

Abstract
Small molecules, namely, coactivator binding inhibitors (CBIs), that block estrogen signaling by directly inhibiting the interaction of the estrogen receptor (ER) with coactivator proteins act in a fundamentally different way than traditional antagonists, which displace the endogenous ligand estradiol. To complement our prior efforts at CBI discovery by de novo design, we used high-throughput screening to identify CBIs of novel structure and subsequently investigated two hits by analog synthesis, finding many compounds with low micromolar potencies in cell-based reporter gene assays. We examined structure-activity trends in both series, using induced-fit computational docking to propose binding poses for these molecules in the coactivator binding groove. Analysis of the structure of the ER-steroid receptor coactivator (SRC) complex suggests that all four hydrophobic residues within the SRC nuclear receptor box sequence are important binding elements. Thus, insufficient water displacement as the smaller CBIs bind at the expansive complexation site may be limiting the potency of compounds in these series, which suggests that higher potency CBIs might be found by screening compound libraries enriched in larger molecules.
Keywords (those from keyword list underlined): coactivator binding inhibitor, estrogen antagonist, estrogen receptor, molecular modeling, structure-activity relationships
1. Introduction
The estrogen receptor (ER), a member of the superfamily of ligand-regulated nuclear transcription factors, mediates the action of estrogens, including the primary endogenous ligand 17β-estradiol (E2).[1] ERs exist as two subtypes, ERα and ERβ, and because they are found in both reproductive (uterus, ovary, and breast) and non-reproductive (bone, brain, and the cardiovascular system) tissues, they have emerged as attractive therapeutic targets for the treatment of breast cancer, the prevention of osteoporosis, and the mitigation of menopausal symptoms via hormone replacement.[2] The binding of an estrogen to the ligand binding domain (LBD) of the ER forms a complex that interacts with specific DNA binding sites and recruits coregulators of the p160 class of steroid receptor coactivators (SRCs).[3] Because the SRC proteins mediate important alterations in chromatin structure and facilitate assembly of the transcriptional machinery, the recruitment of these proteins to the ER is essential for expression of ER-regulated genes.
Traditionally, the inhibition of ER activity has been achieved using antagonist molecules that bind to the ligand binding pocket in place of estradiol and trigger a conformational change that indirectly blocks coactivator binding by an intra-receptor or allosteric process.[4] An alternative and yet underexploited approach to blocking estrogen action involves small molecules that disrupt the interaction between agonist-activated ER and the SRC by an extra-receptor or direct competitive process. Such molecules are termed coactivator binding inhibitors (CBIs).[5]
SRCs, which exist as three subtypes (SRC-1, 2, and 3), possess multiple copies of a conserved, signature sequence motif, LXXLL (L is leucine and X is any amino acid), known as a nuclear-receptor interaction box (NR-box). X-ray crystal structures of several nuclear hormone receptor-agonist complexes bound to protein fragments of p160 coactivators or to peptides having one or more NR boxes have been solved. The coactivators bind to the nuclear receptor LBD through a two-turn amphipathic α-helical motif encompassing the NR box LXXLL signature sequence, with the ER-coactivator complex being further stabilized by interactions between the intrinsic dipole moment of the helical coactivator peptide backbone and charged residues from the ER at either end of the binding groove. The X-ray structure of the ERα complex with the second NR box of SRC-2 shows this interaction in detail (Figure 1a).[6] From this image it is evident that the first and third leucine residues of the SRC-2 NR-2 box ILHRLL peptide project downward into a short, but deep “hydrophobic groove” made up of several residues from helices 3, 4, 5, and 12 of the LBD. Notable as well, the second leucine and the preceding isoleucine residue (ILHRLL) rest on a largely “hydrophobic shelf” adjacent to the groove. All of these interactions are likely contributors to the high affinity binding of the SRC to the ER.
Figure 1.

(a) Crystal structure of GRIP1 peptide (red) on the surface of the ERα (brown = hydrophobic, green/blue = neutral to hydrophilic); (b) HTS hits of ERα coactivator binding inhibitors identified by a TR-FRET assay.
In spite of this detailed molecular portrayal of the site of receptor-coactivator interaction, only a few small-molecules have been found that bind to this hydrophobic surface groove-shelf region of the ER and block the interaction with coactivator (i.e., act as CBIs).[5a, 5b, 5e, 6b, 6c] With one exception,[5b] the ER CBIs reported thus far have been discovered using de novo design, and they have only micromolar affinities for ER. Given the recent availability of chemical libraries and screening facilities to academic researchers,[7] we were hopeful that we might use high throughput screening (HTS) to discover CBIs of novel structures having higher affinities that might be more biologically useful.
To this end, we developed and optimized a time-resolved fluorescence resonance energy transfer (TR-FRET) assay to screen large compound libraries for non-peptidic compounds that would show ERα CBI activity.[8] In this assay, the interaction between a europium-labeled ERα LBD and a Cy5-labeled fragment of SRC-3, induced upon estradiol binding to the ER, was monitored by TR-FRET, and an 86,000-member library of small molecules was screened for the ability to disrupt this interaction, monitored by a decrease in TR-FRET signal. This activity, followed by confirmatory assays we have described,[8] identified four distinct ERα-CBI scaffolds (1–4) with IC50 values of 5–30 μM that were selected for follow-up chemistry and structure-activity relationship (SAR) development (Figure 1b). All four compounds were re-synthesized and re-evaluated in the primary TR-FRET assay.
Curiously, samples of 2, 3 and 4 resynthesized in our laboratories showed no activity in the TR-FRET assay. The activity of re-synthesized 1 diminished somewhat compared with the original library sample, but it nevertheless showed distinct activities in both the TR-FRET assay and in a reporter gene assay (see below). Gratifyingly, analogs prepared in parallel with the resynthesis of 4 showed activity, even when the resynthesized version of the original hit compound was inactive within the concentration limits of our assay.
In the present work, we describe the optimization of two new series of CBIs, namely those based on the scaffolds of 1 and 4. In probing the structure-activity relationships in these series, we have utilized a cell-based ERα-mediated luciferase reporter gene assay to demonstrate that the compounds are both cell-permeable and active in a more biologically relevant assay. In addition, we used two different concentrations of estradiol in the reporter gene assay to indirectly confirm that the inhibitors do not bind at the ligand-binding pocket, thereby supporting our proposed mechanism for the action of these compounds. We found that the structural changes we made to the 1 and 4 scaffolds in developing these two series have significant effects on their potencies as CBIs.
We then performed in silico analysis of the interaction of these molecules with the hydrophobic coactivator binding groove-shelf region of the ER coactivator interaction site, using induced-fit based modeling methods, and we obtained binding interaction models that provided a satisfying rationale for our observed SAR. In further analysis of the molecular size and the spatial extent of the interaction of these CBIs with ERα, compared to that of the ILHRLL sequence of the SRC NR-box peptide, we noted that the interaction surface area of our CBIs was much less than that of the peptide ligand. This suggests that the potencies of the CBIs explored thus far are limited by the entropic component of free energy, due to the less complete displacement of bound surface waters by these smaller molecules. It further suggests that higher potency CBIs might be found by screening compound libraries enriched in larger molecules.
2. Results and Discussion
2.1. Analog Design
In deciding which building blocks to use to prepare analogs of 1 and 4, we chose hydrophobic alkyl and aryl substituents we believed would be well-suited for binding in the coactivator binding groove-shelf region, based on both the natural leucine and isoleucine residues of the NR Box motif and other work that has shown the coactivator binding groove capable of binding other aliphatic and aromatic substituents.[5e, 6b, 9] Because we were not certain of the binding orientation of 1 or 4 within the coactivator binding groove, we chose various hydrophobic substituents to append to the cores. We hoped that using an array of substituents would give differential biological results that would, in turn, reveal the binding orientation of the molecules within the groove through induced-fit computational modeling of ligand binding poses.
2.2. Chemical Synthesis
2.2.1. Quinazolinone Series 1
Modifications of CBI quinazolinone hit 1 were explored in four sectors, namely within chlorophenyl A, piperazine B, linker C, and thioquinazolinone D (Figure 2).
Figure 2.

