lloyd MD, Lee H-J, Harlos K, Zhang Z-H, Baldwin JE, Schofield CJ, Charnock JM, Garner CD, Hara T, Terwisscha van Scheltinga AC, Valegård K, Viklund JAC, Hajdu J, Andersson I, Danielsson Å, Bhikhabhai R. J Mol Biol. 1999;287:943–960. doi: 10.1006/jmbi.1999.2594. [DOI] [PubMed] [Google Scholar]
- 48.Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE. Nature. 1995;375:700–704. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 49.Yu B, Edstrom WC, Benach J, Hamuro Y, Weber PC, Gibney BR, Hunt JF. Nature. 2006;439:879–884. doi: 10.1038/nature04561. [DOI] [PubMed] [Google Scholar]
- 50.Yang C-G, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]