

Abstract
Transcription factors (TFs) are essential for the regulation of gene expression and often form emergent complexes to perform vital roles in cellular processes. In this paper, we focus on the parallel Max and Mlx networks of TFs because of their critical involvement in cell cycle regulation, proliferation, growth, metabolism, and apoptosis. A basic-helix-loop-helix-zipper (bHLHZ) domain mediates the competitive protein dimerization and DNA binding among Max and Mlx network members to form a complex system of cell regulation. To understand the importance of these network interactions, we identified the bHLHZ domain of Max and Mlx network proteins across the animal kingdom and carried out several multivariate statistical analyses. The presence and conservation of Max and Mlx network proteins in animal lineages stemming from the divergence of Metazoa indicate that these networks have ancient and essential functions. Phylogenetic analysis of the bHLHZ domain identified clear relationships among protein families with distinct points of radiation and divergence. Multivariate discriminant analysis further isolated specific amino acid changes within the bHLHZ domain that classify proteins, families, and network configurations. These analyses on Max and Mlx network members provide a model for characterizing the evolution of TFs involved in essential networks.
Keywords: protein evolution, basic-helix-loop-helix-leucine zipper (bHLHZ) domain, Myc/Max/Mad network, Mlx and Mondo Network, phylogenetic tree, discriminant analysis
Introduction
Organism development requires the coordination of complex biological processes involving gene regulatory, protein interaction, and metabolic networks (Barabási and Oltvai 2004; Siegal et al. 2006). Transcription factors (TFs) form important links in such networks by responding to cellular signals, recruiting cofactors to promoter regions, and regulating the transcription of target genes that determine cell function and fate. Hence, protein and DNA interactions that comprise TF networks are fundamental for proper cellular regulation.
Understanding the evolutionary dynamics of TF networks is critical for discerning the essential components regulating key pathways among organisms. Changes to TF networks are known to appreciably contribute to morphological and developmental differences observed between related species (Fujimoto et al. 2008; Maerkl and Quake 2009). Such network evolution is characterized by natural selection acting on individual members as well as their interacting partners. Consequently, different patterns of variability and conservation occur, which can alter network interactions and result in functional divergence. The ability for a TF network to withstand such perturbations over large evolutionary distances indicates the network is functionally robust and likely vital for important cellular processes (Alberghina et al. 2009).
One large superfamily of TFs characterized by the basic-helix-loop-helix (bHLH) DNA binding and dimerization domain is critical for development in almost all eukaryotes (Jones 2004). Individual bHLH proteins form dimer complexes that recognize the 5′-CANNTG-3′ E-box binding motif in promoter regions to regulate transcription of diverse gene targets. bHLH proteins are well known to contribute to neurogenesis, myogenesis, heart development, hematopoiesis, cell proliferation, and cell lineage determination (Atchley and Fitch 1997; Massari and Murre 2000; Robinson and Lopes 2000; Jones 2004; Kewley et al. 2004).
Through modular evolution, multiple domain shuffling events coupled bHLH and other domains to create a functionally heterogeneous set of TFs (Morgenstern and Atchley 1999; Moore et al. 2008). Furthermore, gene duplications, gene deletions, and changes to the bHLH domain have modified bHLH TF network interactions and altered the complexity of transcriptional regulation (Levine and Tjian 2003; Van Dam et al. 2008). For example, some bHLH proteins have a leucine zipper region (Z) adjacent to the carboxyl end of the bHLH region that stabilizes dimerization and subsequently restricts interaction between basic-helix-loop-helix-zipper (bHLHZ) proteins (Dang et al. 1989; Orian et al. 2003).
Using Max and Mlx Networks as a Model
Herein, we focus on members of the Max and Mlx networks, which form two parallel bHLHZ TF networks that are critically involved in regulating cell growth, metabolism, apoptosis, proliferation, and differentiation (table 1) (Lüscher 2001). Extensive studies in model organisms such as Mus musculus, Drosophila melanogaster, and Caenorhabditis elegans demonstrate that the Max and Mlx networks have maintained functional similarity over extensive evolutionary time, although they have evolved considerably in terms of their sequences, network membership, and complexity (Lüscher 2001).
Table 1.
Max and Mlx Network Members
| Max Network | Potential Overlapping Members | Mlx Network | |||
| Core | |||||
| Myc | Max | Mxd | Mnt | Mlx | Mondo |
| Diptera | |||||
| Myc (dMyc, dm) | Max (dMax) | Mnt (dMnt) | Mlx (dMlx) | Mondo (dMondo, Mio) | |
| Nematode | |||||
| Mxl-1 | MDL-1 | Mxl-2 | MML-1 (T20B12.6) | ||
| Mxl-3 | |||||
| Vertebratea | |||||
| c-Myc (Myc2, Niard, Nird) | Max (Myn) | Mxd1 (Mad1) | Mnt (Rox, Mad6, Mxd6) | Mlx (BigMax) | MondoA (bHLHe36, KIAA0867, MIR, MLXIP) |
| N-Myc (N-Myc1,N-Myc2, MycN) | Mxd2 (Mad2, Mxi1, Mxi) | MondoB (ChREBP, WBSCR14, MLXIPL) | |||
| L-Myc (MycL1, LMyc1) | Mxd3 (Mad3, Myx) | ||||
| Mga (KIAA0518, Mad5, Mxd5) | Mxd4 (Mad4, MSTP149, MST149) | ||||
NOTE.—Network components are listed according to their presence in the four main animal networks. Columns represent orthologous proteins between networks and paralogous proteins within. Known aliases for each protein are provided in parentheses.
Rodents have an additional N-Myc duplicate termed S-Myc, whereas primates have an L-Myc duplicate named L-Myc2. Mga has unknown origin within the vertebrate network.
Max and Mlx network members, including Max, Myc, Mnt, Mxd, Mlx, and Mondo proteins, are defined by a highly conserved C-terminus bHLHZ domain that specifies dimerization with either Max or Mlx proteins. Their bHLHZ region is defined by a 13 residue basic region (b1–13), 2 α-helices each consisting of 15 residues (H101–115, H201–215), a variable length loop (L), and a 28 residue leucine zipper (Z1–Z28) (Atchley and Fernandes 2005). Each bHLHZ monomer forms two asymmetric α-helices (bH and HZ) that can dimerize and fold into a globular left-handed four-helix bundle that can bind DNA. Still, additional dimerization restrictions and DNA-binding preferences exist for each bHLHZ protein within the interaction network.
The fruitfly D. melanogaster exhibits a minimal network consisting of single copies of dMax, dMlx, dMnt, dMyc, and dMondo genes (table 1 and fig. 1) (Peyrefitte et al. 2001). Nematodes are distantly related to flies and other arthropods in the Ecdysozoa lineage (Budd and Telford 2009), and C. elegans, for example, has a markedly different yet clearly orthologous network. This is presumably due to massive gene reduction and rearrangement that occurred in nematodes (Witherspoon and Robertson 2003; Denver et al. 2004; Coghlan 2005). In C. elegans, two Max orthologs (Mxl-1 and Mxl-3) and a single Mlx ortholog (Mxl-2) act as central dimerization partners for the Mad-like ortholog MDL-1 and Myc and Mondo-like protein MML-1, respectively (table 1 and fig. 1) (Yuan et al. 1998; Gallant 2006; Pickett et al. 2007).
FIG. 1.—

Max and Mlx network protein distribution. (A) Species tree determined by Flybase, Ensembl, and Tree of Life resources. Circled numbers correspond to the emergence of the labeled network as shown in figure 2. (B) Outlined and Gray cells indicate thatthe protein is expected to be present or absent, respectively, within the organism. “X” means the bHLHZ was found within a protein or expressed sequence tag, “*” means part of the sequence was found or all were found within a genetic region, and 0 means the protein is known to be absent.
In contrast, Max and Mlx networks in Homo sapiens and M. musculus contain several members, with paralogous families for Myc (c-, L-, and N-Myc), Mxd (Mxd1-4, formerly Mad1, Mxi1, Mad3, and Mad4), and Mondo (MondoA and MondoB), along with single copies of Max, Mlx, Mnt, and Mga genes (table 1 and fig. 1) (Gallant 2006). Rodents and humans possess additional Max interacting proteins S-Myc and L-Myc2, respectively, indicating that they sustained separate evolutionarily ancient duplication events (Depinho et al. 1987; Doskocil 1996). Another c-Myc homolog, B-Myc, exists in the murine lineage and lacks the C-terminal bHLHZ sequence. Consequently, it cannot interact with Max or bind DNA (Burton et al. 2006).
Despite differences in network structure, Max and Mlx network member domains and functions remain stable among species (Yuan et al. 1998; Gallant 2006; Steiger et al. 2008). In general, Myc and Mondo family proteins promote gene transcription by interacting with Max and Mlx, respectively, and recruiting a histone acteylase complex to their N-terminus transactivation domain (McMahon et al. 1998; Dang 1999; Billin et al. 2000; de Luis et al. 2000; Cairo et al. 2001). In an antagonistic fashion, Mnt and Mxd family proteins competitively dimerize with Max and recruit a histone deacytelase complex through an N-terminus Sin3 interaction domain that represses transcription (Hurlin et al. 1997). Although there are contradicting results regarding Mnt and Mlx dimerization (Meroni et al. 1997, 2000; Cairo et al. 2001), vertebrate Mxd1 and Mxd4 proteins can also heterodimerize with Mlx and potentially antagonize Mondo function (Billin and Ayer 2006). Because Max and Mlx have no intrinsic transcriptional activity, they ostensibly serve as obligate dimerization partners during transitions in transcriptional signaling. Hence, Max and Mlx networks differentially regulate gene transcription according to competitive dimerization and reciprocal behavior of protein members (Grinberg et al. 2004).
Mnt and Mad antagonize Myc in a general and cell-specific manner, respectively, by differentially regulating transcription for overlapping gene targets (Hurlin et al. 1997; Orian et al. 2003). Such DNA-binding specificity arises from protein-specific residues that interact with flanking regions of the canonical “CACGTG” motif. Myc shows a preference for 5′-GC, 5′-CG, or 5′-AG prior to the E-box (Lüscher and Larsson 1999), Mxd1:Max heterodimers prefer an extended “CCACGTGG” E-box (Rottmann and Lüscher 2006), whereas MondoB recognizes the carbohydrate response element (ChORE) designated by two CACGTG E-boxes separated by exactly five nucleotides (Shih and Towle 1992; Shih et al. 1995). Moreover, the synthetic lethal interaction of D. melanogaster orthologs dMondo and dMyc indicate both are necessary to regulate at least one essential gene involved in cell growth (Billin and Ayer 2006). This orchestration of Max and Mlx network members enables cells to refine the regulation of shared gene targets through a complex system of activation and repression.
The coordinated expression and dimerization of Max and Mlx network members are essential for normal development (Blackwood et al. 1992; Charron et al. 1992; Amati et al. 1993; Grandori et al. 2000; Shen-Li et al. 2000; Walker et al. 2005). Mnt and Myc family proteins are essential for proper cell growth (Pierce et al. 2004; Toyo-Oka et al. 2004; Benassayag et al. 2005; Loo et al. 2005; Pierce et al. 2008), whereas Mnt, Myc, and Mad family proteins are important for cell cycle progression (Amati and Land 1994; Hanson et al. 1994; Hurlin et al. 1995; Zhou and Hurlin 2001). In parallel, Mlx and Mondo family proteins are important in growth and energy homeostasis (Billin et al. 2000; Ma et al. 2005; Sans et al. 2006; Stoltzman et al. 2008). Although not individually essential, MondoA and MondoB are important for proper glucose metabolism and formation of triglycerides (Ma et al. 2006; Peterson et al. 2010).
The relative abundance and activity of member proteins in these two networks are tightly controlled due to the substantial effects of even some minor perturbations (Grandori et al. 2000; Hooker and Hurlin 2006). Most notably, deregulation of Myc is directly associated with oncogenesis and attributes to over 70,000 human deaths a year in the United States (Nesbit et al. 1999; Dang et al. 2006). Loss of Mnt can also result in tumor formation (Hurlin et al. 2004; Nilsson et al. 2004; Hooker and Hurlin 2006), although no significant observations have been able to classify Mad or Mnt as tumor suppressors (Schreiber-Agus et al. 1998; Rottmann and Lüscher 2006). Moreover, the central role of MondoB in lipid synthesis and glucose response implicates it as a possible contributing factor in fatty liver, obesity, and Type II diabetes (Postic et al. 2007).
Parallel and essential regulation of the Max and Mlx networks show that these TFs exhibit distinct characteristics necessary for proper cell development. The homologous bHLHZ domain is integral in distinguishing the preference of protein interactions, complex structure, and gene targets that direct downstream effects of these TFs. Still, the importance and evolution of Max and Mlx interactions are relatively unknown. The function and origin of Max-interacting protein Mga have not been formally addressed, distinctions in Mxd function and binding have yet to be determined, and ramifications of Max and Mlx network gene loss in C. elegans and D. melanogaster are uncertain.
Herein, we investigate how networks involving TFs essential for organism development change during organismal diversification over extensive evolutionary time and distances. Using phylogenetic and multivariate statistical analyses, we characterize Max and Mlx network interactions in animals by comparing the bHLHZ domain of its members across diverse species. In particular, we address several questions regarding the evolution of network structure and the bHLHZ interaction domain. Did network structure diverge in bursts of diversification or through several incremental evolutionary events? Is the DNA binding and protein–protein interaction bHLHZ domain conserved among orthologous members or in particular lineages? Finally, what residues in the bHLHZ domain restrict and distinguish potential dimerization and DNA-binding patterns?