SAR strategy for 1
The synthesis of the thioxo-hydroquinazolinone 1 was initiated by combining 2-methoxycarbonyl phenyl isothiocyanate and γ-amino butyric acid 5a and analogs to obtain thiourea derivative 6, followed by base-promoted cyclization to give quinazolinone 7. Acid 7 was further coupled with 1-(3-chlorophenyl)piperazine to provide thioxo-quinazolinone 1 and its derivatives. (Scheme 1)
Scheme 1.

General Synthetic Method for the Preparation of Thioxo-quinazolinone 1 and Derivativesa
aReagents and conditions: (a) EtOH, reflux, >48h, 40–72%; (b) KOH, EtOH; (c) 1N HCl, quantitative; (d) EDCI, HOBt, CH2Cl2-DMF
Modification of the chlorophenyl region A and linker region C
In the first stage of hit optimization, we introduced a variety of substituted phenyl rings as 3-chlorophenyl replacements (sector A). Compounds 1a–n were readily prepared by the same procedure as shown in Scheme 1 by coupling various phenyl-piperazines 8 with acid 7a. At the same time, a series of linkers (Sector C) was introduced to bridge the phenyl piperazine and quinazolinone moieties. Compounds 1o–x incorporate longer 5-carbon chains; 1y, 1z, and 1aa employ a seven-carbon linker, and 1bb has only a one-carbon chain between the two rings (Table 1).
Table 1.
Optimization of Sector A: Luciferase Activity of 1, 1a–z, 1aa and 1bb in Reporter Gene Assay.
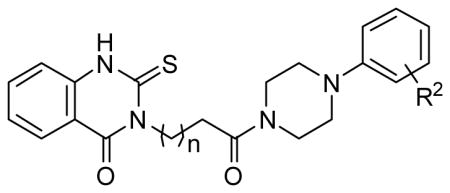 | |||
|---|---|---|---|
| Comp. | R2 | n | Luc(IC50, μM) |
| Peptide | - | - | |
| 1 | 3-Cl | 2 | 14.8a |
| 1a | 4-Cl | 2 | 7.1 |
| 1b | 3,4-dichloro | 2 | 5.5 |
| 1c | 4-OMe | 2 | 7.4 |
| 1d | 4-OH | 2 | Inactiveb |
| 1e | 2-OH | 2 | 4.8 |
| 1f | 3-OH | 2 | 10.4 |
| 1g | 4-CF3 | 2 | 2.3 |
| 1h | 3-CF3 | 2 | 35a |
| 1i | 2-CF3 | 2 | 11.9 |
| 1j | 3-CF 3, 4-Cl | 2 | Inactiveb |
| 1k | 4-iPr | 2 | 26a |
| 1l | 3-NO2 | 2 | 8.2c |
| 1m | 4-CN | 2 | Inactiveb |
| 1n | 2-CN | 2 | 7.5c |
| 1o | 3, 4-dichloro | 4 | 4.2 |
| 1p | 4-OH | 4 | 10.8a |
| 1q | 4-CF3 | 4 | 4.4 |
| 1r | 4-CN | 4 | 8.0 |
| 1s | 2-CN | 4 | 3.5 |
| 1t | 3-CN | 4 | 9.9 |
| 1u | 3-NO2 | 4 | 6.6 |
| 1v | 3-Cl | 4 | 5.2a |
| 1w | 4-Cl | 4 | 9.3a |
| 1x | 3-CF3 | 4 | 25a |
| 1y | 3-Cl | 6 | 4.2 |
| 1z | 2-CN | 6 | 2.7d |
| 1aa | 3, 4-di-Cl | 6 | Inactiveb |
| 1bb | 4-CF3 | 0 | Inactiveb |
Compound showed partial inhibition in reporter gene assay;
No activity at 20 μM.
Some toxicity in β-galactosidase assay (never more than 50% at 20 μM);
Significant toxicity in β-galactosidase assay.
The piperazine rings of 1k, 1t and 1u were constructed by reaction of a substituted aniline (3-nitrile or 3-nitro aniline) with bis-(2-chloroethyl)amine hydrochloride. Piperazine 8 was further coupled with quinazolinone acid 7 to afford derivatives 1k, 1t and 1u with 3-isopropyl, 3-cyano-and 3-nitro- substituted groups, respectively (Scheme 2).
Scheme 2.

Synthesis of Thioxo-quinazolinone Derivatives 1k, 1t and 1ua
aReagents and conditions: (a) K2CO3, chlorobenzene, reflux; (b) EDC, HOBt, 7, CH2Cl2, DMF.
Modification of phenyl piperazine sectors A and B
A common synthetic procedure was used to prepare a small library of compounds assembled from a diverse series of aromatic piperazines (9a–f), as shown in Figure 3. In addition, a second series of substituted piperidines replacing the piperazines has been synthesized by reductive amination, followed by coupling with acid 7. Reductive amination of tert-butyl 4-oxopiperidine-1-carboxylate with aniline gave phenyl amine 11, which was alkylated with benzyl bromide to generate compound 12. Boc-deprotection of 12 delivered piperidine 13, which was coupled with acid 7 by the procedure described above to obtain piperidines 10a–c (Scheme 3).
Figure 3.

Replacing substituted phenyl ring with other aromatic rings
Scheme 3.

Synthesis of Thioxo-quinazolinone Derivatives 10a–da
paReagents and conditions: (a) aniline, NaB(OAc)3H, CH2Cl2, 92%; (b) benzyl bromide, iPr2EtN, THF, 86%; (c) TFA-CH2Cl2 (1:4), 70%; (d) EDC, HOBt, CH2Cl2, DMF, 65–86%.
Modification of phenyl quinazolinone region D
In addition to examining the effect of differentially substituted piperazine and piperidine rings on sector B, we synthesized a small group of compounds with substituted phenyl quinazolinone rings by decorating the Sector D phenyl ring with -OMe, -COOMe and -COOH groups. (16a–e and 17, Figure 4, Scheme S1)
Figure 4.

Modification of phenyl quinazolinone
2.2.2. Benzothiazole Series 4
Two obvious points for introducing diversity into the benzothiazole scaffold can be seen in the library hit 4: introduction of various sulfides and amides at each of the two α-thioamides in the molecule. Analogs of benzothiazole 4 were synthesized beginning with the nitration of commercially available 2-mercaptobenzothiazole, followed by reduction to give 6-amino-2-mercaptobenzothiazole 18 (Scheme 4).[10] Alkylation of the thiol with N-alkyl α-chloroamides (prepared from the simple addition of amine and chloroacetyl chloride) gave 17,[11] which was followed by an acylation reaction with chloroacetyl chloride to give α-chloroamide 20. Nucleophilic displacement of the chloride with thiols gave the desired benzothiazoles 4 (see Table 3), usually in limited yields due to their often sparing solubilities in many organic solvents.
Scheme 4.

General Synthetic Method for the Preparation of Benzothiazole 4 and Derivativesa
aReagents and conditions: (a) HNO3, H2SO4; (b) Na2S, NaSH, elemental sulfur, H2O, reflux; (c) ClCH2CONHR1, NEt3, 50 °C; (d) chloroacetyl chloride, NEt3; (e) R2SH, NEt3,50 °C.
Table 3.
