

Abstract
Renewable neurosphere formation in culture is a defining characteristic of certain brain tumor initiating cells. This retrospective study was designed to assess the relationship between neurosphere formation in cultured human glioma, tumorigenic capacity, and patient clinical outcome. Tumor samples were cultured in neurosphere conditions from 32 patients with glioma, including a subpopulation of 15 patients with primary glioblastoma. A subsample of renewable neurosphere cultures was xenografted into mouse brain to determine if they were tumorigenic. Our study shows that both renewable neurosphere formation and tumorigenic capacity are significantly associated with clinical outcome measures. Renewable neurosphere formation in cultured human glioma significantly predicted an increased hazard of patient death and more rapid tumor progression. These results pertained to both the full population of glioma and the subpopulation of primary glioblastoma. Similarly, there was a significant hazard of progression for patients whose glioma had tumorigenic capacity. Multivariate analysis demonstrated that neurosphere formation remained a significant predictor of clinical outcome independent of Ki67 proliferation index. In addition, multivariate analysis of neurosphere formation, tumor grade and patient age, demonstrated that neurosphere formation was a robust, independent predictor of glioma tumor progression. While the lengthy duration of this assay may preclude direct clinical application, these results exemplify how neurosphere culture serves as a clinically relevant model for the study of malignant glioma. Furthermore, this study suggests that the ability to propagate brain tumor stem cells in vitro is associated with clinical outcome.
Keywords: Brain tumor stem cell, human glioma, glioblastoma (GBM), neurosphere, progression free survival, cancer
Introduction
Glioma is the most common malignant brain tumor and glioblastoma multiforme (GBM) is the most common class of primary glioma 1. Despite advances in surgery, radiotherapy, and chemotherapy, median survival from time of diagnosis for GBMs remains 15 months 2.
Neural stem cells were originally characterized and identified by their growth as neurospheres in minimal medium containing growth factors 3, 4. Numerous studies have utilized this culture system to cultivate neural stem-like cells from primary brain tumors 5-9. These cells have the capacity to self-renew, produce neurospheres, and differentiate into cell types present within the tumor of origin. These neurosphere forming cells are often termed ‘brain tumor stem cells’ because of their capacity, upon xenotransplantation into immunodeficient mice, to form gliomas which reflect the histopathological heterogeneity of the parental tumor 5, 10.
Neurosphere assays are commonly used to uncover more relevant brain tumor biology than traditional culture conditions. However, the link between this biology and clinical behavior has yet to be established. Here, we demonstrate that the ability of brain tumors to give rise to multi-passaged neurosphere cultures ex vivo is significantly associated with clinical outcome in a cohort of 32 glioma patients as well as a subpopulation of 15 patients with primary glioblastoma. Similarly, there was a significant hazard of progression for patients with glioma whose cultured tumors had tumorigenic capacity as compared to those whose tumors either did not grow or did not form tumors. Neurosphere formation remained a significant predictor of clinical outcome, independent of the Ki67 labeling index. In addition, multivariate analysis demonstrated that neurosphere formation was a predictor of glioma tumor progression, independent of tumor grade and patient age. These data demonstrate that in vitro growth of neurospheres reflects the clinical severity of gliomas as measured by patient outcome.
Material and Methods
Clinical Data and Tumor Collection
Brain tumor samples and follow-up data for 32 patients with glioma were collected following surgical resection under institutional review board-approved protocols between 1999-2008 and graded by the neuropathologist in accordance with World Health Organization-established guidelines 11. The cohort included 27 primary tumors and 5 recurrent tumors with 17 grade 4 (glioblastoma), 8 grade 3, 4 grade 2, and 3 grade 1 (for patient characteristics, see Table 1). The percentage of tumor cells labeled by Ki67 immunostaining was estimated semi-quantitatively in the areas of highest staining (maximum observed percentage) by two neuropathologists.
Table 1.