Materials and Methods
Obtaining and Aligning Max and Mlx Network bHLHZ Sequences
Approximately 100 eukaryotic species were surveyed for Max and Mlx network members. Initial amino acid (AA) sequences were obtained from Ensembl (Flicek et al. 2010) and NCBI (Sayers et al. 2010) annotations, whereas sequences of unannotated species were gathered from eukaryotic genomic databases, that is, Joint Genome Institute (JGI 2010), Baylor, Dana Farber (Quackenbush et al. 2001), Metazome, Flybase (Tweedie et al. 2009), Vectorbase (Lawson et al. 2009), Sanger (Sanger 2010), Broad (McCarthy 2005), Washington University (2010), Wormbase (Harris et al. 2010), and Kegg (Kanehisa et al. 2010) databases (table 2). When no known ortholog was available, we performed TBlastN and BlastP (Altschul et al. 1990) queries on relevant databases using known protein sequences of similar species. Validated expressed sequence tag and predicted transcripts were given priority, followed by blast hits on scaffolds and unassembled whole genome shotgun reads. A protein was considered absent within a species if distinguishing features in the bHLHZ domain could not be identified manually (Atchley and Fernandes 2005). Note that absence in the database does not necessarily indicate absence within the organism. Rather, it could reflect inadequate sampling or sequencing of the genome.
Table 2.
Sampled Genomes
| Genus Species | Common Name | Source | Status | Published Genome | |
| Vertebrate | Homo sapiens | Human | Complete | Venter et al. (2001) | |
| Rattus norvegicus | Rat | Assembly | Gibbs et al. (2004) | ||
| Bos taurus | Cow | Assembly | Consortium Bovine Genome Sequencing and Analysis et al. (2009) | ||
| Canis familiaris | Dog | Broad | Assembly | Lindblad-Toh et al. (2005) | |
| Monodelphis domestica | Opossum | Assembly | Mikkelsen et al. (2007) | ||
| Ornithorhynchus anatinus | Duckbill platypus | WashU | Assembly | Warren et al. (2008) | |
| Gallus gallus | Chicken | Assembly | Consortium International Chicken Genome Sequencing (2004) | ||
| Anolis carolinensis | Green Anole Lizard | Assembly | |||
| Xenopus tropicalis | Western Clawed Frog | JGI | Assembly | ||
| Xenopus laevis | African Clawed Frog | ||||
| Xenopusn nigroviridis | Green pufferfish | Broad | Assembly | ||
| Danio rerio | Zebrafish | Sanger | Assembly | ||
| Callorhinchus milii | Elephantfish | Assembly | Venkatesh et al. (2007) | ||
| Core | Branchiostoma floridae | Florida lancet (Amphioxus) | JGI | Assembly | Putnam et al. (2008) |
| Ciona savignyi | Sea squirt | Broad | Assembly | ||
| Ciona intestinalis | Sea squirt | JGI | Assembly | Dehal et al. (2002) | |
| Molgula tectiformis | Sea grapes | ||||
| Strongylocentrotus purpuratus | Purple sea urchin | Baylor | Assembly | Consortium Sea Urchin Genome Sequencing et al. (2006) | |
| Asterias vulgaris | Sea star | ||||
| Diptera | Drosophila melanogaster | Fruitfly | Adams et al. (2000) | ||
| Culex pipiens | Southern house Mosquito | Broad | Assembly | ||
| Aedes aegypti | Yellow fever mosquito | TIGR | Assembly | Nene et al. (2007) | |
| Anopheles gambiae | Malaria mosquito | Complete | Sharakhova et al. (2007) | ||
| Core | Bombyx mori | Silkworm moth | Assembly | Consortium International Silkworm Genome (2008) | |
| Tribolium castaneum | Red flour beetle | Baylor | Assembly | Consortium Tribolium Genome Sequencing et al. (2008) | |
| Apis mellifera | Honeybee | HGSC | Assembly | Consortium Honeybee Genome Sequencing (2006) | |
| Nasonia vitripennis | Jewel wasp | Baylor | Assembly | Werren et al. (2010) | |
| Acyrthosiphon pisum | Pea aphid | Baylor | Assembly | Consortium International Aphid Genomics (2010) | |
| Pediculus humanus | Human louse | ||||
| Daphnia pulex | Waterflea | JGI | Progress | ||
| Boophilus microplus | Southern cattle tick | ||||
| Ixodes scapularis | Deer tick | Assembly | Hill and Wikel (2005) | ||
| Nematode | Caenorhabditis elegans | Roundworm | Complete | Hillier et al. (2008) | |
| Caenorhabditis briggsae | Roundworm | Sanger | Assembly | Gupta and Sternberg (2003); Stein et al. (2003) | |
| Brugia malayi | Filariod worm | Sanger | Assembly | Scott and Ghedin (2009) | |
| Core | Schistosoma mansoni | Trematode | Assembly | ||
| Capitella capitata | Polycheate worm (Annelida) | JGI | Complete | ||
| Helobdella robusta | Leech (Annelida) | ||||
| Aplysia californica | California sea hare | Broad | Assembly | ||
| Lottia gigantea | Owl limpet (sea snail) | Complete | |||
| Nematostella vectensis | Starlet sea anemone | JGI | Assembly | Putnam et al. (2007) | |
| Hydra magnipapillata | Hydra | Venter | Assembly | Chapman et al. (2010) | |
| Trichoplax adhaerens | Placazoa | JGI | Assembly | Srivastava et al. (2008) | |
| Amphimedon queenslandica | Sponge | JGI | Progress | ||
| Monosiga brevicollis* | choanoflagellate | JGI | Complete | King et al. (2008) |
To adequately represent the distribution of species across the Metazoa, our analyses were restricted to a subset of 45 diverse species (19 Deuterostomes: 14 Chordates, 3 Urochordates, 2 Echinodermes; 21 Protostomes: 16 Ecdysozoans, 4 Lophotrochozoans, 1 Trematode; 2 Cnidarian, 1 Placozoan, 1 Porifera, and 1 Choanoflagellate). Although the Choanoflagellida lineage is not part of the Metazoa, it is closely related and serves as an outgroup for the animal lineage. ClustalW (Larkin et al. 2007), Muscle (Edgar 2004), and Dialign (Subramanian et al. 2008) algorithms provided similar AA alignments of the bHLHZ domain with small deviations in gap location within the loop region. Morgenstern and Atchley (1999) previously described issues with gaps during phylogenetic reconstruction of bHLH sequences and the inability to determine proper homology between proteins for the loop region. To circumvent these problems, we removed the middle and nonhomologous portion of the loop and optimized over the bHHZ sequence when comparing different protein families.
Phylogenetic Reconstruction Using the bHHZ Domain
Max and Mlx network members belong to the DNA-binding class B 5′-CACGTG-3′ E-box binding group, which is suggested to represent the ancestral HLH sequence (Atchley and Fitch 1997). Since the history of divergence among Max and Mlx members is uncertain, we included several additional class B bHHZ sequences as outgroup sequences for comparison in each phylogenetic analysis of the 352 taxa. These outgroup sequences included H. sapiens, D. melanogaster, and C. elegans orthologs of SREBF1, USF2, TCF3, MYOD, and HES1.
When determining phylogenetic relationships within the bHHZ domain among Max and Mlx network proteins, we used multiple tree reconstruction algorithms including Bayesian, maximum likelihood (ML), and distance methods. This was done to ensure adequate representation of evolutionary models and expose any potential algorithm-specific idiosyncrasies. Table 3 lists parameter combinations and programs used for each method.
Table 3.
Phylogenetic Reconstructions
| Typea | Method | Qc | Site Rate | Log Lk | Treeb |
| Bayesian | MrBayes | Mixed | Fixed | −23,893.743 | C* |
| Bayesian | MrBayes | Mixed | Γ, estimate pinvar | −23,834.159 | B |
| ML | PAML | JTT | Pinvar | −21,602.4493 | B |
| ML | ProML | JTT | Fixed | −21,777.7348 | B |
| ML | ProML | JTT | Γ:α = 1.3, C = 4 | −20,788.22426 | B |
| ML | ProML | JTT | Γ:α = 1.3, C = 4, pairwise correlation | −20,696.53095 | B |
| ML | PhyML | JTT | Fixed (pinvar = 0) | −21,614.3738 | A |
| ML | PhyML | WAG | Fixed (pinvar = 0) | −21,548.877 | B |
| ML | PhyML | WAG | Estimate pinvar | −21,548.88396 | B |
| ML | PhyML | JTT | Estimate pinvar | −21,612.50602 | A |
| ML | PhyML | JTT | Γ:C = 4, α, pinvar = 0 | −20,550.54622 | A* |
| ML | PhyML | WAG | Γ:C = 4, α, pinvar = 0 | −20,675.15114 | A |
| ML | PhyML | JTT | Γ:C = 4, α = 2, pinvar = 0 | −20,582.61548 | A |
| Distance | NJ (HyPhy) | PC | Fixed | A | |
| Distance | NJ (HyPhy) | PC_RV | Fixed | A | |
| Distance | NJ (HyPhy) | JTT | Fixed | A | |
| Distance | NJ (HyPhy) | JTT | Γ:C = 4 | B | |
| Distance | NJ (HyPhy) | JTT + F | Fixed | B | |
| Distance | BioNJ | JTT | Fixed | A | |
| Distance | BioNJ | JTT | Γ:α = 1 | A | |
| Distance | BioNJ | PMB | Fixed | B* | |
| Distance | ProtPars | Ordinary parsimony | B | ||
| Distance | NeighborNet | JTT | A |
Bayesian, ML, and distance methods for reconstructing the bHHZ tree.
Trees fall under three main topologies (A, B, and C) shown in figure 4, where an asterisk (*) indicates the tree shown.
Q, AA substitution matrix; PC, Poisson correction; PC_RV, Poisson corrected with rate variation; WAG, Whelan Goldman model; JTT, Jones Taylor Thornton Model; PMB, Probability Matrix from Blocks; +F, with empirical character frequencies; Γ, Gamma rate distribution; C, number of rate categories; Pinvar, proportion of invariant sites; Mixed, Mixed Fixed Rate model explores rate matrices, such as JTT and WAG, where each contributes to the rate in proportion to its posterior distribution of the converged model.
We estimated several neighbor joining trees (Saitou and Nei 1987) based on different models of selection using HyPhy (Kosakovsky Pond et al. 2005). We also used BioNJ (Gascuel 1997), which iteratively reduces the variance of distance estimates for a minimum evolution tree by applying weighted averages. The initial distance matrix required for BioNJ was created using ProtDist of the Phylip package (Felsenstein 2005). Protpars, a parsimony method developed by Felsenstein (2005) was also used for comparison.
Further, we applied a Bayesian approach and several ML methods for statistical comparison of phylogenies. PAML provides a framework for complex models during ML phylogenetic reconstruction (Yang 2007). Comparatively, ProML (Felsenstein 2005) and PhyML (Guindon and Gascuel 2003) use an internal BioNJ method to build an initial tree prior to optimizing topologies and ML estimates. PhyML couples stepwise addition with topology rearrangement to simultaneously optimize branch lengths and likelihood probabilities for each iteration of its hill-climbing algorithm. This method claims to reduce computational time while maintaining comparable accuracy levels with other ML approaches. We also used MrBayes for a comparable Bayesian phylogenetic reconstruction (Ronquist and Huelsenbeck 2003).
To further evaluate the stability and robustness of estimated phylogenetic trees, we implemented a bootstrap analysis. The bootstrap method creates a consensus tree that reflects the confidence of the tree topology at each clade. Since missing data can confound bootstrap sampling, we restricted our data set to the 299 taxa with complete bHHZ sequences and performed 100 bootstrap replicates using PhyML.
Entropy as a Conservation Score
In the context of protein sequence analysis, entropy measures the amount of information or conservation at a site by the observed distribution of AAs (Shannon 1948). We calculated the Shannon Entropy for all sites, where 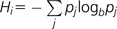 is the entropy for site i with probability pj of being in state j. Entropy can be standardized so H ∈ [0,1] by setting b equal to the number of possible states. AA entropy assumes independence among AA states and standardizes by log base b = 20. However, treating each AA independently does not reflect the similarity in physicochemical properties. To accentuate changes in physicochemical properties at a site, Atchley et al. (1999) developed a functional entropy measure that groups AAs into eight functional categories, that is, acidic (D, E), basic (K, R, H), aromatic (F, Y, W), aliphatic (A, G, I, L, V, M), amidic (N, Q), hydroxylated (S, T), cysteine (C), and proline (P), then standardizes values by log base b = 8. Hence, a site with a low functional entropy but high AA entropy suggests that it is conserved for a particular physicochemical property but not a particular residue.
is the entropy for site i with probability pj of being in state j. Entropy can be standardized so H ∈ [0,1] by setting b equal to the number of possible states. AA entropy assumes independence among AA states and standardizes by log base b = 20. However, treating each AA independently does not reflect the similarity in physicochemical properties. To accentuate changes in physicochemical properties at a site, Atchley et al. (1999) developed a functional entropy measure that groups AAs into eight functional categories, that is, acidic (D, E), basic (K, R, H), aromatic (F, Y, W), aliphatic (A, G, I, L, V, M), amidic (N, Q), hydroxylated (S, T), cysteine (C), and proline (P), then standardizes values by log base b = 8. Hence, a site with a low functional entropy but high AA entropy suggests that it is conserved for a particular physicochemical property but not a particular residue.
Transforming AA Sequences into Metric Data Using Factor Scores
Statistically rigorous analyses of AA variability procedures typically require a numeric representation of alphabetic AA codes in protein sequence data. To interpret structural and functional attributes of bHLHZ sites, we transformed each AA sequence into the five multivariate physicochemical metrics proposed by Atchley et al. (2005) that independently describe multidimensional characters of the various AAs. Atchley et al. (2005) used factor analysis to distinguish the common and unique variance of approximately 500 AA indices. They found five basically orthogonal factors adequately summarized the latent variable structure and denoted these vectors by polarity, accessibility, and hydrophobicity (PAH); propensity for secondary structure (PSS); molecular size (MS); codon composition (CC); and electrostatic charge (EC). Each column in the AA alignment is then represented by a five element vector of converted PAH, PSS, MS, CC, and EC values.