Activities and structures of benzothiazole 4 analogs in a cell-based luciferase assay.
| Comp. | R1 | R2 | Luc IC50(μM) |
|---|---|---|---|
| 4 | c-Pr | N-MeIm | 20–50 |
| 4a | c-Pr | Im | >50 |
| 4b | c-Pr | o-CF3Ph | 6.3 |
| 4c | c-Pr | m-CF3Ph | 20–50 |
| 4d | i-Bu | 2,5-diClPh | 9.1 |
| 4e | c-Pr | o-MePh | >50 |
| 4f | i-Bu | o-MePh | >50 |
| 4g | i-Bu | o-i-PrPh | >50 |
| 4h | i-Bu | o-t-BuPh | 5.8 |
| 4i | i-Bu | o-ClPh | 20–50 |
| 4j | i-Bu | m-ClPh | 20–50 |
| 4k | i-Bu | o-CF3Ph | 20–50 |
| 4l | i-Bu | m-CF3Ph | >50 |
| 4m | t-Bu | o-EtPh | 20–50 |
| 4n | i-Pr | o-ClPh | 20–50 |
| 4o | i-Pr | m-ClPh | 6.3 |
| 4p | i-Pr | o-CF3Ph | 20–50 |
| 4q | i-Pr | m-CF3Ph | >50 |
| 4r | c-Pr | p-MePh | 20–50 |
| 4s | i-Pr | p-MePh | >50 |
| 4t | i-Bu | p-MePh | >50 |
| 4u | c-Pr | p-EtPh | 20–50 |
| 4v | i-Bu | p-i-PrPh | >50 |
| 4w | c-Pr | p-t-BuPh | 20–50 |
| 4x | i-Pr | p-t-BuPh | >50 |
| 4y | i-Bu | p-t-BuPh | >50 |
| 4z | c-Pr | p-OMePh | 20–50 |
| 4aa | i-Pr | p-OMePh | >50 |
| 4bb | i-Bu | p-OMePh | >50 |
| 4cc | i-Pr | p-ClPh | 20–50 |
| 4dd | i-Bu | p-ClPh | >50 |
| 4ee | c-Pr | p-CF3Ph | 6.0 |
| 4ff | i-Pr | p-CF3Ph | 15 |
| 4gg | i-Bu | p-CF3Ph | 17 |
| 4hh | c-Pr | p-BrPh | 20–50 |
| 4ii | i-Pr | p-BrPh | 20–50 |
| 4jj | i-Bu | p-BrPh | >50 |
| 4kk | c-Pr | 2-Naph | 8.7 |
| 4ll | i-Pr | 2-Naph | 10 |
| 4mm | i-Bu | 2-Naph | 12 |
| 4nn | i-Pr | 3,4-diClPh | >50 |
| 4oo | i-Bu | 3,4-diClPh | 20–50 |
| 4pp | i-Bu | 2,4-diMePh | 20–50 |
| 4qq | i-Pr | 2,4-diMePh | 20–50 |
| 4rr | i-Pr | p-EtPh | 20–50 |
| 4ss | i-Bu | p-EtPh | >50 |
| 4tt | c-Pr | p-i-PrPh | 20–50 |
| 4uu | i-Pr | p-i-PrPh | >50 |
| 4vv | i-Pr | 2,5-diClPh | 6.1 |
2.3. Biological Activities
2.3.1. Verification of Locus of CBI Interaction with the ER LBD
The inhibition of coactivator binding by a small molecule, as measured by our time-resolved FRET assay, could involve either direct competition with the coactivator peptide for the ER surface (i.e., by a CBI mechanism) or binding within the ligand binding pocket and restructuring the ER in a way that prevents coactivator recruitment, that is, by the allosteric-type mechanism through which conventional antagonists function.[5e, 8] To rule out the latter mechanism, we first performed careful control experiments using the reporter gene assay. A similar investigation of the alternative mode of activity in our CBI assays was also made by molecular modeling.
By Reporter Gene Assays
A reporter gene assay was developed to evaluate the inhibitory potential of these compounds in a cellular context and confirm that they are, in fact, cell-permeable.[12] To accomplish this, a human endometrial cancer (HEC-1) cell line expressing endogenous nuclear receptor coactivators but not ER was transfected with a plasmid set comprising a full-length ERα expression vector, a luciferase reporter gene plasmid fused to an estrogen response element (2ERE Luc), and pCMV β-galactosidase (β-gal; internal control). The transfected cells were incubated with 1 nM estradiol together with increasing concentrations of CBI; luciferase activity was then measured. Active compounds were identified as those that inhibited estrogen receptor-mediated transcription of the reporter gene, measured at 1 nM estradiol, and effected a concentration-dependent decrease in the luciferase output.
To verify that activity in this assay was not due to a conventional antagonist mechanism, a CBI titration was performed using two concentrations of the agonist ligand, estradiol, 1 nM and 100 nM. This 100-fold increase in estradiol concentration should have no effect on the IC50 of a compound acting by a coactivator binding inhibition mechanism, because it is directly competing with the SRC, not the ligand. On the other hand, the 100-fold increase in estradiol concentration would cause a marked right shift in the inhibition curve of a compound that was operating as a conventional antagonist.
Using these assays, we found no significant change in inhibitory potency of the best two compounds in both series: at 100 and 1 nM estradiol, IC50 values were, respectively, 2.4 and 2.0 μM for 1g; 3.6 and 6.0 μM for 4o (See Figure 5 for competition curves and Tables 1 and 3 for IC50 values). By contrast, as we have previously shown, conventional antagonists such as tamoxifen are subject to competition by estradiol and show the expected 100-fold right shift in IC50 when evaluated in this format.[13] These results indicate that the inhibition of reporter gene transcription in cells that we are observing with our new compounds is occurring by a coactivator binding inhibition mechanism, not by conventional antagonism.
Figure 5.

Luciferase Reporter Gene Assay. Representative compounds 1g and 4o show dose-dependent inhibition of full-length ligand binding site.
By Molecular Modeling
As a prelude to our induced-fit docking studies by molecular modeling, described below, we performed an additional probe of the site of action of our active molecules by modeling. To test the possibility that these compounds might be competing with estradiol and functioning as conventional antagonists, we examined their fit into the ligand-binding pocket of the X-ray structure of ERα complexed with the classical antiestrogen 4-hydroxytamoxifen (HT). HT occupies the ER ligand-binding site and stabilizes an antagonist conformation of the LBD in which helix 12 is repositioned into the coactivator site, blocking coactivator binding (PDB code 3ERT).[3] Since HT and the high potency test compound 1g have very similar molecular volumes, in principle, both could separately occupy the expansive ligand-binding pocket in ER. Glide docking of 1g into the emptied HT site suggests that the phenyl piperazine-quinazolinone scaffold is able to penetrate the site, but the best binding pose is quite different from that of HT. The predicted molecular orientation occupies the pocket only partially, while much of the structure resides in solvent (Figure 6) avoiding contact with the receptor.
Figure 6.
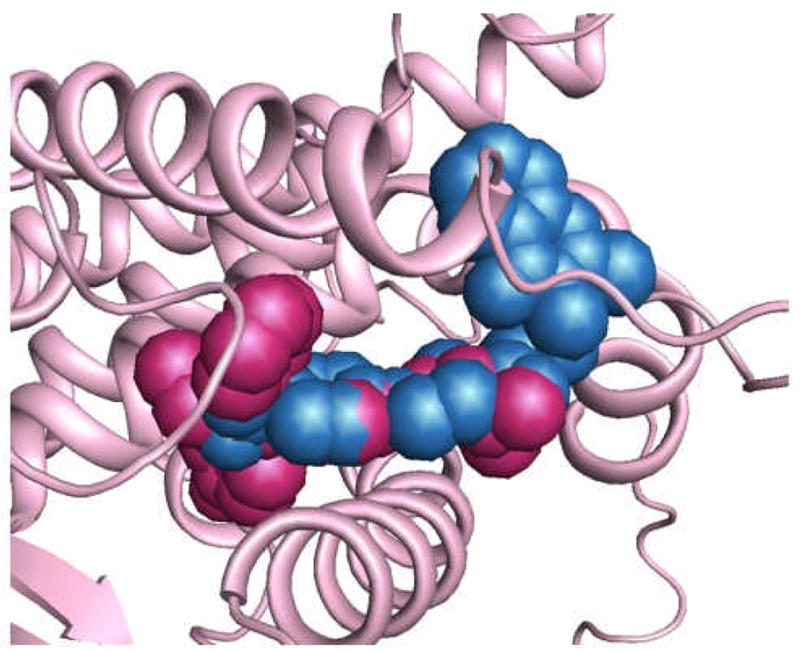
Alignment of 1g (blue) and 4-hydroxy-tamoxifen (HT) (maroon) in 3ERT (hERα-HT complex) as determined by Glide docking. Much of 1g is predicted to bind well outside the ligand binding site.