Patient Characteristics
| Sample ID | Age | Gender | Grade | Treatment Prior To Surgery* | Treatment Post Surgery* | Neurosphere (0=no, 1=yes) | Tumorigenic (0=no, 1=yes) |
|---|---|---|---|---|---|---|---|
| 1600 | 33.75 | M | 4 | R, C (Tamoxifen) | 1 | 1 | |
| NS 94 | 43.83 | M | 3 | R,C(Temodar) | C(Carboplatin) | 1 | ? |
| NS105 | 33.33 | F | 3 | R,C(Temodar) | C(Lomustine) | 0 | 0 |
| NS 171 | 46.08 | M | 4 | R | C(Temodar) | 0 | 0 |
| NS68 | 64 | M | 4 | R,C(Carboplatin) | 0 | 0 | |
| NS 85 | 60.5 | M | 4 | R,C(Temodar) | C(Lomustine) | 1 | ? |
| NS 111 | 59 | M | 3 | R,C(Temodar) | 0 | 0 | |
| NS74 | 62.58 | M | 4 | R,C(Temodar) | 0 | 0 | |
| NS 76 | 70.92 | M | 4 | R,C(Temodar) | 1 | ? | |
| NS 78 | 38.25 | F | 2 | C(Temodar) | 0 | 0 | |
| NS 98 | 60.42 | M | 4 | R,C(Temodar) | 1 | ? | |
| NS 122 | 51.58 | M | 2 | 0 | 0 | ||
| NS 127 | 42.58 | F | 2 | 0 | 0 | ||
| NS 134 | 77.83 | M | 4 | R,C(Temodar) | 0 | 0 | |
| NS 135 | 48.83 | M | 3 | R,C(Temodar) | 0 | 0 | |
| NS 141 | 24.92 | M | 3 | R | R,C(Temodar) | 0 | 0 |
| NS 139 | 34 | M | 3 | R,C(Temodar) | 0 | 0 | |
| NS 150 | 16.92 | M | 1 | 0 | 0 | ||
| NS 175 | 30.83 | M | 2 | 0 | 0 | ||
| NS 165 | 28.83 | M | 4 | C(GLIADEL Wafer) | R,C(Temodar) | 0 | 0 |
| NS 45 | 19.75 | M | 1 | R | 0 | 0 | |
| NS 174 | 35.42 | F | 3 | 0 | 0 | ||
| NS 146 | 56.33 | M | 4 | R,C(Temodar) | C(Erlotinib HCl) | 1 | 1 |
| NS 144 | 54.08 | F | 4 | R,C(Temodar) | 1 | 1 | |
| NS 151 | 31.5 | M | 3 | R,C(Temodar) | 1 | ? | |
| NS 157 | 55.33 | F | 4 | R,C(Temodar) | 1 | 1 | |
| NS 163 | 68 | F | 4 | R,C(Avastin) | 1 | ? | |
| NS 167 | 76.25 | M | 4 | R,C(Temodar) | 1 | 1 | |
| NS 176 | 60.75 | F | 4 | R,C(Avastin) | 1 | 1 | |
| NS 177 | 47.5 | M | 4 | R,C(Temodar) | 1 | 0 | |
| NS 64 | 10 | M | 1 | R | 0 | 0 | |
| NS 107 | 64 | M | 4 | R,C(Temodar) | 1 | 0 |
R= Radiation therapy, C= Chemotherapy.
All patients received surgical resection.
Cell Culture
Tumors were cultured as described previously 6 in DMEM/F12 medium supplemented with B27 (GIBCO), 20 ng/ml bFGF (Peprotech), 50 ng/mL EGF, penicillin/streptomycin (Invitrogen), L-glutamine (Invitrogen) and 5 ug/ml heparin (Sigma-Aldrich). Only the first seventeen cultures were also treated with leukemia inhibitory factor (20 ng/ml) as it was determined to be neither effective nor necessary. Heparin, bFGF and EGF were added twice a week. Every 7 to 10 days, spheres were passaged into fresh media following either enzymatic dissociation with TryplE or chopping using an automatic chopper (Geneq. Inc.) (8). Nine of the cell culture lines underwent clonal analysis at clonal density of 100 cells/ml to avoid clumping. Staining for nestin, neuronal (TuJ1) and glial (GFAP) markers were performed as previously described 6.