Discriminant Analysis of Proteins, Networks, and Binding Partners
To identify the structure of variation in physicochemical properties among proteins, we statistically ranked sites according to their ability to distinguish protein groups by using stepwise linear discriminant analysis (DA) (Fisher 1936). DA is a widely used robust statistical method for discriminating variables among a priori defined groups. While canonical DA considers all variables simultaneously when building the discriminatory model, the stepwise method discriminates groups by iteratively incorporating variables that maximize the between- versus within-group variance after conditioning on prior variables included.
In order to reveal the impact of natural selection among orthologs in different network configurations, we first grouped all orthologous species sequences by their network topology (fig. 2 and table 2): 1) core, 2) nematode, 3) Diptera, or 4) vertebrate. Because paralogs may be under different selection pressures, they were considered distinct proteins, for example, c-Myc, N-Myc, L-Myc, S-Myc, and L-Myc2 were all grouped separately. However, DA performs poorly on discrete data (Dillon and Westin 1982). Hence, we independently used each of the five factor score transformations of the AA sequences to annotate sites according to these distinct physicochemical properties. Gaps in the alignment were replaced by zeros, although imputing missing residues gave comparable results (data not shown). Stepwise DA, implemented using SAS software, produced an ordered list of best discriminant sites along with the average square canonical correlation (ASCC), which signifies the cumulative amount of among class variance documented by the included sites (table 4).
FIG. 2.—

Max and Mlx network topologies. Rectangles activate and trapezoids repress transcription of unique and overlapping sets of gene targets when heterodimerized with obligate dimers (ovals). For example, c-Myc:Max and MondoB:Mlx activate whereas Mnt:Max represses transcription. Solid lines indicate known dimerizations, whereas debated or unknown interactions are shown by a dotted line. Circled numbers indicate 1) Core, 2) Nematode, 3) Diptera, and 4) Vertebrate topologies determined according to figure 1. Mga in vertebrates has unknown function, with both repressive and active capabilities and is represented by a trapezoid. Rodents also contain S-Myc and humans have L-Myc2, which interact with Max.
Table 4.
DA of Max and Mlx Network Proteins
| Ident | Step | PAH (ASCC) | PSS | MS | CC | EC | Discriminate |
| (A) Max | |||||||
| b5 | 1 | H203 (0.2757) | H106 (0.2489) | H206 (0.3012) | b1 (0.2726) | H206 (0.3089) | b1 (2ab) |
| b6 | 2 | b2 (0.3290) | H101 (0.3110) | H203 (0.5707) | b4 (0.3333) | L5 (0.3725) | b4 (2a) |
| b8 | 3 | H113 (0.3494) | b7 (0.3274) | H101 (0.5865) | H108 (0.5658) | H106 (0.6052) | b7 (2b) |
| b9 | 4 | L6 (0.3542) | b1 (0.3405) | L8 (0.5948) | Z19 (0.6206) | b7 (0.6358) | H102 (2a) |
| b10 | 5 | H210 (0.4665) | Z19 (0.5206) | H111 (0.6246) | Z5 (0.6358) | H102 (0.6589) | H106 (2ab) |
| b12 | 6 | H211 (0.5101) | Z22 (0.6409) | Z4 (0.6544) | Z16 (0.6646) | L6 (0.6654) | H108 (4) |
| b13 | 7 | b11 (0.5146) | b11 (0.6679) | Z25 (0.6793) | Z2 (0.6877) | Z16 (0.6945) | L6 (2a) |
| H103 | 8 | H209 (0.6085) | H209 (0.6810) | L13 (0.6840) | Z14 (0.6928) | L8 (0.7012) | H203 (2) |
| H104 | 9 | Z20 (0.6751) | H210 (0.7269) | H208 (0.7221) | Z7 (0.7016) | H209 (0.7025) | H206 (4) |
| H110 | 10 | Z22 (0.7069) | b3 (0.7500) | Z1 (0.7392) | H208 (0.7417) | Z27 (0.7903) | H209 (2ab) |
| H115 | 11 | L9 (0.7121) | H109 (0.7586) | Z27 (0.8069) | H213 (0.7518) | Z22 (0.7908) | H211 (2a) |
| L14 | 12 | Z8 (0.7293) | Z12 (0.7716) | Z3 (0.8249) | H211 (0.7624) | L10 (0.8230) | Z1 (2) |
| H201 | 13 | L3 (0.7467) | Z10 (0.7735) | L5 (0.8281) | Z8 (0.7828) | H211 (0.8499) | Z3 (2a) |
| H202 | 14 | Z11 (0.7537) | Z13 (0.7820) | H209 (0.8286) | L3 (0.7913) | L1 (0.85.02) | Z5 (4) |
| H205 | 15 | L7 (0.7642) | H206 (0.8025) | b2 (0.8303) | L7 (0.8209) | Z18 (0.8578) | Z7 (2a) |
| H212 | 16 | Z1 (0.7853) | Z6 (0.8394) | b3 (0.8496) | Z17 (0.8256) | Z15 (0.8819) | Z15 (2a) |
| 17 | Z26 (0.7896) | L7 (0.8709) | Z17 (0.8648) | H210 (0.8778) | Z9 (0.8912) | Z19 (2b4) | |
| 18 | Z18 (0.7934) | L3 (0.9002) | L11 (0.8716) | L10 (0.8908) | B2 (0.9217) | Z27 (2ab) | |
| 19 | H213 (0.8160) | Z7 (0.8793) | Z15 (0.8922) | ||||
| 20 | Z13 (0.8216) | H204 (0.8872) | L5 (0.9146) | ||||
| 21 | Z23 (0.8480) | H106 (0.8909) | |||||
| 22 | H109 (0.9061) | Z18 (0.8976) | |||||
| 23 | L10 (0.8981) | ||||||
| 24 | L9 (0.8987) | ||||||
| 25 | Z15 (0.9297) | ||||||
| (B) Mlx | |||||||
| b5 | 1 | L8 (0.2968) | H206 (0.3186) | H206 (0.2861) | Z24 (0.2659) | H206 (0.3297) | b6 (2) |
| b9 | 2 | H206 (0.5835) | H201 (0.3333) | H201 (0.3333) | L16 (0.4298) | H202 (0.3333) | H111 (2) |
| b12 | 3 | H201 (0.6280) | Z3 (0.5391) | Z3 (0.5771) | H202 (0.5636) | Z2 (0.5868) | L8 (4) |
| b13 | 4 | b6 (0.6657) | H213 (0.6979) | Z15 (0.8027) | Z3 (0.7415) | Z15 (0.7880) | L16 (2) |
| H103 | 5 | Z24 (0.9016) | H209 (0.7591) | Z5 (0.8489) | H214 (0.7912) | Z4 (0.8431) | H201 (2) |
| H106 | 6 | H111 (0.8017) | Z4 (0.8745) | Z16 (0.8082) | H102 (0.8786) | H206 (2) | |
| H110 | 7 | Z24 (0.8906) | Z16 (0.8930) | H111 (0.8885) | b6 (0.9062) | H213 (3) | |
| H205 | 8 | Z16 (0.9650) | Z24 (0.9696) | H107 (0.8991) | H214 (2) | ||
| Z21 | 9 | Z14 (0.9259) | Z2 (4) | ||||
| Z3 (24) | |||||||
| Z15 (23) | |||||||
| Z16 (2) | |||||||
| Z24 (23) | |||||||
| (C) Myc | |||||||
| b5 | 1 | H103 (0.1594) | H102 (0.1277) | b6 (0.1599) | b6 (0.1606) | b6 (0.1568) | b3 (3) |
| b9 | 2 | H206 (0.2740) | H206 (0.2419) | H102 (0.2812) | H108 (0.2771) | b10 (0.1752) | b4 (4ae) |
| b12 | 3 | H114 (0.2857) | H109 (0.3319) | b10 (0.2929) | L6 (0.3664) | H106 (0.3017) | b6 (4e) |
| b13 | 4 | b3 (0.4072) | H103 (0.4367) | H106 (0.3916) | H111 (0.4781) | H103 (0.3796) | b7 (4cd) |
| H110 | 5 | H107 (0.4315) | b10 (0.4716) | L7 (0.4955) | H103 (0.5584) | H107 (0.4078) | H102 (34a) |
| H115 | 6 | 102 (0.5527) | H107 (0.5034) | H103 (0.5716) | b10 (0.5953) | L7 (0.5122) | H103 (4de) |
| H205 | 7 | Z22 (0.6511) | b6 (0.5372) | H107 (0.6123) | H107 (0.6111) | H104 (0.6101) | H108 (4a) |
| 8 | Z24 (0.7224) | L7 (0.6224) | b11 (0.6565) | H104 (0.6785) | b11 (0.6494) | H111 (3) | |
| 9 | b11 (0.7552) | b4 (0.6518) | b8 (0.6823) | H206 (0.7173) | H202 (0.6779) | L7 (4) | |
| 10 | H104 (0.7760) | H111 (0.7026) | H104 (0.7139) | L7 (0.7504) | H112 (0.7093) | H203 (3) | |
| 11 | b7 (0.7943) | H203 (0.7291) | H111 (0.7831) | b1 (0.7830) | Z1 (0.7240) | H206 (4c) | |
| 12 | L6 (0.8118) | L5 (0.7470) | Z1 (0.8047) | b8 (0.7999) | H111 (0.7825) | Z22 (4a) | |
| 13 | Z5 (0.8166) | Z8 (0.7583) | H202 (0.8264) | H102 (0.8198) | b3 (0.8048) | Z24 (3) | |
| 14 | H201 (0.8350) | H108 (0.7776) | H201 (0.8368) | H112 (0.8417) | H207 (0.8286) | ||
| 15 | L8 (0.8483) | Z9 (0.7883) | H108 (0.8563) | b11 (0.8528) | L5 (0.8406) | ||
| 16 | Z1 (0.8630) | L8 (0.8042) | H112 (0.8684) | H202 (0.8638) | Z13 (0.8653) | ||
| 17 | b4 (0.8696) | Z10 (0.8223) | b7 (0.8741) | H106 (0.8783) | H102 (0.8857) | ||
| 18 | H214 (0.8834) | H113 (0.8362) | Z15 (0.8877) | H203 (0.8910) | Z7 (0.8925) | ||
| 19 | Z26 (0.8881) | H114 (0.8395) | L5 (0.8974) | b7 (0.8972) | Z22 (0.9088) | ||
| 20 | H109 (0.8913) | H112 (0.8468) | Z16 (0.9076) | H213 (0.9049) | |||
| 21 | H202 (0.9003) | b2 (0.8552) | |||||
| 22 | Z20 (0.8625) | ||||||
| 23 | Z19 (0.8656) | ||||||
| 24 | H207 (0.8748) | ||||||
| 25 | Z22 (0.8952) | ||||||
| 26 | Z14 (0.8995) | ||||||
| 27 | b11 (0.9033) | ||||||
| (D) Mondo | |||||||
| b9 | 1 | b11 (0.2418) | b11 (0.2401) | Z25 (0.1989) | Z25 (0.2499) | H201 (0.1851) | b11 (2) |
| b12 | 2 | H103 (0.2716) | H204 (0.4423) | Z14 (0.3014) | Z14 (0.2980) | Z25 (0.3608) | H102 (2) |
| b13 | 3 | H213 (0.4601) | L6 (0.6095) | L7 (0.4525) | H102 (0.4769) | Z14 (0.4945) | H105 (2) |
| 4 | H201 (0.5732) | B10 (0.6176) | H109 (0.5837) | H211 (0.4839) | L7 (0.6248) | H109 (2) | |
| 5 | b2 (0.6395) | H202 (0.6252) | H113 (0.6241) | L2 (0.5915) | H113 (0.6724) | L5 (2) | |
| 6 | Z6 (0.7290) | Z28 (0.7380) | H105 (0.7043) | L4 (0.6606) | H204 (0.7681) | L6 (2) | |
| 7 | L8 (0.7614) | L7 (0.7899) | Z4 (0.7141) | Z15 (0.7483) | L4 (0.7820) | L8 (4b) | |
| 8 | H209 (0.8022) | H205 (0.8084) | Z1 (0.7884) | H112 (0.7771) | L11 (0.8048) | L11 (2) | |
| 9 | H208 (0.8208) | Z15 (0.8578) | Z27 (0.8486) | Z11 (0.8171) | H207 (0.8356) | H201 (2) | |
| 10 | H203 (0.8583) | Z17 (0.8814) | L5 (0.8833) | b1 (0.8428) | L5 (0.8597) | H204 (24) | |
| 11 | Z11 (0.8720) | L8 (0.9137) | Z22 (0.9030) | Z21 (0.8825) | L6 (0.8875) | H208 (4) | |
| 12 | Z17 (0.9053) | b7 (0.8926) | H112 (0.9020) | H211 (2) | |||
| 13 | b5 (0.9097) | H201 (0.8922) | Z6 (4a) | ||||
| 14 | Z22 (0.9098) | Z25 (3) | |||||
| Z28 (23) | |||||||
| (E) Mnt | |||||||
| b5 | 1 | H115 (0.3237) | H113 (0.3861) | H204 (0.4244) | H204 (0.4889) | Z23 (0.3944) | L1 (3) |
| b8 | 2 | H212 (0.4596) | L3 (0.7095) | H201 (0.5000) | H208 (0.5000) | H204 (0.7556) | L3 (3) |
| b9 | 3 | Z27 (6955) | H212 (0.8186) | Z21 (0.7443) | H115 (0.8241) | H201 (0.8729) | H204 (4) |
| b10 | 4 | H112 (0.8102) | H206 (0.8483) | H212 (0.8572) | H212 (0.9674) | Z2 (0.9539) | Z21 (34) |
| b12 | 5 | L7 (0.8675) | H213 (0.8717) | Z4 (0.9056) | Z23 (3) | ||
| b13 | 6 | L10 (0.8623) | H204 (0.9430) | Z27 (4) | |||
| 7 | L1 (0.9112) | ||||||
| (F) Mxd | |||||||
| b2 | 1 | H102 (0.1885) | H106 (0.2000) | H106 (0.1874) | H106 (0.1874) | H106 (0.1874) | b4 (24c) |
| b9* | 2 | H113 (0.3611) | H105 (0.3434) | b8 (0.3525) | b8 (0.3524) | b8 (0.3594) | b8 (4a) |
| b10 | 3 | Z11 (0.5064) | Z7 (0.4748) | H211 (0.4864) | Z11 (0.4812) | H102 (0.4932) | H102 (24cd) |
| b12 | 4 | H115 (0.5839) | L8 (0.5859) | Z16 (0.6016) | H211 (0.6034) | H114 (0.6135) | H105 (24c) |
| b13 | 5 | Z8 (0.6665) | H115 (0.6850) | H114 (0.6654) | H201 (0.6704) | Z16 (0.6911) | H106 (4d) |
| H101* | 6 | H106 (0.6742) | Z23 (0.7355) | H102 (0.7570) | H214 (0.7120) | H115 (0.7354) | H113 (4c) |
| H110* | 7 | Z22 (0.7462) | b11 (7724) | H115 (0.8081) | H113 (0.7443) | Z11 (0.7758) | H114 (4b) |
| L9 | 8 | b4 (0.7721) | b8 (0.7949) | Z11 (0.8239) | Z7 (0.7634) | Z1 (0.8028) | L8 (2) |
| H202* | 9 | L4 (0.7980) | Z5 (0.8043) | Z18 (0.8374) | H109 (0.7802) | H214 (0.8290) | H206 (4b) |
| H205* | 10 | H210 (0.8167) | b5 (0.8316) | b11 (0.8642) | Z3 (0.7998) | H108 (0.8498) | H211 (4) |
| H212 | 11 | H201 (0.8284) | H103 (0.8463) | H203 (0.8729) | H208 (0.8287) | H209 (0.8553) | H214 (4) |
| Z21 | 12 | H213 (0.8458) | b3 (0.8646) | L2 (0.9011) | L4 (0.8470) | Z14 (0.8633) | Z7 (4b) |
| 13 | Z4 (0.8544) | L3 (0.8778) | H115 (0.8610) | H203 (0.8720) | Z8 (4d) | ||
| 14 | L2 (0.8594) | Z27 (0.8839) | Z9 (0.8711) | Z19 (0.8779) | Z11 (4b) | ||
| 15 | H206 (0.8896) | Z24 (0.8887) | L1 (0.9002) | b6 (0.8825) | Z13 (4c) | ||
| 16 | Z13 (0.8918) | Z19 (0.8928) | L7 (0.8854) | Z18 (4d) | |||
| 17 | H114 (0.8952) | H203 (0.9064) | H105 (0.9007) | ||||
| 18 | H209 (0.9075) | ||||||
NOTE.—Stepwise DA classifying each protein by network according to its bHLHZ sites. Stepwise DA was performed separately for each protein (A–F) and factor transformation (PAH, PSS, MS, CC, and EC) where each step incorporates the next most discriminating site. Variance explained is represented by the average squared canonical correlation (ASCC). Invariant sites for each protein are listed in the “Ident” column, and conserved synapomorphies are highlighted and listed under the “Discriminate” column. Networks are designated in parantheses 1) Core, 2) Nematode, 3) Diptera, and 4) Vertebrate. Paralogs are treated as individual subcategories. Possible protein categories are (A) Max (1,2a: Mxl-1, 2b: Mxl-3,3,4), (B) Mlx (1,2,3,4), (C) Myc (1,3,4a: c-, 4b: N-, 4c: L-, 4d: S-, 4e: L-Myc2), (D) Mondo (1,2,3,4a:MondoA, 4b: MondoB), (E) Mnt (1,3,4), and (F) Mxd (1,2,4a:Mxd1, 4b:Mxi1, 4c:Mxd3, 4d:Mxd4). *Mxd2 in Bos taurus contains multiple substitutions and was not considered for identifying identical or synapomorphic sites.