In addition, MM-GBSA calculations[14] were carried out to compare estimated binding free energies (ΔGbinding = Ecomplex(minimized) − Eligand(minimized) − Ereceptor) for HT and 1g. The latter complex, as pictured in Figure 6, is posited to be much less stable than that for HT (ΔΔGbinding ~ 17 kcal/mol). These calculations are in qualitative agreement with conclusions from the mechanistic studies in the reporter gene assays, described above. This consistency between biology and modeling offers a measure of confidence that the CBI docking results, reported later, might be useful for guiding future design strategies.
2.3.2. Structure-Activity Relationships From Cell-Based Reporter Gene Assays
Quinazolinone Series 1
The key structural features of hit 1 are the phenyl quinazolinone and the substituted phenyl piperazine moieties connected by a carbon chain linker (Figure 1b). Hit optimization focused on these two heterocyclic units. A number of hydrophobic and hydrophilic substituents at various positions of the aromatic ring representing Sector A were introduced, and compounds with variable carbon chain linkers (Sector C) were also examined (Table 1).
Most of the compounds in this series showed activities in the low micromolar range (~5–10 μM). Although hit 1 with 3-chlorophenyl substitution and the analog with a five-carbon linker (1v) both showed only weak activity, the analog with a seven-carbon linker (1y) had good activity with an IC50 value of 4.2μM. Movement of the chloro group from the meta- (compound 1) to the para-position (1a) increased activity, with compound 1a showing full inhibition with an IC50 value of 7.1μM; however, the corresponding longer chain compound 1w had decreased activity. Further substitution of the phenyl ring with two chloro groups (1b) increased activity slightly, but longer chain length led to complete loss of activity (1aa). Introduction of a hydrophilic OH group at the ortho-position (1e) increased activity 2 fold compared to the meta OH compound (1f), but all activity was lost with a para OH group (1d). Accordingly, the longer linker analog with a para-OH substitution (1p) had poorer activity.
Since the CBI binding site is highly hydrophobic, we anticipated that compounds bearing a CF3-group would fit into the hydrophobic coactivator groove more effectively than those bearing hydrophilic groups. Indeed, an analog incorporating a para-CF3 group (1g) had good activity, but potency decreased progressively with the ortho (1i) and meta (1h) analogs. The 6-carbon chain meta-CF3 compound 1x showed very weak partial inhibition, but compound 1q with a linker of only two carbons longer than 1g had only slightly poorer activity. On the other hand, reducing the linker flexibility by two carbons (1bb) led to complete loss in activity. These results imply that CBI binding to the hydrophobic groove involves a subtle interplay of effects sensitive to small alterations in substituent placement and composition. In the modeling section below, we suggest that reorganization of the side chains of the NR-box helix and displacement of water from the coactivator binding groove are two important factors contributing to CBI binding.
To our surprise, incorporation of both meta-CF3 and para-Cl substituents on the same phenyl ring (1j) led to complete loss of activity. This is consistent with activity-domination by CF3 and a binding penalty resulting from directing the hydrophobic m-CF3 moiety into solvent (cf. Figure 8 binding model). Substituting the phenyl ring with a cyano-group in the para- (1r), ortho- (1s), and meta- (1t) positions (n = 4) led to analogs with good cellular assay activities, as indicated by IC50 values ranging from 3–10 μM. Interestingly, 1s, with ortho-substitution, is the most active isomer (IC50 of 3.5 μM). This series complies with the binding model in which an edge of this substituted phenyl ring bearing the o-CN group is directed into the aqueous shell around the receptor (See molecular modeling section below).
Figure 8.
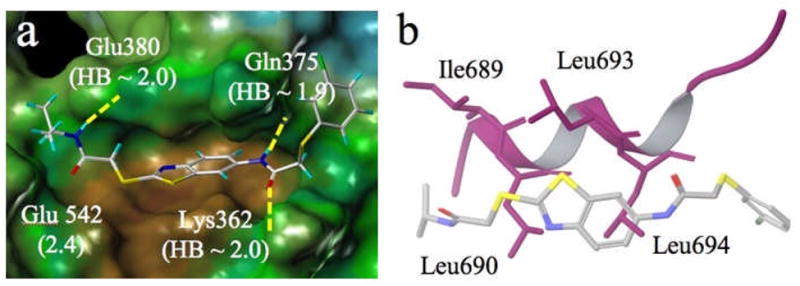
a) Docked pose of 4o showing three hydrogen bond anchors and side chain movement upon docking in Å in parentheses; b) Alignment of 4o (stick) and coactivator peptide (purple ribbon).
The linker-shortened hydroxy series 1d–1f (n = 2) shows similar behavior and can be rationalized equally well (cf., the model in Figure 9). Unfortunately, the similar ortho-CN analogs with a shorter carbon chain linker (1n) and the longer carbon chain linker (1z) both cause nonspecific cellular toxicity, shown by a decrease in the internal control β-galactosidase reporter gene. A strong electron withdrawing influence as represented by the 3-NO2 analog (1u) delivered good cellular activity (IC50 = 6.6 μM), but the corresponding shorter chain analog (1l) showed some toxicity. Installation of the hydrophobic isopropyl substituent (1k) represents a limited mimic of the LXXLL helix backbone, but this compound showed only partial inhibition in the cell-based assay (Table 1).
Figure 9.

Number of overlapped waters within 2 Å of coactivator peptide and the three ligands 1b, 1e and 4o. Reporter gene assay IC50 values in parentheses.
To examine tolerance of aromatic diversity within the binding site, a small library of compounds was prepared that combined different aromatic rings and heterocycles (Figure 3), for instance, naphthalene, (9a, 9b), pyridine (9d), pyrimidine (9e) and pyrazine (9f). Three compounds in this series (9a, 9c and 9d) delivered fairly good activities (Table 2), though not superior to previous analogs. To explore the SAR around the phenyl piperazine moiety, several derivatives were prepared with a piperidine ring instead of piperazine, either increasing the hydrophilicity (10a) or introducing an extra potential hydrogen bonding center (e.g., N-phenylpiperidine-4-hydroxy derivative, 10c) as shown in Scheme 3, but neither substance showed appreciable activity (Table 2).
Table 2.
Activities of analogs with substituted piperazines (9a–f), piperidines (10a–c), quinazolinones (16a–e) and 17 in a cell-based reporter gene assay.
| Comp. | reporter gene (IC50, μM) | Comp. | reporter gene (IC50, μM) |
|---|---|---|---|
| 9a | 8.6 | 10c | Inactivea |
| 9c | 10.9 | 16a | 7.3 |
| 9d | 6.6 | 16b | 21b |
| 9e | Inactivea | 16c | Inactivea |
| 9f | 7.0b | 16d | 22b |
| 10a | Inactivea | 16e | Inactivea |
| 10b | 23.5 | 17 | Inactivea |
No activity at 20 μM.
Compound showed partial inhibition in reporter gene assay.
A small set of compounds were prepared with substituents on the quinazolinone benzene ring, and their activities are best understood when compared to the unsubstituted analogs 1 and 1g (Table 1). Compound 16b with di-methoxy substitution (Table 2) experienced a severe loss of activity compared to 1g (Table 1), as did compounds 16c, 16d and 16e, having di-methoxy substitution. These results suggest there is limited tolerance for bulk at this site. A methyl ester at C7 elicits an improvement in activity (16a, Table 2); however, a carboxylic acid at the same position ablates activity (17, Table 2), illustrating the hydrophobicity requirement of the CBI binding site. The 3D binding models described below suggest that the quinazolinone benzene ring is sandwiched by a small hydrophobic protein patch and solvent. Thus, introducing methoxy groups in the ring appears to add hydrophobicity in a solvent bath, leading to reduced potency.
Benzothiazole Series 4
Upon resynthesis of the original benzothiazole hit 4, we discovered that the resynthesized compound was inactive (IC50 >50 μM) in the TR-FRET assay but showed modest activity (IC50 = 20–50 μM) in the reporter gene assay. HPLC analysis of the original library hit showed it to be only ca. 85% pure, and thus it is believed that the impurities contributed to the false positive activity of the library compound in the TR-FRET assay. The NMR spectrum of a commercial sample of this compound, which we assume to be from the same source that provided the sample in the original library, was consistent with the α-chloroamide precursor of compound 4 that would be too cysteine-reactive to make a useful probe. Analogs prepared in parallel with our efforts to confirm that activity of compound 4 did, however, show activity in both TR-FRET and cell-based assays; thus, they were examined further.
The benzothiazole scaffold 4 contains two α-thioamides, each of which can be diversely functionalized with different amines and thiols. R1 was functionalized with different alkyl amines, and there is no clear preference for any of them. R2 was functionalized with a number of arenethiols. Some compounds containing substituents at the ortho and meta positions on the benzene ring showed increased activity over the resynthesized hit (Table 3, 4b); however, those compounds containing a para-oriented substituent, particularly hydrophobic ones, were among the most consistently potent compounds assayed in this series. For instance, the CF3-substituted compounds showed consistently lower IC50 values when this substituent was in the para-position, as opposed to the ortho or meta position (Table 3, 4ee–4gg). Surprisingly, the CF3-containing compounds were much more potent than the CH3-containing compounds. In fact, all compounds with ortho or para methyl groups experienced complete loss of activity.
Introduction of halogens (Cl and Br) at the para position gave compounds showing moderate activity, but placing a Cl substituent in the meta position of the ring also led to good to moderate binders (Table 3, IC50 values for 4n and 4o = 20–50 μM and 6.3 μM, respectively). Other potent compounds incorporated a 2-naphthyl ring, rather than a substituted phenyl, at R2, and all these showed IC50 values of 9–12 μM (Table 3, 4kk–4mm). Replacing the naphthyl group with the bioisosteric 3,4-dichlorophenyl compound gave a marked reduction in activity (Table 3, 4nn–oo). Since there are neither pi-pi nor pi-cation interactions in the binding model for benzothiazoles (see model in Figure 8), the origin of the difference may arise from selective desolvation (see below).
The binding site seems to be tolerant of various alkyl groups introduced at the R1 position, as changing the alkyl substituent in the best compounds did little to change activities. In general, however, the c-Pr substituent at R1 tends to increase the potency of the compounds, sometimes by two-fold, relative to i-Bu and i-Pr (Table 3, 4b compared with 4k and 4p; 4ee compared with 4ff and 4gg). While R1 is most likely deep in the hydrophobic CBI binding cleft, R2 may be outside of it. This has unexpected consequences for the observed SAR that is noted below in the context of binding site water displacement.
2.4. Induced-Fit Molecular Modeling to Identify Binding Poses for CBIs
To identify the putative binding poses of the compounds in the phenyl piperazine and benzothiazole series, we employed the crystal structure of the human estrogen receptor α complexed by both diethylstilbestrol (ligand-binding pocket) and a peptide from the NR box II region of the coactivator GRIP1 (CBI site; pdb code 3ERD). The peptide was deleted, and docking of the ligands to the coactivator site was performed with the Prime induced-fit docking method.[15] This procedure allows for flexibility of both ligand molecule and protein side chains within the target coactivator binding groove. The energetically favored pose is subsequently derived with MM-GBSA,[14] a method that provides ligand binding energies with reasonable accuracy;[16] the same method was used to evaluate the interaction of compound 1g with the ERα ligand binding pocket (Figure 6). For additional details, see Methods, Molecular Modeling.
2.4.1. Phenyl Piperazine Scaffold
All active compounds in Table 1, with the exception of 1e and 1f (each of which bears an OH hydrogen donor), exhibit similar docked poses at the coactivator binding groove. These show good alignment with the X-ray structure of the bound ERα-coactivator peptide and simultaneously form productive hydrophobic contacts and H-bonds with Gln375, Lys362 or Val368 (Figure 7). In addition, as a result of the induced-fit docking, each is accompanied by side chain movements of Glu380, Glu542, Met543. Ile358 and/or Leu372 in order to better match dipoles of the functional groups on the phenyl ring associated with the piperazine ring or hydrophobic contacts with the benzene center of the quinazolinone ring. It is noteworthy that the center of mass of 1b does not fall along the axis of the peptide helix, but below it and deeper into the groove (Figure 7b). The docking has provided an intuitively reasonable spatial overlap by superposing the dichlorobenzene ring onto Leu690 and the piperazine ring onto Leu694, hydrophobic onto hydrophobic.
Figure 7.
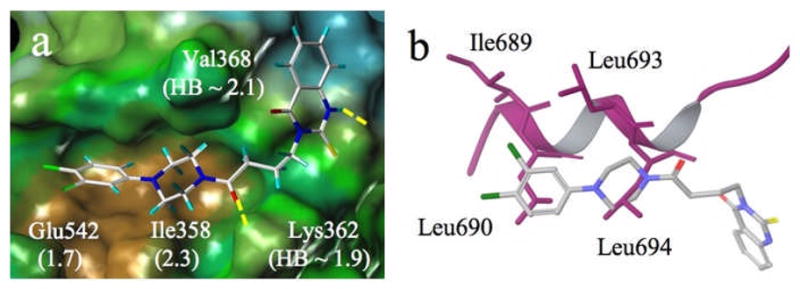
a) Induced-fit docked pose for 1b at the coactivator binding site of the ERα receptor (PDB X-ray structure code 3ERD). Displacements of Glu542 and Ile358 side chains resulting from docking and H-bond distances are indicated in Å in parentheses; brown = hydrophobic; green = neutral; blue = polar; b) Alignment of 1b (stick) and the coactivator peptide (purple ribbon; 3ERD). Hydrophobic residues Leu690 and Leu694 are matched by ligand hydrophobes.
To test the possibility that docking might insert 1b arbitrarily deep within the groove, the coactivator peptide from 3ERD was rigidly docked into the binding site. In none of the resulting poses do the Leu and Ile side chains penetrate more deeply into the pocket than observed in the X-ray structure, indicating that the hydrophobic basin of the protein is most likely the arbiter of hydrophobe location.
Pronounced side chain relocation can also occur for Lys362, a cationic charge clamp residue. For example, H-bonding to Lys362 by the piperazine-associated carbonyl group in 1b (3-carbon linker, n = 2) can contribute to modifying the locus of the side chain. Figure 7 depicts the docked pose of 1b in which the A-sector 3,4-dichlorophenyl piperazine moiety slips deeply into the hydrophobic groove, while the D-sector phenyl quinazolinone resides outside of it. During ligand fitting, Glu542 and Ile358 were relocated by 1.7 and 2.3 Å, respectively, to accommodate the two chlorines and the piperazine ring. In addition to the Lys362----O=C H-bond (1.9 Å), the quinazolinone NH and the Val368 C=O interact similarly at 2.1 Å.
For certain active CBI analogs in this series, such as 1q (IC50 4.4 μM), induced-fit docking leads to displacements of the surrounding amino acid side chains by 2–4 Å. By and large, however, compounds in this family appear to exhibit quite similar binding poses by adopting compensating local adjustments resulting from linker length variation. An exception is compound 1e (IC50 4.8 μM), which assumes a U-shaped pose overlapping Leu690 and Leu694 side chains and a section of the coactivator peptide backbone. Details of the modeled binding poses and side chain relocations for 1q and 1e are described in the Supporting Information (Figures S1 and S2).
2.4.2. Benzothiazole Scaffold
Induced-fit modeling of compound 4o, one of the most active members of this series, provides a satisfying CBI binding pose, as illustrated by Figure 8. The structure not only fits well in the hydrophobic groove, but also forms good hydrogen bonding interactions with the charge clamp residues Lys362, Glu380 and Gln375 (2.0 Å). Like inhibitors 1b and 1e (Figures 7 and S2, respectively), this structure offers hydrophobic features that appear to serve as Leu690 and Leu694 side chain surrogates (i.e., CH2S, one edge of the thiazole ring, and the centrally placed benzene ring).
The model accommodates a number of the SAR trends discussed above. For example, activities appear to be relatively unperturbed by replacement of the terminal i-Pr group in 4o with other hydrophobes. Figure 8 illustrates the deep apolar pocket of the CBI site in which the NHR1 is situated. At the other end of the molecule, the SR2 moiety extends beyond this pocket so as to become exposed to both a rather hydrophobic protein surface and aqueous solvent, suggesting that amphiphilic substitution might best serve binding. Indeed, the CF3 group, tolerant of both polar and nonpolar environments, appears to be a superior substituent.[17] Replacement of CF3 with CH3 or OMe depletes activity in accord with water layering. One puzzling observation is the complete loss of activity when the methyl group of 4 is removed (i.e., 4a). In the context of the model of Figure 8, this might have its origin in the observed loss of efficacy for carboxylic acid 17 relative to its methyl ester 16a. Both the quinazolinone COOH and NH are exposed to solvent (Figure 7b). Just as we speculated that 17 may be extracted into solvent and out of the binding cleft, such a phenomenon may operate for 4a as well.