Results and Discussion
Myc, Max, Mnt, Mxd, Mlx, and Mondo proteins comprise the basic members of the Max and Mlx interaction networks found throughout the Metazoa. The presence of at least one identifiable bHLHZ sequence belonging to a Max and Mlx network member in all animals surveyed emphasizes the importance of these ancient TFs (fig. 1). Using phylogenetics and statistical concepts like entropy and DA, we identified protein-specific residues within the bHLHZ domain that potentially restrict DNA-binding and influence patterns of transcriptional regulation.
Max and Mlx Network Protein Presence/Absence in Metazoa
Protein sequences from approximately 100 species were obtained from an array of genome databases using sequence annotations, predicted transcripts, and significant blast hits (see Materials and Methods). To concisely yet adequately represent the diversity of Max and Mlx network members in animals, we restricted our analysis to 352 sequences coming from 45 diverse species. We used well-defined and highly conserved sequences of the bHLHZ domain of Max and Mlx network members to ascertain if the various proteins occurred in a given organism. As shown in figure 1, we identified core network members (“X”) in almost all surveyed species and predict their existence (outlined) even if a particular member was only partially found (“*”) or unidentifiable (blank). Exceptions occur when a gene has been experimentally validated as missing (“0”) (Yuan et al. 1998; Gallant 2006), and we conjecture that consecutive absences are deletions (gray). However, absence of a given protein in the database does not denote absence in the organism because several of the queried genome assemblies are still in draft or assembly phase with low coverage (table 2).
Lineage-specific radiation and deletion of Max and Mlx network components resulted in four main network configurations in animals (figs. 1 and 2). At the stem of Bilateria and Radiata divergence, six core proteins represent the ancestral Max and Mlx network topology. This core topology consists of Max, Mlx, Myc, Mxd, Mnt, and Mondo proteins, for which all animals surveyed contain at least one identifiable network member, as determined by the bHLHZ sequence (fig. 1). However, lower order organisms may have fewer members, whereas nematodes, flies, and vertebrates have distinct topologies and derived configurations (fig. 2).
Organisms that diverged near the root of the Metazoa can provide significant insight into the origin and evolution of network members. Trichoplax adhaerens of the Placazoa lineage is the simplest known animal with the smallest known genome (Srivastava et al. 2008), whereas the choanoflagellate Monosiga brevicollis is one of the closest single-celled organisms related to animals (King et al. 2008). The presence of Myc and Max in both Trichoplax and Monosiga strongly implies that these proteins have ancient roots and are important for basic cellular function. Max, Myc, Mxd, Mlx, and Mondo bHLHZ sequences were recovered in Trichoplax, whereas the first identifiable instance of Mnt occurs within the Cnidaria and Bilateria lineages. Hence, the origin of the Max and Mlx networks dates to over 500 Ma and predates the origin of animals.
Flies (Diptera) and nematodes are the only known organisms to be missing a core network member (Gallant 2006). Previous reports of yeast two-hybrid assays, interaction screens, and genome searches indicate that flies lack a Mxd gene, whereas nematodes are missing both Mnt and Myc orthologs (Yuan et al. 1998; Gallant 2006). We observe that fruitfly D. melanogaster and mosquitoes Aedes aegypti, Anopheles gambiae, and Culex pipens lack an identifiable Mxd sequence, whereas moth Bombyx mori possesses an orthologous bHLH sequence. The absence in lower as well as higher Diptera reinforces the idea that Mxd loss is specific to the entire Diptera lineage. We could not find Mxd in ticks Boophilus microplus or Ixodes scapularis, indicating ticks, which are part of the Arachnida lineage, may have also independently lost Mxd. Similarly, nematodes C. elegans, C. briggsae, and Brugia malayi do not have Myc or Mnt orthologs. Instead, these species along with the trematode Schistosoma mansoni contain similar yet divergent orthologs for Max, Mlx, Mxd, and Mondo.
In contrast, two whole genome duplication (WGD) events, which occurred either prior to or during vertebrate divergence (Dehal and Boore 2005), ostensibly resulted in the radiation of Myc, Mxd, and Mondo proteins. Only a single copy of Max, Mlx, and Mnt exists in vertebrates despite multiple duplication events, suggesting that the regulation of these proteins is highly controlled by natural selection. In contrast, Myc has experienced additional independent duplication events. Approximately 35–50 Ma new and old world primates, but not prosimians, exhibit a duplication of L-Myc denoted L-Myc2 (Morton et al. 1989; Arnason et al. 1998). Because L-Myc2 is intronless, it presumably arose via a reverse transcriptase event. The murine lineage, including mouse and rat, also exhibits a duplication of N-myc, forming Myc family member S-Myc. The presence of both the 5′ and 3′ untranslated region (UTR) and absence of conventional N-Myc introns suggest that S-Myc was formed by an N-Myc cDNA sequence reintegrating into the genome.
Another Max network member, Mga, also arose during vertebrate divergence. Mga is predicted to be an Myc family member because its bHLHZ domain is most similar to c-Myc (Hurlin et al. 1999). However, the origin of Mga is ambiguous due to issues with genome coverage and prediction for the 12,189 bp transcript.
Like other network members, Mga has a C-terminus bHLHZ domain with conserved sites in the basic region responsible for E-box recognition. However, it also contains a second DNA recognition domain in its N-terminus that recognizes the DNA Brachyury T-box motif (Hurlin et al. 1999). Unlike the characteristic exon structure in other T-box proteins, the T-domain in Mga lacks introns, implying that it was inserted via reverse transcription.
Branchiostoma floridae (lancelet or amphioxus) of the cephalochordate lineage contains a sequence with 33.8% identity and 53.8% similarity to Mga in humans over its bHLHZ domain. However, the B. floridae sequence does not contain a T-domain. Instead, this 11,851 bp hypothetical transcript contains a second N-terminus bHLHZ domain. Since the divergence of Branchiostoma was prior to the vertebrate WGD events (Putnam et al. 2008; Kawashima et al. 2009), Mga may have arisen independently whereby the T-domain insertion into this ancestral duplicate altered the transcript 5′ end. Alternatively, Mga truly arose during the radiation in vertebrates and is a divergent member of the Myc family.
Although Diptera and Nematoda lineages represent experimentally validated gene loss, other instances of member absence may simply be the result of missing data. For example, our criterion for protein identification reports the chicken Gallus gallus ortholog Mxd3 as absent, although it is likely to exist in the genome. We found that the 5′ UTR of Mxd3 overlaps the 3′ UTR of the Prelid1 gene in all vertebrates sampled (fig. 3). Although sequencing in this region in Gallus is of poor quality with nonoverlapping contigs, conservation of identifiable Mxd3 sequence fragments within bacterial artificial chromosome clone AC195499 provides strong evidence that Mxd3 exists and is functional in chicken.
FIG. 3.—

Mxd, Myc, and Mondo synteny. Cartoon depiction of genetically linked homologs for H: human, O: opossum, C: chicken, X: Xenopus, and T: Tetraodon. Synteny among paralogous gene families (shaded boxes) suggest a common origin, whereas orthologs (white boxes) confirm orientation and structure. Tree structure displays proposed order of duplication prior to divergence. Solid lines between species indicate conserved orthology, dashed lines indicate intermediate species have a missing or unlinked ortholog, and hashes between genes show breaks in contig sequences. Gene sizes and distances are not to scale. (A) The Mxd family is linked with ADD paralogs. Tetraodon carries two copies of Prelid1, and ZFYVE28* is an unnamed duplicate of ZFYVE28. A gap in the chicken genome coverage suggests Mxd3 is conserved yet unavailable. (B) The Myc family is linked to Fam84 and Fam49 paralogs. Translocations surrounding N-Myc in opossum potentially resulted in its loss. (C) The Mondo family is flanked by BCL7, Clip, and VPS37 paralogs. MondoB was unidentifiable in Xenopus. BCL7B, VPS37D, and Clip2 were all found on different contigs for Xenopus and Tetraodon.
Myc, Mxd, and Mondo Family Genes Exhibit Synteny in Vertebrates
The syntenic region around paralogs gives evidence for regional conservation of duplications and suggests an order of divergence. As shown in figure 3, Mxd3 is genetically linked with mitochondrial precursor protein Prelid1 (Fox et al. 2004) and an unannotated protein similar to zinc finger protein ZFYVE28. Similarly, Mxd4 is associated with ZFYVE28 in Monodelphis domesticus (opossum), G. gallus (chicken), and Xenopus tropicalis (clawed frog) and also linked with the second copy of Prelid1 in the pufferfish Tetraodon nigroviridis. This synteny suggests that Mxd3 and Mxd4 are within similarly conserved and paralogous genetic regions. Mxd1, Mxd2, and Mxd4 paralogs are also genetically linked with the three member ADD family of cytoskeleton proteins (Anong et al. 2009). The relative orientation of these genes supports evidence that these families radiated during the two WGD events that occurred either prior to or during vertebrate divergence.
The Myc family of proteins is syntenic with the FAM84(A, B) and FAM49(A, B, C) families associated with DNA repair and unknown functions, respectively (McDonald et al. 2003). Mga has been proposed to be a Myc family member (Hurlin et al. 1999), although we found no paralogous families that corroborate this supposition. c-Myc and N-Myc are genetically linked with FAM84 and FAM49 homologs, whereas L-Myc is in proximity to only FAM49C. Because L-Myc is not essential for viability (Hatton et al. 1996), dispensable promoter elements may affect the selective pressure on surrounding genes.
Although knockout studies in mice indicate both c-Myc and N-Myc are essential for growth (Charron et al. 1992; Davis et al. 1993; Moens et al. 1993; Sawai et al. 1993), we were unable to identify N-Myc in opossum. Chromosomal rearrangements show that N-Myc is no longer flanked by FAM84A and FAM49A, which are located within 4 Mb of the distal end of opossum Chromosome 1 and 20 Mb upstream, respectively. Hence, opossum N-Myc may have been lost during this translocation, and N-Myc may be conditionally dispensable.
In contrast to Myc and Mxd protein families, the Mondo family contains only two paralogs despite their coincidental emergence during vertebrate divergence. The origin of MondoA and MondoB duplication can be extrapolated from their genetic linkage with BCL7(A, B, C), CLIP(1, 2, 3, 4), and VPS37(A, B, C, D) protein families (fig. 3). The most recent common ancestor of the four paralogs in CLIP and VPS37 dates to the origin of vertebrates (Flicek et al. 2010). However, no combination of VPS37A, VPS37C, CLIP3, CLIP4, and BCL7C are genetically linked in pufferfish, clawed frog, chicken, opossum, or human.
Max and Mlx Network bHHZ Domains Show Clear Phylogenetic Relationships
Variable selective pressures among homologs in different lineages may cause inferred evolutionary relationships to differ from the order of divergence. Phylogenetic trees display the association of multiple taxa by grouping sequences according to a measure of similarity (Hedges 2002). Using phylogenetic reconstructions, we infer the relationship and divergence of the homologous bHHZ domain to determine the relative importance of DNA binding and dimerization among Max and Mlx network proteins.
We used several Bayesian, ML, and distance-based phylogenetic methods to diversify reconstruction strategies and compare the resulting optimal phylogenetic trees (fig. 4 and table 3). In addition, we estimated a bootstrap consensus tree based on 100 replicates to assess the stability of each clade (fig. 5). Since the root of the tree is unknown, we also included orthologous bHHZ sequences for SREBF1, USF2, TCF3, MYOD, and HES1 to compare the relationship with outgroup sequences.
FIG. 4.—

Phylogeny of the bHHZ domain. Phylogenetic reconstruction of bHHZ domain for all Max and Mlx network members. Three major tree topologies emerge (A–C); each are individually scaled with branch lengths proportional to the expected number of changes per unit time. (A) PhyML algorithm using JTT rate matrix with four site rate categories estimated from a discretized Gamma distribution. (B) BioNJ algorithm using PMB rate matrix and a single site rate. (C) MrBayes algorithm using Gamma distribution of rate categories over 2 million generations. Specific parameterizations described in table 3.
FIG. 5.—

bHHZ PhyML tree with bootstrap values. PhyML tree based on 100 bootstrap replicates. Bootstrap values are reported above each branch, for example, 82 of the 100 trees similarly group N-Myc sequences, whereas 89 distinctly group L-Myc. Some clades have been collapsed for visualization, and the number of taxa within that lineage is noted in parentheses. Mxd (orange), Mnt (red), Myc (light green), Max (light blue), Mlx (dark blue), Mondo (dark green), and Mga (magenta). Human, Drosophila, and Caenorhabditis elegans orthologs of SREBF, USF, TCF3, MYOD, and Hairy were used as outgroups (yellow).
For all tree methods, orthologous sequences for each protein formed distinguishable clades in all phylogenetic reconstructions (fig. 4). Each protein clade includes sequences from species spanning the Metazoa, emphasizing distinct conservation among orthologous proteins within the bHHZ domain. The grouping of these protein clades is highly robust, which is remarkable considering the diversity of organisms represented and variety of phylogenetic models implemented.
Still, the relationship between protein groups showed slight variability among methods, which we classify into three highly related types of tree topologies (table 3 and fig. 4). The first type of bHHZ tree reconstruction (A: fig. 4A) forms a distinct Mga clade that is closely related to Mlx and Mondo. In comparison, the second topology type (B: fig. 4B) associates the Mga clade with Myc. Most tree reconstructions resemble these topologies, where the Mnt and Mxd clade and Mondo and Mlx clade are distantly related, whereas Max, Myc, Mga, and outgroup sequences are at intermediate distances. In the third, and less common, topology type (C: fig. 4C), Mga and Myc share a clade, whereas all outgroup sequences are within a single clade with Mlx and Mondo. Despite these distinctions, the similarity of these tree topologies attests to the robustness of protein groups and stability of the overall topology.
Bootstrap values also support the distinct classification of protein groups and give confidence estimates for each protein clade. In particular, bootstrap values (in parentheses) for Max (60), Mxd (60), Mondo (49), and Mga (46) protein groups indicate clear sequence similarity, whereas Mnt (17) and Myc (4) are likely to have fewer distinguishing sites (fig. 5). Interestingly, Mlx forms an individual clade in all tree reconstructions, yet Mxl-2 clades separately from Mlx and Mondo in the bootstrap analysis. Accordingly, Mondo and Mlx form distinct sister clades in all tree reconstructions, suggesting that Mondo and Mlx bHHZ domains have similar sequence constraints. This is also the case for Mnt and Mxd proteins, which consistently form sister clades and have a bootstrap value of 44. However, low bootstrap support for more ancestral nodes and variability among tree reconstructions prevents us from determining the relationship among the other protein groups. Still, outgroup sequences including TCF3, MYOD, and HES proteins were consistently separate from Max and Mlx network member clades suggesting a close relationship among these network proteins, although USF2 and SREBF1 also grouped alongside Max in some topologies.
Consistent branching patterns and bootstrap support values also depict distinct groupings among paralogs for vertebrate protein families (fig. 5). Mxd1 and Mxd2 form sister clades (bootstrap value 47) as does Mxd3 and Mxd4 (47) and MondoA and MondoB (62). L-Myc and N-Myc are closer paralogs than c-Myc, which agrees with previous findings (Atchley and Fitch 1995), and all are distinguishable from invertebrate orthologs. Mga bHHZ sequences are tightly grouped, although their relationship with other proteins varies among tree constructions and largely defines the distinction between type A and B topologies. Hence, the concordance among many different algorithms and bootstrap estimates gives credence to the distinctions among protein paralogs and the general relationship among protein groups.
Sequences within each protein group also exhibit branching patterns largely analogous to speciation events. Orthologous protein sequences from vertebrates, chordates, insects, and more ancestrally divergent species generally branch according to their proposed order of divergence represented in figure 1. Branching of nematode sequences, however, do not correspond with the order of taxon divergence. MML-1, Mxl-1, Mxl-2, Mxl-3, and MDL-1 bHHZ sequences show large divergence from Max and Mlx network members despite being clearly orthologous proteins. We identified one Max ortholog in Schistosoma that is related to Mxl-3 and a Mlx ortholog similar to Mxl-2. Mxd in S. mansoni is an outgroup of both Mxd and Mnt clades. Thus, Mnt may truly be lost in this lineage whereby Mxd contains binding functions attributable to both proteins. Nematode MDL-1 orthologs are more closely related to Mxd, which signifies a potential loss of Mnt function in this lineage. Moreover, the bHHZ domain of MML-1 is most similar to Mondo proteins while its binding partner Mxl-2 is an outgroup for Mlx. Hence, the Mlx network is conserved in nematodes, and the antagonistic behavior of Myc and Mnt transcriptional regulation is presumably lost.
The bHLHZ Domain Exhibits Site-Specific Constraint
To quantify AA variability at sites, we compare Shannon Entropy values (Shannon 1948), where low entropy signifies site conservation and high values represent variation. This standardized AA (HAA) entropy treats all changes equally to stress conservation of a particular AA. However, some AAs are functionally and structurally similar and confer comparable functional attributes, for example, leucine, isoleucine, and valine. Hence, we also use a functional group (HFG) entropy value developed by (Atchley et al. 1999) based on eight groups of AAs, which accentuates similarity between AAs and variability in functional changes.
We find several highly conserved sites within the bHLHZ domain known to be responsible for DNA binding and stable dimer formation. As seen in figure 6 (black bars), sites b5, b9, b12, b13, H110, and H205 have HAA entropy values close to zero and are thus highly conserved in all Max and Mlx network members. This is in accordance with known c-Myc, Max, and Mxd1 crystal structures, where sites b5, b9, and b13 make base contacts with DNA that restrict binding to the class B 5′-CACGTG-3′ E-box motif (Ferré-D'Amaré et al. 1993; Nair and Burley 2003), whereas the helical structure creates a surface consisting of sites b1, b2, b6, b10, b12, and b13 that make phosphodiester backbone contacts (Lüscher and Larsson 1999; Nair and Burley 2003). Moreover, site H110 is a buried site that interacts with H204 and H205, whereas H114 packs against sites H212 and H213 in Max.
FIG. 6.—

BHHZ entropy for Max and Mlx network members. Black columns represent standardized AA entropy. Gray bars represent standardized functional entropy (Atchley et al. 1999). All network proteins were included in calculation.
Low HFG entropy values at sites b2, H103, H104, and H215 denote particular AA attributes are important for these sites, although a specific AA is not required (fig. 6, gray lines). Hence, the structural restrictions on buried site H103 and phosphate backbone contacts by H104 slightly vary between proteins and may distinguish binding abilities (Atchley and Zhao 2007). Crystal structures further show that H215 interacts with its symmetry mate in Max (Ferré-D'Amaré et al. 1993). Similarly, the conservation of leucine heptad repeats necessary for stable dimerization is shown by the relative decrease in entropy for sites Z14 and Z21 within the zipper.
Site conservation and distinguishing residues are clearly seen in the predicted HMMER sequences shown in figure 7 (Durbin et al. 1998). HMMER uses a profile hidden Markov model to probabilistically infer the most likely residue at each site. The majority of conservation (bold) is within the basic region as well as sites that flank the loop.
FIG. 7.—

HMMER sequence of bHLHZ domain. Highly conserved residues that occur with over 90% probability are bold, those invariant in vertebrates are uppercase, and other lower case letters show the most explanatory residue for that site. Invariant sites are italicized and underlined to emphasize their importance. Dots are simply placeholders for the loop alignment and are not included in loop numbering for individual proteins.
bHLHZ Sites Can Distinctly Classify Max and Mlx Network Proteins
Distinctly conserved sites within the basic region potentially distinguish binding constraints among proteins and determine their overlapping or distinct gene targets. Site b10 shows discriminatory power among Max and Mlx members; Mxd and Mnt have lysine (K); Mga, Max, and Myc have arginine (R); and Mondo and Mlx possess a glutamine (Q). According to crystal structures, sites b3, b7, b10, and b11 point away from the DNA major groove and interact with regions outside the E-box (Nair and Burley 2006). These sites are distinctly conserved among the Myc, Mxd, Mnt, and Max sequences in vertebrates, and O’Hagan et al. (2000) found that they can differentially influence cellular transformation. Interestingly, Myc b3 and b7 are variable in invertebrates with site b3 predominantly consisting of small and tiny AAs (SNA). This is in contrast to site b3 in human c-Myc, which is known to impose additional DNA-binding restrictions due to its large molecular size (MS) (Solomon et al. 1993).
Residues outside the basic region and higher order conformations also affect DNA-binding restrictions. It is hypothesized that two tandemly arranged MondoB:Mlx heterodimers are required to stabilize binding with the ChORE element (Ma et al. 2007). Mutation experiments verified that the loop region of Mlx but not MondoB specify this interaction. Large hydrophobic residues L8:Phe (F) and L10:Ile (I) are predicted to create a favorable protein interaction interface, whereas basic residue L14:Lys (K) neutralizes ECs with the DNA backbone (Ma et al. 2007). Although L14:Lys (K) is highly conserved, only vertebrates have L8:Phe (F) in their extended 15-residue loop. Instead, arthropods have a 13-residue Mlx loop, the Mxl-2 loop has only 11 sites, and the Mlx loop is variable in other invertebrates. Hence, this higher order interaction may be lost or depend on alternative residues in other species.
Although the zipper region also exhibits variability, multiple mutation studies have found that it confers interaction preferences and is essential for dimerization (Reddy et al. 1992; Arsura et al. 1995; Orian et al. 2003). Sites Z17 and Z18 form antiparallel contacts between monomers during Max dimerization and were found to deviate significantly in human Mxd1 and c-Myc (Nair and Burley 2003). The neutral charges of Z17:Gln-Z18:Asn (QN) in human Max allow homodimerization, yet cause flaring compared with the more stable interaction with positively charged residues Z17:Arg-Z18:Arg (RR) of c-Myc and complementary hydrogen bond interactions with Z17:Glu (E) of Mxd1. Hence, Max more readily dimerizes with c-Myc or Mxd1 instead of homodimerizing (Nair and Burley 2003; Grinberg et al. 2004).
Sites Z17 and Z18 are invariant in Max, except for Trichoplax Max and nematode Mxl-1 and Mxl-3. Similarly, Mxd Z17:Glu-Z18:Gln (EQ) is largely conserved in all Mxd sequences, although Mxd4 Z18 is conserved for His (H), and Mxd3 Z17 varies between positively (KR) and negatively (ED) charged residues. In human c-Myc, Z11:Glu (E) forms polar contacts with Z15:Arg (R) and Z18:Arg (R) (Nair and Burley 2003). Although most species have polar residues at these sites, they are not highly conserved and human c-Myc is the only sequence to have a negatively charged residue at Z11. Generally, Myc Z17 is composed of positively charged residues and Z18 is polar. In contrast, Z10:Asp-Z15′:Glu (DE') and Z17:Lys-Z22′:Arg (KR′) repulsive forces in C. elegans Mxl-1 prevent homodimerization, where ′ marks the opposing monomer (Yuan et al. 1998). Hence, the charge and polarity of Z10, Z15, Z17, and Z18 may appreciably influence the binding affinities among Max and Mlx network proteins. These patterns of conservation imply Myc, Max, and Mxd dimerization preferences are largely conserved among all species apart from deviations in nematode interactions.
Still, several residues within Mxd bHLHZ have previously been documented as unique to the Mxd family (Yuan et al. 1998). These distinctly conserved residues include H106:Cys (C), L16:Thr-H201:Thr-H202:Leu (TTL), H211:His-H212:Ile (HI), and Z17:Glu-Z18:Gln (EQ). However, site H6:Cys is not Mxd specific as Mnt is invariant for cysteine and Mxd4 contains a tyrosine (Y) in all sampled species. Additionally, conservation of H211:His-H212:Ile (HI) applies only to Mxd duplicates in vertebrates because Phe-Ile (FI) is conserved among arthropods and variable otherwise. Our results confirm conservation of L16:Thr-H201:Thr-H202:Leu (TTL) in all Mxd orthologs including MDL-1, with comparable conservation of L16:Ser-H201:Asn-H202:Leu (SNL) in Mnt. This differs from Myc variability between alanine and proline at L16 and invariability of lysine and valine at sites H201 and H202, respectively. Similarly, Mondo and Mlx are highly conserved at sites H107 (F/Y) and H202 (A/A). Strict AA conservation at these sites conveys their specific role in structure and function, such as the van der Waals contacts site H107 forms with H201 and H204 (Atchley and Fernandes 2005). Together, sites H107, H201, and H202 discriminate the Max and Mlx protein groups and reveal their potential involvement in distinguishing protein structures.