2.4.3. What the Current CBIs Offer and What They Lack: Entropic Contribution to Binding Free Energy Originating from Water Displacement
In our models, compounds 1b, 1e and 4o populate the coactivator peptide binding groove with differential but productive hydrogen bonding and favorable hydrophobic contacts. According to induced-fit docking, these structural similarities are achieved by a reasonable spatial match of the organic structures and the part of the coactivator peptide that binds most deeply in the groove. In spite of these common and favorable features, all three compounds derived by different strategies fail to deliver potencies having IC50 values of 1 μM or below in the reporter gene assay. This could be the result of poor penetration of the cellular membrane, although an estimate of Caco-2 membrane permeabilities suggests that the values for 1b, 1e and 4o do not differ significantly from those for the potent ER ligands DES (diethylstilbestrol) and HT (See Methods, Molecular Modeling). More likely, the CBIs are simply too small to compete effectively with the α-helical coactivator peptide.
The docked poses of the synthetic inhibitors reveal an important characteristic which speaks to this point. Assuming that compounds 1b (Figure 7b), 1e (Figure S2b) and 4o (Figure 8b) are representative of each class, they are predicted to fill important parts of the groove, particularly those occupied by the Leu690 and Leu694 side chains of the coactivator peptide. However, none of the present analogs provide structural elements that contact the protein shelf on which the Ile689 and Leu693 hydrophobes reside. Given the importance of entropic contributions to the free energy of binding, the principal focus on groove-only inhibitors may be misplaced.
Many studies have shown that ligand binding to receptors is frequently entropy-driven,[18] and that the source of the large – TΔS contribution to ΔG arises by the release of water molecules from the binding pocket during the ligand complexation event.[19] For very tightly bound waters, the contribution has been estimated to be as much as 2 kcal/mol per water at 300 K,[20] with lesser values for bound waters having a greater degree of freedom. With the goal of developing a relative semi-quantitative estimate of the importance of this phenomenon in the present case, the coactivator-bound X-ray structure (PDB code 3ERD) was relieved of the coactivator peptide, surrounded by a box containing 8742 SPC waters 10 Å from the ERα protein, and then subjected to 1 ns molecular dynamics at 300 °C with the Desmond protocol (see Experimental Section).[21] The X-ray structure of the coactivator peptide and the docked structures of 1b, 1e and 4o were separately superposed on the solvated binding site and, for each structure, all waters were deleted except those that overlapped the ligand severely or displayed oxygen atom-ligand atom distances below the sum of the van der Waals radii.[22] The superpositions are depicted in Figure 9.
Clearly, the peptide is suggested to displace 5-fold more water molecules than the non-peptidic CBI systems: The 13 waters that line the shelf adjacent to the binding groove occupied by the bound coactivator side chains of Ile689 and Leu693 are displaced by the peptide, but they are modeled to be unaffected by the binding of our CBIs (See Figures S3 and S4 of the Supporting Information). If a conservative but average 0.2 kcal/mol were assigned to each displaced water molecule at ambient temperatures, the peptide then contributes about 14 kcal/mol more ERα binding free energy from – TΔS than do 1b, 1e or 4o. It is equally significant that Ile689 and Leu693 residues of the peptide (i.e., those that interact with the hydrophobic shelf, not the groove) appear able to displace almost as many water molecules (13 total) as each of the three micromolar active inhibitors.
Libraries available for HTS often comprise molecules that conform to specific size and lipophilicity criteria associated with typical drug-like molecules,[23] and while these libraries have obviously given rise to many successful screening campaigns, there have been recent calls for a re-examination of this process for targets not conventionally sighted by HTS (e.g., inhibition of protein-protein interactions, as we are attempting to do with our CBIs).[24] Based on this seeming limitation of our screening library and on modeling data, we suggest that insufficient water displacement by the low molecular weight CBIs at the coactivator binding groove is likely to be responsible for the limited potency of our compounds.
3. Conclusions
The development of compounds that can block the interaction between the estrogen-activated ER and important coactivator proteins could provide unique pharmacological tools for interrupting the signal transduction cascade by which this transcription factor regulates gene activity and might provide a lead for novel therapeutic agents. Nevertheless, inhibition of protein-protein interactions with small molecules remains a significant challenge.[25] In this report, we described the synthesis and follow-up cell-based assays of a selection of hits that came from a high-throughput screening effort to search for small molecule coactivator binding inhibitors (CBIs) in a large compound library. The screening protocol employed a TR-FRET assay that we have described previously for inhibition of the interaction of ERα with the important coactivator SRC3.[8]
Compounds 1 and 4 were identified as the most promising hits, and optimization through analog synthesis and further biological evaluation in a cell-based reporter gene assay yielded several compounds that were active in the low micromolar range (e.g., 1b, 1g, and 4o). The mechanism of action of the potential CBIs was further examined, both experimentally and by modeling, to verify that the inhibitory activity of these compounds results from direct competition with coactivator for binding rather than by competition with estradiol at the ligand binding site.
Compounds 1b, 1q, 1e and 4o were subjected to extensive induced-fit docking experiments. Depicted in Figures 7, 8, S1 and S2, the resulting binding models are characterized by protein side chain movements tailored to each ligand, a situation parallel to that suggested for side chain rearrangements involving different ligands that perturb G-protein coupled receptors.[26] The models not only provide insights into a variety of aspects of the evolving CBI structure-activity relationships, but illuminate that a key aspect of SRC blockade is mimicry of the two most deeply buried hydrophobic leucine side chains (Leu690 and Leu694).
To the extent that water release is a contributor to the free energy of binding, it is clear from Figure 9 that the coactivator peptide is far more effective than the CBIs we have explored, and that the remaining two shelf-oriented apolar side chains (Ile689 and Leu693) of the peptide that displace 13 water molecules have no counterparts in our new compounds. These results suggest that the next generation of small molecule CBIs should span more of the peptide space, particularly on the shelf adjacent to the deep binding groove bordered by helices H-3, H-4, H-5 and H-12. This might well come at the cost of inhibitor molecular weights beyond the Lipinski ideal of 500,[23] but if other molecular properties are satisfactory, the potency gain could certainly compensate.[24]
The only other two CBIs that have been discovered through an HTS approach are an ERα CBI[5b] and a thyroid hormone receptor (TR) CBI.[27] Both compounds have IC50 values comparable to those reported here, and follow-up medicinal chemistry has produced only modestly more potent compounds.[13, 28] Based on these results and on those of our modeling studies, we believe that a different approach to discovering potent CBIs by HTS is warranted—an approach that utilizes targeted libraries enriched in higher molecular weight compounds.
5. Experimental Section
Chemistry General
Mass spectrometric analysis was provided by the Emory University Mass Spectrometry Center. Routine proton and carbon NMR spectra measured during synthesis were obtained on a Varian Inova-400 (400 MHz). Solvents for NMR were deuteriochloroform (CDCl3) (residual shifts: δ 7.26 for 1H and δ 77.7 for 13C) and deuteriomethyl sulfoxide (DMSO-d6; residual shift: δ 2.5 for 1H). The residual shifts were taken as internal references and reported in parts per million (ppm). TLC and preparative thin-layer chromatographies (PTLC) were performed on precoated, glass-backed plates (silica gel 60 F254; 0.25 mm thickness) from EM Science and were visualized by UV lamp. Column chromatography was performed with silica gel (230–400 mesh ASTM) using the “flash” method. Elemental analyses were performed by Atlantic Microlab Inc. Norcross, Georgia. All solvents and other reagents were purchased from Aldrich Chemical Co., Milwaukee. The reagents were used as received. All reactions were performed under anhydrous nitrogen atmosphere in oven-dried glassware. All compounds for which CHN analysis data is not reported were judged at least 95% pure by HPLC (Waters 4.6 mm × 150 mm C18 5 μm (WAT045905) with UV detection at 254 nm; 1.00 mL/min of various mixtures of acetonitrile and water).