Network Topologies Have Distinct bHLHZ Sequences
Variations in network topology may also impose disparate restrictions on Max and Mlx network members. To infer potential structural or functional differences among major species groups, we examine protein orthologs in 1) core, 2) nematode, 3) Diptera, and 4) vertebrate networks and identify discriminating sites among network topologies. Because the alphabetic nature of AA sequences does not provide a basis for rigorous statistical procedures, we transformed each aligned protein sequence into five biologically relevant physicochemical metrics (See Materials and Methods) (Atchley et al. 2005). This permits the residues within each AA sequence to be compared according to their multidimensional physicochemical properties, that is, PAH, PSS, MS, CC, and EC. Stepwise DA was performed on orthologous proteins using each metric separately to identify the best discriminating sites among networks (table 4).
Nematode sequences showed the greatest amount of divergence for all orthologous proteins. Using the protein structure prediction program 3DJigsaw (Bates et al. 2001), we predicted the structure for Mxl-1:MDL-1, Mxl-2:MML-1, and Mxl-3:Mxl-3 dimers based on Protein Data Bank structures 1NLW, 1NKP, and 1HLO, respectively (Brownlie et al. 1997; Nair and Burley 2003). This allowed us to view the relative location of invariant residues and nematode-specific sites within the dimer complex (fig. 8). The proximity of hydrophobic residues H106:His (H) and H203′:Leu (L) in Mxl-3 may strengthen monomer interactions as compared with the polar H106:Ser (S) and H203:Gln (Q) residues conserved in Max sequences of other species. Discriminating sites in Mxl-3 appear to face away from the DNA and dimer interface, whereas distinct changes in Mxl-1 occur throughout the DNA- and protein-binding region. This further suggests that Mxl-1 is divergent from Max, possibly from relaxed selection after duplication, which may result in variable binding affinities with dimerization partner MDL-1. MDL-1 experienced only a few changes, which are also present in other vertebrate Mxd family members. Specifically, MDL-1 b4:Ala (A) and Mxd3 b4:Val (V) similarly changed to nonpolar residues, whereas MDL-1 H102:Asn (N) and Mxd3 H102:Gln (Q) replaced positively charged residues.
FIG. 8.—

Nematode bHLHZ structure. Caenorhabditis elegans dimers recognizing DNA (yellow). Sites distinguishing nematode orthologs are colored green, whereas identical sites for all orthologs are red. Backbone and side chain atoms for these sites are displayed. (A) Mxl-1 (blue) and MDL-1 (purple) heterodimer. Identical Max sites are not shown for this structure. (B) Full Mxl-2 sequence (blue) and MML-1 bHLHZ (purple) heterodimer. (C) Mxl-3 (blue) homodimer.
Nematodes also exhibit distinctions in Mxl-2 and MML-1 interacting partners. Nematode Mlx-2 shows disparity at nearby sites H111:Lys (K), H201:Asn (N), and H206:Phe (F) compared with the otherwise conserved H111:Gln (Q) and H201:Lys (K) sites observed in other Mlx sequences. Meanwhile, MML-1 has a contrasting surface consisting of sites b11:Asn, H102:Ala, H105:Asp, and H109:Gln (N, A, D, Q) that faces away from the dimer complex. In other bHLHZ proteins, such as SREBP and PHO4, sites b11, H102, and H201 are known to contact the phosphate backbone (Atchley and Zhao 2007), suggesting that nematodes may have altered DNA-binding patterns. These differences in nematode orthologs account for the majority of variability among network members (table 4).
In contrast, Max bHLHZ is highly conserved, with an expected 0.003 AA difference per million years, which is 16 times lower than that for Myc bHLHZ (Atchley and Fitch 1995). Interestingly, both Max and Myc bHLHZ domains required numerous sites to explain at least 90% of variability between network configurations. Because Max is a highly conserved sequence with minimal variation and Myc contains multiple changes that overlap network topologies, there was little structured variability upon which DA could easily distinguish classes. No sites were able to directly discriminate Max in the Diptera network, and only sites H108:His (H), H206:Asp (D), Z5:His (H), and Z19:Ala (A) showed any power in discriminating Max vertebrate sequences due to their changes in codon composition and charge. Although these sites have not been previously annotated for conserved structure or function, the proximity of negatively charged H108:His (H) and positively charged H206′:Glu (E) on opposing Max monomers may form stable contacts in vertebrates. In other species, the charge of Max H108 is largely neutral, whereas Max H206 is positive.
Myc also exhibited only minor differences between networks, with the Diptera lineage mostly divergent by changes in hydrophobicity. Drosophila melanogaster Myc b3:Asn (N) and H203:Asn (N) lost, whereas H111:Lys gained hydrophobic properties compared with almost all other species. Site H102 differed in both Diptera Myc and vertebrate c-Myc compared with the otherwise conserved Asp (D) residue, where c-Myc H102:Glu (E) is bigger and dMyc H102:Gly (G) is smaller and not negatively charged. Sites b4:Thr (T), H108:Phe (F), and Z22:Lys (K) also discriminated c-Myc, whereas L-Myc displayed differences in aromatic b7 and neutrally charged H206. Interestingly, N-Myc showed overlapping similarities with either L-Myc or c-Myc at these residues and had no significantly discriminating sites of its own. Although most of these sites are not structurally or functionally annotated, they are in close proximity to the DNA and may affect binding abilities and hence alter patterns of transcriptional regulation.
Primarily, residues within loop and zipper regions discriminated Mlx and Mondo orthologs among core, Diptera, and vertebrate networks. Vertebrate paralogs MondoA and MondoB have H215:Ser (S) instead of proline that characteristically kinks and terminates the first α-helix in Max network members. MondoA also has a shorter loop consisting of only seven residues, whereas MondoB resembles ancestral Mondo loop sequence with 11 residues and a proline at L6. As seen with Mlx:MondoB interactions, variability in the loop sequence is likely to have a prominent role in determining dimer and higher order conformations. However, vertebrates may have slightly different conformations due to the acquired charge at Mlx sites Z2:Lys (K) and Z3:Glu (E) and polar residues H204:Thr (T) and H208:Thr (T) for both MondoA and MondoB. Other changes in the Diptera lineage include Mlx Z15 that is not positively charged, Mlx Z24 that is aliphatic, and distinct aliphatic residues at Z25 and Z28 in Mondo.
Do Mlx Interacting Proteins Have Distinct bHLHZ Attributes?
Dimerization experiments have not been performed in an organism from the core network and must be inferred from orthologous network interactions. Mnt:Max, Myc:Max, and Mondo:Mlx heterodimers have been verified in both vertebrates and Drosophila, implicating their interactions are ancestral. The Mxd:Max interaction is also assumed to be ancestral because all Mxd family proteins can heterodimerize with Max and MDL-1 can interact with both Max orthologs (Baudino and Cleveland 2001).
Dimerization properties restricting Mlx interactions are currently unknown. Notably, the interaction between Mnt and Mlx is unresolved due to conflicting evidence (Meroni et al. 1997, 2000; Cairo et al. 2001; Billin and Ayer 2006). If Mnt does not interact with Mlx, Max and Mlx networks are decoupled in both fly and nematode lineages, and Mondo lacks a known repressor counterpart within the Mlx network. In vertebrates, Mxd1 and Mxd4 can heterodimerize with Mlx, whereas Mxd2 and Mxd3 cannot. MDL-1 cannot interact with Mlx ortholog Mxl-2 in nematodes (Cairo et al. 2001; Billin and Ayer 2006), suggesting that Mxd can dimerize only with Max in the core network and the interaction between Mxd1 and Mxd4 with Mlx is derived. Because the Mxd bHLHZ domain has a strictly defined loop consisting of nine residues, these binding restrictions are likely the result of specific residue changes within homologous sites.
To predict if Mxd can heterodimerize with Mlx in species belonging to the core network, we used Mxd protein family members to identify sites that discriminate Mlx-binding properties. Using vertebrate Mxd sequences grouped according to Mlx binding ability, we applied DA on the factor-transformed sequences to identify sites that maximally discriminate between binding groups (see Materials and Methods). DA weights sites to standardize variability within groups and maximize among group variation. The resulting linear discriminant function gives the greatest separation among a priori defined groups. In this case, DA estimates site coefficients to discriminate Mxd proteins able (Mxd1, Mxd4) and unable (Mxd2, Mxd3) to bind Mlx.
In vertebrates, 25 of the 80 Mxd bHLHZ sites are invariable (fig. 7, capital letters). Of the remaining variable sites, the size of Z15, quantified by the factor score transformation, explains 90% of variability between Mxd-and Mlx-binding groups. Factor scores quantifying secondary structure, codon composition, and charge of Z15 also contribute to Max- and Mlx-binding discrimination by also accounting for a large portion of variability between binding groups. Mxd1 and Mxd4 Z15 are invariant for Gln (Q), whereas Mxd2 Glu (E) and Mxd3 Arg (R) are charged. Site Z8 PAH also shows discriminatory power, although it is not conserved in Mxd1 (ILVMQ), Mxd2 (QR), or Mxd3 (QRKE) and has overlapping properties. Site Z15 and Z8 are variable among invertebrates with the observed AAs SCNQRKAEY and VINTAMLEDQ, respectively, showing no clear pattern of size, charge, or hydrophobicity conservation.
Based on these DA results, we then predicted the binding ability of unclassified Mxd sequences by their posterior probability of membership to a particular Mlx-binding group. That is, we let the discriminant function classify unknown data. Although the linear discriminant function completely and correctly classifies known binding partners, binding of nonvertebrate Mxd members is indeterminate. PAH (47.83%), PSS (50%), MS (30.43%), CC (30.43%), and EC (39.13%) metrics predict less than half of Mxd sequences within the core network can dimerize with Mlx. This indicates Mxd in invertebrates is unlikely to dimerize with Mlx, although it cannot be firmly established.
Differences within Mxd and Mnt sequences prevent adequate prediction of Mlx binding. However, Mnt is largely conserved among all species sampled, which indicates Mnt:Mlx binding is consistent among all species. Sites L1, L3, and Z23 differentiate the Diptera lineage, although D. melanogaster shows additional variability with no distinct conservation among AA attributes. Mnt H204:Val (V) discriminates vertebrates from the otherwise conserved isoleucine in other species, whereas vertebrate Z21:Thr (T) is larger than residues in sequences from Diptera and core networks. As H204 and Z21 are involved in dimerization interactions, they may further specify Mnt-binding restrictions and abilities among species.
Conclusions and Summary
Max and Mlx network members are found in the earliest known precursor organisms to animals and throughout the animal kingdom. Retention of these proteins over a billion years of evolution in such a diverse array of organisms suggests that the Max and Mlx networks have vital roles in cell regulation and organismal development. The presence of Myc and Max in choanoflagellate M. brevicollis further verifies their evolution is both ancient and highly constrained.
Clear points of radiation and deletion shape the four major network configurations found in animals. However, some species exhibit losses for certain members of the Max and Mlx networks. This can be attributed to 1) lineage-specific duplication or deletion, 2) gene pseudogenization, 3) low coverage or unassembled genomes, and 4) unidentifiable orthology due to gene divergence. Although we are unable to identify the bHLHZ domain of some network members, it is still plausible that they exist, even in ancient lineages, such as Trichoplax and Monosiga. Other cases, including chromosomal translocations surrounding N-Myc of M. domestica and absence of Mxd in ticks, imply a lineage-specific gene loss may have occurred.
Although the ancestral divergence of trematodes is uncertain (Carranza et al. 1997), we provide evidence that nematodes and trematodes shared a common ancestor prior to arthropod divergence. The absence of an identifiable Myc or Mnt ortholog in Schistosoma and similar patterns in divergence for Max-, Mlx-, and Mondo-like sequences suggests that nematodes and trematodes experienced a major reconfiguration of the Max and Mlx networks. Moreover, the absence of a second Max ortholog in both Schistosoma and B. malayi and similarity between Mxl-3 and Max suggests that Mxl-1 originated from a duplication of Mxl-3 in Caenorhabditis. Likewise, nematode Mxl-2 and trematode Mlx divergence occur at similar sites, yet both still demonstrate clear sequence orthology to Mlx. We predict the nematode-specific divergence at packed sites H111:Lys (K) and H201:Asn (N) in Mlx-2 exhibit compensatory changes for the otherwise conserved H111:Gln (Q) and H201:Lys (K) Mlx sites. In addition, inconsistent changes in hydrophobicity, accessibility, and size suggest the region around b11, H102, H105, and H109 in nematode MML-1 either lost or altered its involvement with an interacting partner.
Stability in clade structure among phylogenetic reconstructions indicates the bHHZ sequences of Max, Mlx, Myc, Mondo, Mnt, Mxd, and Mga have distinguishable sequence attributes that contribute to their dimerization and DNA binding with similar patterns of conservation that have been retained over millions of years of evolution. We think that the similarity between Mondo and Mlx bHHZ domains probably results from dimerization constraints and unique gene targets within the parallel network. Because Mnt and Mxd bHHZ domains do not interact, we anticipate their similarity relates to their role in gene repression. In contrast, the dissociation of Mondo and Myc proteins with transactivation activity denotes independent dimerization and DNA-binding attributes. The lack of a distinct Myc clade further highlights its diversity and insinuates Myc orthologs have different propensities in dimerization and transcriptional regulation. Mga is a “wandering taxon” that is phylogenetically unstable and not consistently grouped with any outgroup sequences. Thus, we predict Mga rapidly diverged after duplication of a Max or Mlx network member and was subsequently conserved. Furthermore, paralogs in vertebrate protein families formed separate clades and sequences generally bifurcated in order of species divergence, demonstrating strong selective forces are acting on these sequences.
Several sites exhibit common and unique characteristics of the bHLHZ domain that depict the divergence of Max and Mlx network members in animals. Sites b5, b9, b12, b13, H110, and H205 are largely invariant among network members due to site-specific restrictions in E-box DNA binding and dimerization stability. Likewise, sites b2, H103, H104, and H215 have low functional entropy values presumably due to their role in contacting the DNA phosphate backbone and involvement in protein conformation. Although the zipper is required for stable dimerization, the relatively low entropy of Z14 and Z21 suggests that these leucine repeats are important contact points between monomers.
Using DA, we statistically identified specific sites that distinctly classify proteins, network topologies, and potential dimerization patterns. Sites H107, H201, and H202 completely discriminate Max and Mlx network proteins. While site H202 is not annotated, site H201 forms van der Waals contacts with H107 and anchors the second helix to DNA (Atchley and Zhao 2007). Such variability in important residues likely alters both DNA- and protein-binding abilities that determine gene target recognition and protein function.
Similarly, changes among orthologs may display evolutionary adaptations. Specifically, site b3 in Myc is unconserved in invertebrate sequences, which can affect DNA recognition and transformation capabilities. Interestingly, N-myc had overlapping similarities with c-Myc and L-Myc discriminating sites with no distinct sites of its own, suggesting that these changes have cumulative or compensatory effects among Myc family members. Protein dimerization may also differ among species due to variability between Max and Mlx network members at sites Z17 and Z18, which were found to attribute Max dimerization preferences. Similarity in loop length and conservation between MondoB and invertebrate Mondo sequences suggests that they have corresponding dimerization and DNA-binding restrictions. However, heterotetramer conformation may differ in invertebrates due to the lack of L8:Phe (F). Instead, this higher order structure may rely on negatively charged L7:Asp (D) and hydrophobic L11:Gly (G) residues, which are highly conserved among animal species. Although our results emphasize the potential importance of particular sites in the bHLHZ domain, mutation experiments are still necessary to validate the contribution of these residues and other discriminating sites in DNA and protein interactions.
Dimerization properties among Max and Mlx network members have been investigated in vivo for C. elegans, D. melanogaster, and M. musculus (Blackwood et al. 1992; Amati and Land 1994; Arsura et al. 1995; Yuan et al. 1998; Hurlin et al. 1999; Billin et al. 2000; Meroni et al. 2000; Cairo et al. 2001; Orian et al. 2003). However, interactions between members in the core network are unknown. Although our predictions for invertebrate Mxd binding using DA were indeterminable, we anticipate Mxd1 and Mxd4 binding with Mlx is derived and results from independent changes within the bHLHZ domain. Furthermore, conflicting reports on Mnt and Mlx heterodimerization raise several questions concerning the extent of Mnt repression and Mondo regulation.
For example, do Mad or Mnt competitively dimerize with Mlx to regulate Mondo? How does the loss of Mxd2 and Mxd3 or gain of Mxd1 and Mxd4 binding with Mlx affect Mondo regulation in vertebrates? Does loss of Mad in flies change dMnt function? Although Mxd is dispensable in flies and individual knockouts in mice have minor changes in phenotype, the persistence of Mxd in most other species including nematodes indicates that it has a basic and important role in cell maintenance.
These evolutionary analyses provide a basis for understanding important aspects of Max and Mlx network interactions and function in animals. Although no direct ortholog of Myc or Max has been found in yeast (Brown et al. 2008), yeast contains interacting homologs Sin3 and GCN5 as well as E-boxes and may still be harboring unidentified Max and Mlx network orthologs. Using the protein distinctions we have described, it is now possible to distinguish Max and Mlx network member bHLHZ domains, search for unannotated sequences in highly divergent species, and attribute structural and functional differences among these proteins. Hence, these results will enable the refinement of protein annotation within an evolutionary context of network interactions and facilitate the functional analysis of important proteins, such as the Myc proto-oncogene.
Supplementary Material
Supplementary figure is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank Dr. Richard Gibbs and the Honey Bee Genome Sequencing Consortium for making their data publicly available and the BCM-HGSC for providing the genome and BLAST service. This work was supported in part by the National Institutes of Health (R01GM070806).
References
- Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Alberghina L, Höfer T, Vanoni M. Molecular networks and system-level properties. J Biotechnol. 2009;144(3):224–233. doi: 10.1016/j.jbiotec.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amati B, Land H. Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Genet Dev. 1994;4(1):102–108. doi: 10.1016/0959-437x(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Amati B, Littlewood TD, Evan GI, Land H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993;12(13):5083–5087. doi: 10.1002/j.1460-2075.1993.tb06202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anong WA, et al. Adducin forms a bridge between the erythrocyte membrane and its cytoskeleton and regulates membrane cohesion. Blood. 2009;114(9):1904–1912. doi: 10.1182/blood-2009-02-203216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnason U, Gullberg A, Janke A. Molecular timing of primate divergences as estimated by two nonprimate calibration points. J Mol Evol. 1998;47(6):718–727. doi: 10.1007/pl00006431. [DOI] [PubMed] [Google Scholar]
- Arsura M, Deshpande A, Hann SR, Sonenshein GE. Variant Max protein, derived by alternative splicing, associates with c-Myc in vivo and inhibits transactivation. Mol Cell Biol. 1995;15(12):6702–6709. doi: 10.1128/mcb.15.12.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, Fernandes AD. Sequence signatures and the probabilistic identification of proteins in the Myc-Max-Mad network. Proc Natl Acad Sci U S A. 2005;102(18):6401–6406. doi: 10.1073/pnas.0408964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, Fitch WM. Myc and Max: molecular evolution of a family of proto-oncogene products and their dimerization partner. Proc Natl Acad Sci U S A. 1995;92(22):10217–10221. doi: 10.1073/pnas.92.22.10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, Fitch WM. A natural classification of the basic helix-loop-helix class of transcription factors. Proc Natl Acad Sci U S A. 1997;94(10):5172–5176. doi: 10.1073/pnas.94.10.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, Terhalle W, Dress A. Positional dependence, cliques, and predictive motifs in the bHLH protein domain. J Mol Evol. 1999;48(5):501–516. doi: 10.1007/pl00006494. [DOI] [PubMed] [Google Scholar]
- Atchley WR, Zhao J. Molecular architecture of the DNA-binding region and its relationship to classification of basic helix-loop-helix proteins. Mol Biol Evol. 2007;24(1):192–202. doi: 10.1093/molbev/msl143. [DOI] [PubMed] [Google Scholar]
- Atchley WR, Zhao J, Fernandes AD, Druke T. Solving the protein sequence metric problem. Proc Natl Acad Sci U S A. 2005;102(18):6395–6400. doi: 10.1073/pnas.0408677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A, Oltvai Z. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Bates PA, Kelley LA, MacCallum RM, Sternberg MJ. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins. 2001;(5suppl):39–46. doi: 10.1002/prot.1168. [DOI] [PubMed] [Google Scholar]
- Baudino TA, Cleveland JL. The Max network gone mad. Mol Cell Biol. 2001;21(3):691–702. doi: 10.1128/MCB.21.3.691-702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benassayag C, et al. Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol Cell Biol. 2005;25(22):9897–9909. doi: 10.1128/MCB.25.22.9897-9909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billin AN, Ayer DE. The Mlx network: evidence for a parallel Max-like transcriptional network that regulates energy metabolism. Curr Top Microbiol Immunol. 2006;302:255–278. doi: 10.1007/3-540-32952-8_10. [DOI] [PubMed] [Google Scholar]
- Billin AN, Eilers AL, Coulter KL, Logan JS, Ayer DE. MondoA, a novel basic helix-loop-helix-leucine zipper transcriptional activator that constitutes a positive branch of a max-like network. Mol Cell Biol. 2000;20(23):8845–8854. doi: 10.1128/mcb.20.23.8845-8854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, Lüscher B, Eisenman RN. Myc and Max associate in vivo. Genes Dev. 1992;6(1):71–80. doi: 10.1101/gad.6.1.71. [DOI] [PubMed] [Google Scholar]
- Brown S, Cole M, Erives A. Evolution of the holozoan ribosome biogenesis regulon. BMC Genomics. 2008;9(1):442. doi: 10.1186/1471-2164-9-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlie P, et al. The crystal structure of an intact human Max-DNA complex: new insights into mechanisms of transcriptional control. Structure. 1997;5(4):509–520. doi: 10.1016/s0969-2126(97)00207-4. [DOI] [PubMed] [Google Scholar]
- Budd G, Telford M. The origin and evolution of arthropods. Nature. 2009;457(7231):812–817. doi: 10.1038/nature07890. [DOI] [PubMed] [Google Scholar]
- Burton RA, Mattila S, Taparowsky EJ, Post CB. B-myc: N-terminal recognition of myc binding proteins. Biochemistry. 2006;45(32):9857–9865. doi: 10.1021/bi060379n. [DOI] [PubMed] [Google Scholar]
- Cairo S, Merla G, Urbinati F, Ballabio A, Reymond A. WBSCR14, a gene mapping to the Williams–Beuren syndrome deleted region, is a new member of the Mlx transcription factor network. Hum Mol Genet. 2001;10(6):617–627. doi: 10.1093/hmg/10.6.617. [DOI] [PubMed] [Google Scholar]
- Carranza S, Baguñà J, Riutort M. Are the Platyhelminthes a monophyletic primitive group? An assessment using 18S rDNA sequences. Mol Biol Evol. 1997;14(5):485–497. doi: 10.1093/oxfordjournals.molbev.a025785. [DOI] [PubMed] [Google Scholar]
- Chapman JA, et al. The dynamic genome of Hydra. Nature. 2010;464(7288):592–596. doi: 10.1038/nature08830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron J, et al. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992;6(12A):2248–2257. doi: 10.1101/gad.6.12a.2248. [DOI] [PubMed] [Google Scholar]
- Coghlan A. Nematode genome evolution [Internet] 2005. WormBook. ed. The C. elegans Research Community; 2005 [cited 2011 Aug 29]. Available from: http://www.wormbook.org/chapters/www_genomevol/genomevol.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium Bovine Genome Sequencing and Analysis, et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science. 2009;324(5926):522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium Honeybee Genome Sequencing. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443(7114):931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium International Aphid Genomics. Genome sequence of the pea aphid Acyrthosiphon pisum. Plos Biol. 2010;8(2):e1000313. doi: 10.1371/journal.pbio.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium International Chicken Genome Sequencing. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Consortium International Silkworm Genome. The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol. 2008;38(12):1036–1045. doi: 10.1016/j.ibmb.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Consortium Sea Urchin Genome Sequencing. The genome of the sea urchin Strongylocentrotus purpuratus. Science. 2006;314(5801):941–952. doi: 10.1126/science.1133609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium Tribolium Genome Sequencing. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452(7190):949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19(1):1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, McGuire M, Buckmire M, Lee WM. Involvement of the ‘leucine zipper’ region in the oligomerization and transforming activity of human c-myc protein. Nature. 1989;337(6208):664–666. doi: 10.1038/337664a0. [DOI] [PubMed] [Google Scholar]
- Dang CV, et al. The c-Myc target gene network. Semin Cancer Biol. 2006;16(4):253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7(4):671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- Dehal P, Boore J. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3(10):e314. doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P, et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science. 2002;298(5601):2157–2167. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- de Luis O, Valero MC, Jurado LA. WBSCR14, a putative transcription factor gene deleted in Williams-Beuren syndrome: complete characterisation of the human gene and the mouse ortholog. Eur J Hum Genet. 2000;8(3):215–222. doi: 10.1038/sj.ejhg.5200435. [DOI] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Thomas WK. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature. 2004;430(7000):679–682. doi: 10.1038/nature02697. [DOI] [PubMed] [Google Scholar]
- Depinho R, Hatton K, Tesfaye A, Yancopoulos G, Alt F. The human myc gene family: structure and activity of L-myc and an L-myc pseudogene. Genes Dev. 1987;1(10):1311–1326. doi: 10.1101/gad.1.10.1311. [DOI] [PubMed] [Google Scholar]
- Dillon WR, Westin S. Scoring frequency data for discriminant analysis: perhaps discrete procedures can be avoided. J Mark Res. 1982;19(1):44–56. [Google Scholar]
- Doskocil J. The amplification of oligonucleotide themes in the evolution of the myc protooncogene family. J Mol Evol. 1996;42(5):512–524. doi: 10.1007/BF02352281. [DOI] [PubMed] [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological sequence analysis: probabilistic models of proteins and nucleic acids. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6. Seattle (WA): Distributed by the author. Department of Genome Sciences, University of Washington; 2005. [Google Scholar]
- Ferré-D'Amaré AR, Prendergast GC, Ziff EB, Burley SK. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature. 1993;363(6424):38–45. doi: 10.1038/363038a0. [DOI] [PubMed] [Google Scholar]
- Fisher R. The use of multiple measurements in taxonomic problems. Ann Eugen. 1936;7:179–188. [Google Scholar]
- Flicek P, et al. Ensembl's 10th year. Nucleic Acids Res. 2010;38(Database issue):D557–D562. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EJ, et al. PRELI (protein of relevant evolutionary and lymphoid interest) is located within an evolutionary conserved gene cluster on chromosome 5q34-q35 and encodes a novel mitochondrial protein. Biochem J. 2004;378(3):817–825. doi: 10.1042/BJ20031504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Ishihara S, Kaneko K, Hogeweg P. Network evolution of body plans. PLoS One. 2008;3(7):e2772. doi: 10.1371/journal.pone.0002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant P. Myc/Max/Mad in invertebrates: the evolution of the Max network. Curr Top Microbiol Immunol. 2006;302:235–253. doi: 10.1007/3-540-32952-8_9. [DOI] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14(7):685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- Gibbs R, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428(6982):493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- Grinberg AV, Hu CD, Kerppola TK. Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Mol Cell Biol. 2004;24(10):4294–4308. doi: 10.1128/MCB.24.10.4294-4308.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Gupta BP, Sternberg PW. The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans. Genome Biol. 2003;4(12):238. doi: 10.1186/gb-2003-4-12-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson KD, Shichiri M, Follansbee MR, Sedivy JM. Effects of c-myc expression on cell cycle progression. Mol Cell Biol. 1994;14(9):5748–5755. doi: 10.1128/mcb.14.9.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, et al. WormBase: a comprehensive resource for nematode research. Nucleic Acids Res. 2010;38(Database issue):D463–D467. doi: 10.1093/nar/gkp952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton K, et al. Expression and activity of L-Myc in normal mouse development. Mol. Cell Biol. 1996;16(4):1794–1804. doi: 10.1128/mcb.16.4.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges S. The origin and evolution of model organisms. Nat Rev Genet. 2002;3(11):838–849. doi: 10.1038/nrg929. [DOI] [PubMed] [Google Scholar]