General procedures for synthesis of compound 1 and analogs
Synthesis of compound 1 and analogs
Compound 7 (0.2 mmol, 1.0 eq), EDCI (0.22 mmol, 1.1 eq), HOBt (0.22 mmol, 1.1 eq) in a mixed solvent of dichloromethane (5 ml) and DMF (0.5 ml) were stirred at room temperature for at least 10 h. The product was purified by either filtration followed by washing with solvent or by chromatography to obtain the final product 1 and analogs as white solid.
Compound 1
1H NMR (400 MHz, DMSO-d6) δ: 12.97 (s, 1 H), 7.94 (d, J=8.0Hz, 1 H), 7.73 (t, J=7.2Hz, 1 H), 7.38 (d, J=8.0 Hz, 1 H), 7.32 (t, J=8.0 Hz, 1 H), 7.22 (t, J=8.0 Hz, 1 H), 6.96 (d, J=1.6 Hz, 1 H), 6.90 (dd, J= 8.4, 1.6 Hz, 1 H), 6.80 (d, J=8.0 Hz, 1 H), 4.45 (t, J=6.8 Hz, 2 H), 3.54 (br, 4 H), 3.21 (m, 2 H), 3.13 (m, 2 H), 2.45 (t, J=7.2 Hz, 2 H), 1.96 (t, J=7.2 Hz, 2 H); Anal. calcd for C22H23 ClN4O2S.0.5H2O: C, 58.46; H,5.35; N, 12.40; found: C, 58.42; H, 5.10; N, 12.32.
General Procedure for the Synthesis of Compound 4 and Analogs
A 50-mL round-bottom tube was charged with α-chloroacetamidobenzothiazole 20 (1.0 eq), and dissolved, typically with the aid of heat, in anhydrous DMF. Triethylamine (2.0 eq) was added to the solution, and then the appropriate arenethiol (1.2 eq). The solution was heated for 16 hours at 50 °C and then cooled to room temperature. The solution was added to a 70% saturated brine solution, and extracted with ethyl acetate. The organic layer was dried over MgSO4, filtered and evaporated. The resulting solid was recrystallized from boiling CH3NO2.
Compound 4
mp 168–169 °C; 1H NMR (500 MHz, DMSO-d6) δ: 10.61 (s, 1 H), 8.39 (d, J=3.9 Hz, 1 H), 8.33 (d, J=2.2 Hz, 1 H), 7.74 (d, J=8.8 Hz, 1 H), 7.46 (dd, J=8.8, 2.2 Hz, 1 H), 7.24 (d, J=1.0 Hz, 1 H), 6.96 (d, J=1.2 Hz, 1 H), 4.04 (s, 2 H), 3.88 (s, 2 H), 3.59 (s, 3 H), 2.63 (app octet, J=3.7 Hz, 1 H), 0.62 (td, J=7.0, 4.9 Hz, 2 H), 0.41 (m, 2 H); 13C NMR (125 MHz, DMSO-d6) δ: 168.04, 167.39, 149.46, 140.77, 140.22, 136.38, 136.12, 129.25, 124.26, 121.78, 119.17, 112.05, 39.07, 37.30, 33.70, 23.35, 6.35; HRMS-ESI (m/z): [M + H]+ calcd for C18H20N5O2S3,434.0779; found, 434.0760.
Molecular Modeling
To develop binding models of the ERα coactivator binding inhibitors developed herein and an initial structure activity relationship, we employed the crystal structure of the human estrogen receptor α (hERα) ligand binding domain (LBD) bound to the agonist diethylstilbestrol (DES) and a peptide derived from the NR box II region of the coactivator GRIP1 (2 Å resolution; pdb code 3ERD). The peptide was deleted and flexible docking of the ligands to the coactivator site was performed with the Induced Fit Docking module of Schrödinger Suite (2008).[15]
Since the quality of pose prediction depends strongly on reasonable starting structures, prior to ligand docking the protein LBD was first prepared in a form suitable for docking, subsequent MM-GBSA calculations and MD simulation with the “Protein Preparation Wizard” in Maestro. Thus, coactivator peptide, and tightly bound water molecules were deleted; bond orders, assigned; hydrogens, added; protein termini, capped with ACE (N-acetyl) and NMA (N-methyl amide); structures, fixed; and labeling, systematized. In addition, since several residues with missing side chains were detected within and near loops, these were automatically added and conformations for them were predicted using Schrödinger’s “Prime side-chain prediction” module and the OPLS-2000 protein optimized all-atom force field.[29] The procedure follows. For the prediction of the first residue, the side-chain rotamer library is searched to find the rotamer with the lowest predicted energy, while keeping all other side chains fixed. Then the next residue is considered, and so forth, until all residues have been treated. Once the process is complete, the steps are repeated until no residues change rotamer states, i.e., rotamer convergence. Once this was achieved, structure optimization was run on all of the side-chain atoms while keeping backbone atoms fixed. Finally, restrained minimization of the protein was carried out by the IMPREF utility in “Protein Preparation Wizard”, which performs protein refinement with OPLS-2000 until heavy atom positions deviate from the crystal structure with an RMSD of no more than 0.3 Å.
Frequently, ligands are docked flexibly into the binding site of a rigid protein receptor. However, in many situations this yields misleading results as many proteins experience side chain or backbone movements upon ligand binding. Small-molecule blockade of protein-protein interactions as in the present case is particularly susceptible to such effects. Therefore, although more time-consuming, we incorporated both ligand and protein flexibility in all docking exercises in an effort to maximize accuracy. As a result, binding site residues including Ile358, Lys362, Leu372, Glu380, Glu542, and Met543 experienced movements to accommodate docked blockers. In particular, Lys362 was displaced up to 4 Å towards solvent during the course of induced-fitting of various inhibitors.
QikProp Calculations
To explore a potential basis for the low CBI IC50 values in the reporter gene assay, the Caco-2 cell permeabilities of three key ligands, agonist DES and antagonist HT were predicted with the QikProp facility:[16] 1b, 745; 1g, 488; 4o, 572; DES, 907; HT, 624 (< 25 poor, > 500 great).
MM-GBSA Energy Evaluation
To identify the optimal binding poses generated by docking, MM-GBSA calculations were performed within Prime following induced-fit docking. The method is capable of identifying experimental protein-ligand complexes (i.e., the X-ray structure)[30] over alternative poses by computing an estimate of the free energy of binding. The procedure is superior to GlideScore, the scoring function employed by Prime to rank poses during their generation. Consequently, it is prudently used to re-score the list of ligand/receptor docking geometries to select the energetically most favorable complex.
Desmond Molecular Dynamics Simulations and Determination of Numbers of Displaced Waters
MD simulations were performed using the Schrodinger Desmond module developed by D. E. Shaw Research.[31] The above-described crystal structure (3ERD) prepared for induced-fit docking by the “Protein Preparation Wizard” was chosen for a Desmond simulation as well. The coactivator peptide was removed and the protein was solvated in an orthorhombic SPC water box with a outer boundary of 10 Å from the protein. The system was initially subjected to OPLS-2000 optimization to relax the system to the nearest local energy minimum. Trajectory data were recorded every 1.2 ps for a total of 1ns simulation at 300K and a pressure of 1.0 bar using an NPT ensemble class. In addition, seven sodium cations were added to neutralize the system, and the Ewald method was used to affect efficient and accurate long-range electrostatics. The last structure of the 1 ns simulation was taken for analysis of the waters in the binding site since its energy is very similar to that of the average energy value over the nearly isoenergetic 1 ns time course. To guarantee that the ERα protein has equilibrated faithfully, the 1 ns structure was superposed with the starting complex to show that both the protein backbone and the diethylstilbestrol agonist are in essentially identical spatial locations. After the MD treatment, the crystal structure of the coactivator peptide and the docking poses of 1b, 1e and 4o were individually placed in the binding site water pool by superposing the respective protein complex backbones with that of the solvated and simulated apo-protein. Only waters 2 Å or less from any atoms of coactivator peptide, 1b, 1e and 4o were considered to be ligand-overlapped and, therefore, candidates for extrusion from the binding site upon inhibitor binding.