- Hill CA, Wikel SK. The Ixodes scapularis Genome Project: an opportunity for advancing tick research. Trends Parasitol. 2005;21(4):151–153. doi: 10.1016/j.pt.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Hillier L, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5(2):183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Hooker CW, Hurlin P. Of Myc and Mnt. J Cell Sci. 2006;119(Pt 2):208–216. doi: 10.1242/jcs.02815. [DOI] [PubMed] [Google Scholar]
- Hurlin PJ, Quéva C, et al. Mad3 and Mad4: novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995;14(22):5646–5659. doi: 10.1002/j.1460-2075.1995.tb00252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, Quéva C, Eisenman RN. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 1997;11(1):44–58. doi: 10.1101/gad.11.1.44. [DOI] [PubMed] [Google Scholar]
- Hurlin PJ, Steingrìmsson E, Copeland NG, Jenkins NA, Eisenman RN. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 1999;18(24):7019–7028. doi: 10.1093/emboj/18.24.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, et al. Evidence of mnt-myc antagonism revealed by mnt gene deletion. Cell Cycle. 2004;3(2):97–99. [PubMed] [Google Scholar]
- Joint Genome Institute (JGI) [Internet] 2010 Walnut Creek (CA): Department of Energy Joint Genome Institute [updated 2011 Jun 16; cited 2011 Aug 29]. Available from: http://www.jgi.doe.gov. [Google Scholar]
- Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5(6):226. doi: 10.1186/gb-2004-5-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue):D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, et al. Domain shuffling and the evolution of vertebrates. Genome Res. 2009;19(8):1393–1403. doi: 10.1101/gr.087072.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36(2):189–204. doi: 10.1016/s1357-2725(03)00211-5. [DOI] [PubMed] [Google Scholar]
- King N, et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008;451(7180):783–788. doi: 10.1038/nature06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21(5):676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lawson D, et al. VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res. 2009;37(Database issue):D583–D587. doi: 10.1093/nar/gkn857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424(6945):147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438(7069):803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Loo LW, et al. The transcriptional repressor dMnt is a regulator of growth in Drosophila melanogaster. Mol Cell Biol. 2005;25(16):7078–7091. doi: 10.1128/MCB.25.16.7078-7091.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher B. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene. 2001;277(1–2):1–14. doi: 10.1016/s0378-1119(01)00697-7. [DOI] [PubMed] [Google Scholar]
- Lüscher B, Larsson LG. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene. 1999;18(19):2955–2966. doi: 10.1038/sj.onc.1202750. [DOI] [PubMed] [Google Scholar]
- Ma L, Robinson LN, Towle HC. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem. 2006;281(39):28721–28730. doi: 10.1074/jbc.M601576200. [DOI] [PubMed] [Google Scholar]
- Ma L, Sham YY, Walters KJ, Towle HC. A critical role for the loop region of the basic helix-loop-helix/leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res. 2007;35(1):35–44. doi: 10.1093/nar/gkl987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J Biol Chem. 2005;280(12):12019–12027. doi: 10.1074/jbc.M413063200. [DOI] [PubMed] [Google Scholar]
- Maerkl SJ, Quake SR. Experimental determination of the evolvability of a transcription factor. Proc Natl Acad Sci U S A. 2009;106(44):18650–18655. doi: 10.1073/pnas.0907688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20(2):429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy AA. Broad institute: bringing genomics to real-world medicine. Chem Biol. 2005;12(7):717–718. doi: 10.1016/j.chembiol.2005.07.003. [DOI] [PubMed] [Google Scholar]
- McDonald WH, Pavlova Y, Yates JR 3rd, Boddy MN. Novel essential DNA repair proteins Nse1 and Nse2 are subunits of the fission yeast Smc5-Smc6 complex. J Biol Chem. 2003;278(46):45460–45467. doi: 10.1074/jbc.M308828200. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Van Buskirk HA, Dugan KA, Copland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94(3):363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- Meroni G, et al. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 1997;16(10):2892–2906. doi: 10.1093/emboj/16.10.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meroni G, et al. Mlx, a new Max-like bHLHZip family member: the center stage of a novel transcription factors regulatory pathway? Oncogene. 2000;19(29):3266–3277. doi: 10.1038/sj.onc.1203634. [DOI] [PubMed] [Google Scholar]
- Mikkelsen T, et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature. 2007;447(7141):167–177. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- Moens CB, Stanton BR, Parada LF, Rossant J. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development. 1993;119(2):485–499. doi: 10.1242/dev.119.2.485. [DOI] [PubMed] [Google Scholar]
- Moore A, Björklund Å, Ekman D, Bornberg-Bauer E, Elofsson A. Arrangements in the modular evolution of proteins. Trends Biochem Sci. 2008;33(9):444–451. doi: 10.1016/j.tibs.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Morgenstern B, Atchley WR. Evolution of bHLH transcription factors: modular evolution by domain shuffling? Mol Biol Evol. 1999;16(12):1654–1663. doi: 10.1093/oxfordjournals.molbev.a026079. [DOI] [PubMed] [Google Scholar]
- Morton CC, et al. Mapping and characterization of an X-linked processed gene related to MYCL1. Genomics. 1989;4(3):367–375. doi: 10.1016/0888-7543(89)90344-3. [DOI] [PubMed] [Google Scholar]
- Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112(2):193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- Nair SK, Burley SK. Structural aspects of interactions within the Myc/Max/Mad network. Curr Top Microbiol Immunol. 2006;302:123–143. doi: 10.1007/3-540-32952-8_5. [DOI] [PubMed] [Google Scholar]
- Nene V, et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316(5832):1718–1723. doi: 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18(19):3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- Nilsson JA, et al. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol Cell Biol. 2004;24(4):1560–1569. doi: 10.1128/MCB.24.4.1560-1569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan RC, et al. Gene-target recognition among members of the myc superfamily and implications for oncogenesis. Nat Genet. 2000;24(2):113–119. doi: 10.1038/72761. [DOI] [PubMed] [Google Scholar]
- Orian A, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17(9):1101–1114. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C, Stoltzman C, Sighinolfi M, Han K, Ayer D. Glucose controls nuclear accumulation, promoter binding, and transcriptional activity of the MondoA-Mlx heterodimer. Mol Cell Biol. 2010;30(12):2887–2895. doi: 10.1128/MCB.01613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrefitte S, Kahn D, Haenlin M. New members of the Drosophila Myc transcription factor subfamily revealed by a genome-wide examination for basic helix-loop-helix genes. Mech Dev. 2001;104(1–2):99–104. doi: 10.1016/s0925-4773(01)00360-4. [DOI] [PubMed] [Google Scholar]
- Pickett C, Breen K, Ayer D. A C. elegans Myc-like network cooperates with semaphorin and Wnt signaling pathways to control cell migration. Dev Biol. 2007;310(2):226–239. doi: 10.1016/j.ydbio.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, et al. dMyc is required for larval growth and endoreplication in Drosophila. Development. 2004;131(10):2317–2327. doi: 10.1242/dev.01108. [DOI] [PubMed] [Google Scholar]
- Pierce SB, et al. Drosophila growth and development in the absence of dMyc and dMnt. Dev Biol. 2008;315(2):303–316. doi: 10.1016/j.ydbio.2007.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- Putnam N, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317(5834):86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- Putnam N, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453(7198):1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- Quackenbush J, et al. The TIGR Gene Indices: analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 2001;29(1):159–164. doi: 10.1093/nar/29.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy CD, et al. Mutational analysis of Max: role of basic, helix-loop-helix/leucine zipper domains in DNA binding, dimerization and regulation of Myc-mediated transcriptional activation. Oncogene. 1992;7(10):2085–2092. [PubMed] [Google Scholar]
- Robinson KA, Lopes JM. SURVEY AND SUMMARY: Saccharomyces cerevisiae basic helix-loop-helix proteins regulate diverse biological processes. Nucleic Acids Res. 2000;28(7):1499–1505. doi: 10.1093/nar/28.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rottmann S, Lüscher B. The Mad side of the Max network: antagonizing the function of Myc and more. Curr Top Microbiol Immunol. 2006;302:63–122. doi: 10.1007/3-540-32952-8_4. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sanger Genome Sequencing Projects BLAST [Internet] 2010 Sebrafish and C. elgans group. Hinxton (UK): Wellcome Trust Sanger Institute. [cited 2011 Aug 29]. Available from: http://www.sanger.ac.uk/resources/software/blast/ [Google Scholar]
- Sans C, Satterwhite D, Stoltzman C, Breen K, Ayer D. MondoA-Mlx heterodimers are candidate sensors of cellular energy Status: mitochondrial localization and direct regulation of glycolysis. Mol Cell Biol. 2006;26(13):4863–4871. doi: 10.1128/MCB.00657-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai S, et al. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development. 1993;117(4):1445–1455. doi: 10.1242/dev.117.4.1445. [DOI] [PubMed] [Google Scholar]
- Sayers EW, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010;38(Database issue):D5–D16. doi: 10.1093/nar/gkp967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber-Agus N, et al. Role of Mxi1 in ageing organ systems and the regulation of normal and neoplastic growth. Nature. 1998;393(6684):483–487. doi: 10.1038/31008. [DOI] [PubMed] [Google Scholar]
- Scott AL, Ghedin E. The genome of Brugia malayi—all worms are not created equal. Parasitol Int. 2009;58(1):6–11. doi: 10.1016/j.parint.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379–423. 623–656. [Google Scholar]
- Sharakhova MV, et al. Update of the Anopheles gambiae PEST genome assembly. Genome Biol. 2007;8(1):R5. doi: 10.1186/gb-2007-8-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Li H, et al. Essential role for Max in early embryonic growth and development. Genes Dev. 2000;14(1):17–22. [PMC free article] [PubMed] [Google Scholar]
- Shih HM, Liu Z, Towle HC. Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J Biol Chem. 1995;270(37):21991–21997. doi: 10.1074/jbc.270.37.21991. [DOI] [PubMed] [Google Scholar]
- Shih HM, Towle HC. Definition of the carbohydrate response element of the rat S14 gene. Evidence for a common factor required for carbohydrate regulation of hepatic genes. J Biol Chem. 1992;267(19):13222–13228. [PubMed] [Google Scholar]
- Siegal M, Promislow D, Bergman A. Functional and evolutionary inference in gene networks: does topology matter? Genetica. 2006;129(1):83–103. doi: 10.1007/s10709-006-0035-0. [DOI] [PubMed] [Google Scholar]
- Solomon DL, Amati B, Land H. Distinct DNA binding preferences for the c-Myc/Max and Max/Max dimers. Nucleic Acids Res. 1993;21(23):5372–5376. doi: 10.1093/nar/21.23.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava M, et al. The Trichoplax genome and the nature of placozoans. Nature. 2008;454(7207):955–960. doi: 10.1038/nature07191. [DOI] [PubMed] [Google Scholar]
- Steiger D, Furrer M, Schwinkendorf D, Gallant P. Max-independent functions of Myc in Drosophila melanogaster. Nat Genet. 2008;40(9):1084–1091. doi: 10.1038/ng.178. [DOI] [PubMed] [Google Scholar]
- Stein LD, et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003;1(2):E45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltzman CA, et al. Glucose sensing by MondoA: Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci U S A. 2008;105(19):6912–6917. doi: 10.1073/pnas.0712199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol Biol. 2008;3:6. doi: 10.1186/1748-7188-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyo-Oka K, et al. Loss of the Max-interacting protein Mnt in mice results in decreased viability, defective embryonic growth and craniofacial defects: relevance to Miller-Dieker syndrome. Hum Mol Genet. 2004;13(10):1057–1067. doi: 10.1093/hmg/ddh116. [DOI] [PubMed] [Google Scholar]
- Tweedie S, et al. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37(Database issue):D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam T, Snel B, Pilpel Y. Protein complex evolution does not involve extensive network rewiring. PLoS Comput Biol. 2008;4(7):e1000132. doi: 10.1371/journal.pcbi.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh B, et al. Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 2007;5(4):e101. doi: 10.1371/journal.pbio.0050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Walker W, Zhou ZQ, Ota S, Wynshaw-Boris A, Hurlin P. Mnt-Max to Myc-Max complex switching regulates cell cycle entry. J Cell Biol. 2005;169(3):405–413. doi: 10.1083/jcb.200411013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WC, et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453(7192):175–183. doi: 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington University Genome Center. 2010 Genome Institute BLAST Server [Internet]. St. Louis (MO): Washington University School of Medicine. [cited 2011 Aug 29]. Available from: http://genome.wustl.edu/tools/blast. [Google Scholar]
- Werren JH, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327(5963):343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherspoon DJ, Robertson H. Neutral evolution of ten types of mariner transposons in the genomes of Caenorhabditis elegans and Caenorhabditis briggsae. J Mol Evol. 2003;56(6):751–769. doi: 10.1007/s00239-002-2450-x. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yuan J, Tirabassi RS, Bush AB, Cole MD. The C. elegans MDL-1 and MXL-1 proteins can functionally substitute for vertebrate MAD and MAX. Oncogene. 1998;17(9):1109–1118. doi: 10.1038/sj.onc.1202036. [DOI] [PubMed] [Google Scholar]
- Zhou ZQ, Hurlin PJ. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 2001;11(11):S10–S14. doi: 10.1016/s0962-8924(01)02121-3. [DOI] [PubMed] [Google Scholar]