Supplementary Material
Acknowledgments
We are grateful to the NIH for support of the work (U.S. Public Health Service grant HG003918 to JPS and HF, and DK015556 to JAK) and to Dr. Thota Ganesh (Emory University) for resynthesis of hit 3.
We are grateful for support from the following grants: National Institutes of Health R37 DK015556 and X01 MH078953 (to J.A.K.) and U54 HG003018 (to J.P.S. and H.F.).
Abbreviations
- CBI
coactivator binding inhibitor
- DES
diethylstilbestrol
- E2
estradiol
- ER
estrogen receptor
- HT
hydroxytamoxifen
- HTS
high throughput screening
- LBD
ligand-binding domain
- NR-box
nuclear receptor interaction box
- SAR
structure-activity relationship
- SRC
steroid receptor coactivator
- TR-FRET
time-resolved fluorescence resonance energy transfer
Footnotes
Supporting Information Available. Synthetic procedures, elemental analyses, additional docking experiments and graphics illustrating the water displacement by Leu690, Leu693, Leu694 and Ile689 of the SRC.
Contributor Information
Dr. Aiming Sun, Email: asun2@emory.edu.
Dr. John A. Katzenellenbogen, Email: jkatzene@uiuc.edu.
References
- 1.a) Katzenellenbogen BS, Katzenellenbogen JA. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Katzenellenbogen JA, Katzenellenbogen BS. Chem Biol. 1996;3:529–536. doi: 10.1016/s1074-5521(96)90143-x. [DOI] [PubMed] [Google Scholar]
- 2.a) Jensen EV, Jordan VC. Clin Cancer Res. 2003;9:1980–1989. [PubMed] [Google Scholar]; b) Jordan VC. J Med Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]; c) Jordan VC. J Med Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 3.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 4.Henke BR, Heyer D. Curr Opin Drug Discov Dev. 2005;8:437–448. [PubMed] [Google Scholar]
- 5.a) Moore TW, Katzenellenbogen JA. Annual Reports in Medicinal Chemistry. 2009;44:443–457. [Google Scholar]; b) Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, Gonder D, Winneker RC, Frail DE. J Steroid Biochem Mol Biol. 2004;88:351–360. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]; c) Moore TW, Gunther JR, Katzenellenbogen JA. Bioconjug Chem. 2010;21:1880–1889. doi: 10.1021/bc100266v. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Moore TW, Mayne CG, Katzenellenbogen JA. Mol Endocrinol. 2010;24:683–695. doi: 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Parent AA, Gunther JR, Katzenellenbogen JA. J Med Chem. 2008;51:6512–6530. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Nat Struct Mol Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]; b) Becerril J, Hamilton AD. Angew Chem Int Ed Engl. 2007;46:4471–4473. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]; c) Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski AM, Pike ACW. J Biol Chem. 2002;277:21862–21868. doi: 10.1074/jbc.M200764200. [DOI] [PubMed] [Google Scholar]
- 7.Huryn DM, Cosford NDP. Annu Rep Med Chem. 2007;42:401–416. doi: 10.1016/S0065-7743(07)42026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunther JR, Du Y, Rhoden E, Lewis I, Revennaugh B, Moore TW, Kim SH, Dingledine R, Fu H, Katzenellenbogen JA. J Biomol Screen. 2009;14:181–193. doi: 10.1177/1087057108329349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Geistlinger TR, Guy RK. J Am Chem Soc. 2003;125:6852–6853. doi: 10.1021/ja0348391. [DOI] [PubMed] [Google Scholar]; b) Galande AK, Bramlett KS, Trent JO, Burris TP, Wittliff JL, Spatola AF. Chem Bio Chem. 2005;6:1991–1998. doi: 10.1002/cbic.200500083. [DOI] [PubMed] [Google Scholar]; c) Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. J Med Chem. 2004;47:600–611. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 10.a) Sigmund H, Pfleiderer W. Helv Chim Acta. 2003;86:2299–2334. [Google Scholar]; b) Teppema J, Sebbrell LB. J Am Chem Soc. 1927;49:1779–1785. [Google Scholar]
- 11.Naya A, Kobayashi K, Ishikawa M, Ohwaki K, Saeki T, Noguchi K, Ohtake N. Bioorg Med Chem Lett. 2001;11:1219–1223. doi: 10.1016/s0960-894x(01)00176-7. [DOI] [PubMed] [Google Scholar]
- 12.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Endocrinology. 1999;140:800–804. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 13.LaFrate AL, Gunther JR, Carlson KE, Katzenellenbogen JA. Bioorg Med Chem Lett. 2008;16:10075–10084. doi: 10.1016/j.bmc.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Guimaraes CR, Cardozo M. J Chem Inf Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]; b) Niu H, Kalyanaraman C, Irwin JJ, Jacobson MP. J Chem Inf Model. 2006;46:243–253. doi: 10.1021/ci0502855. [DOI] [PubMed] [Google Scholar]
- 15.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen WL. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 17.Sun A, Yoon JJ, Yin Y, Prussia A, Yang Y, Min J, Plemper RK, Snyder JP. J Med Chem. 2008;51:3731–3741. doi: 10.1021/jm701239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Borea PA, Dalpiaz A, Varani K, Gilli P, Gilli G. Biochem Pharmacol. 2000;60:1549–1556. doi: 10.1016/s0006-2952(00)00368-3. [DOI] [PubMed] [Google Scholar]; b) Gilli P, Gilli G, Borea PA, Varani K, Scatturin A, Dalpiaz A. J Med Chem. 2005;48:2026–2035. doi: 10.1021/jm040842z. [DOI] [PubMed] [Google Scholar]
- 19.a) Abel R, Young T, Farid R, Berne BJ, Friesner RA. J Am Chem Soc. 2008;130:2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Freire E. Drug Discovery Today. 2008;13:869–874. doi: 10.1016/j.drudis2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Young T, Abel R, Kim B, Berne BJ, Friesner RA. Proc Natl Acad Sci U S A. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 20.a) Dunitz JD. Science. 1994;264:670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]; b) Zhang L, Hermans J. Proteins Struc Func Genetics. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 21.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proc. ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida. 2006. [Google Scholar]
- 22.Bondi A. J Phys Chem. 1964;68:441–451. [Google Scholar]
- 23.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 24.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 25.a) Peczuh MW, Hamilton AD. Chem Rev. 2000;100:2479–2494. doi: 10.1021/cr9900026. [DOI] [PubMed] [Google Scholar]; b) Peczuh MW, Hamilton AD. Chem Rev. 2000;100:2479–2494. doi: 10.1021/cr9900026. [DOI] [PubMed] [Google Scholar]; c) Toogood PL. J Med Chem. 2002;45:1543–1558. doi: 10.1021/jm010468s. [DOI] [PubMed] [Google Scholar]
- 26.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 27.Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 28.a) Arnold LA, Kosinski A, Estebanez-Perpina E, Fletterick RJ, Guy RK. J Med Chem. 2007;50:5269–5280. doi: 10.1021/jm070556y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hwang JY, Arnold LA, Zhu F, Kosinski A, Mangano TJ, Setola V, Roth BL, Guy RK. J Med Chem. 2009;52:3892–3901. doi: 10.1021/jm9002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu K, Shirts MR, Friesner RA. Journal of Chemical Theory and Computation. 2007;3:2108–2119. doi: 10.1021/ct700166f. [DOI] [PubMed] [Google Scholar]
- 30.Lyne PD, Lamb ML, Saeh JC. J Med Chem. 2006;49:4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- 31.a) Jensen MO, Dror RO, Xu H, Borhani DW, Arkin IT, Eastwood MP, Shaw DE. Proc Natl Acad Sci U S A. 2008;105:14430–14435. doi: 10.1073/pnas.0802401105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Maragakis P, Lindorff-Larsen K, Eastwood MP, Dror RO, Klepeis JL, Arkin IT, Jensen MO, Xu H, Trbovic N, Friesner RA, Palmer AG, III, Shaw DE. J Phys Chem B. 2008;112:6155–6158. doi: 10.1021/jp077018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.