Xenograft
Animal experimentation was done with institutional approval following NIH guidelines using NOD-SCID mice (Jackson Labs). Eight renewable neurosphere cultures were xenografted into mouse brains. Several weeks before transplantation, cells were infected with a lentivirus expressing enhanced green fluorescent protein (EGFP) driven by the PGK promoter. On the day of transplantation, spheres were dissociated using TrypLE Express (Invitrogen) followed by trituration, washed, and stained with Trypan Blue. Cells were resuspended in DMEM/F12 (Invitrogen) at a density of 100,000 live cells/μl and stored at 4°C until injection. Animals were anesthetized using Isoflurane (1.5-2 Vol%, mixed with Oxygen). 200,000 cells were sterotatically injected over 5 minutes into the neostriatum using the coordinates of 0.5mm anterior from Bregma, 2.25mm lateral from the midline and 3.0mm below the pial surface. Animals were sacrificed if symptomatic or, at the latest, 4 months after transplantation. Following perfusion and post-fixation with 4% paraformaldehyde, brains were sectioned on a cryostat prior to counterstaining nuclei with Hoechst or Ethidium Bromide and visualization under fluorescence microscopy.
Statistical analysis
Time to survival (TTS) was determined by the duration of life from the date of surgery using death certificates and the social security death index. Time to progression (TTP) was determined by the duration of progression free survival from the date of surgery until recurrence, death, or, if progression free, until the last follow up date. Renewable neurosphere formation status and tumorigenic capacity were defined as binary variables as described below. As 23 patients in this open population were still alive at the time of this study, survival analysis was employed to take into consideration the censored nature of the outcomes. TTS and TTP were related to the explanatory variables using a Cox proportional hazards model as implemented in Stata 8.0 (StataCorp). This analysis was performed in the full cohort of glioma and within the subpopulation of glioblastoma. Multivariate Cox regression models were performed to control for grade, age, and Ki67 measurement values. The maximum observed percentage of Ki67 staining was treated as a continuous variable. All p-values were two sided and P<0.05 was considered significant. To visualize the survival distribution we used Kaplan-Meier plots.
Inclusion criteria
Only those patients who underwent full treatment, including chemotherapy and radiation therapy for GBMs, were included. Patients who died of unrelated causes were excluded. Cultures maintained for at least three passages, were considered to be capable of renewable neurosphere formation. Thirty-one of the 32 cultures grew as neurospheres, but one grade 3 tumor sample grew adherently. As this culture could be continuously passaged under “neurosphere” conditions, it was included in the neurosphere formation group. One tumor was obtained and initially cultured for several weeks in 10% fetal calf serum prior to placement in neurosphere media. This tumor was included as it formed neuropheres that could be passaged longterm in culture, indistinguishable from other cultures. Tumorigenic capacity was measured in a subpopulation of 26 gliomas; 8 renewable neurospheres were xenografted. The tumorigenic capacity of 6 xenografts that did form tumors (tumorigenic capacity = yes) was compared to the 2 xenografts that did not form tumors as well as to the 18 gliomas that did not form renewable neurosphere cultures (tumorigenic = no). Patients whose gliomas formed continuous neurosphere cultures but were never xenografted were considered unknowns and were not included in the analysis of tumorigenic capacity.
Results
In this cohort of 32 glioma patients who underwent full treatment, the mean patient survival time was 580 days and the mean progression free survival time was 439 days. Nine out of the 32 patients died before the last date of analysis. Fourteen of the 32 tumor samples formed renewable cultures under neurosphere conditions. In the subpopulation of 15 primary GBM, the mean patient survival time was 416 days and the mean progression free survival time was 295 days. Five of the 15 patients died before the last date of analysis. Eleven out of the 15 tumor samples formed renewable cultures under neurosphere conditions. Consistent with their similarities to neural progenitor cultures and with previous studies 5, 6, 8, 12, the neurosphere cultures we utilized had cells with the capacity to self-renew and undergo at least partial multi-lineage differentiation. We performed quantitative clonal analysis on 9 neurosphere cultures and found them capable of producing new neurospheres with a frequency of 2-24% of the cells plated (not shown). These neurosphere cultures generally expressed the intermediate filament nestin (Figure 1 A and B). When plated on adhesive substrate following removal of growth factors, cells with antigenic properties of neurons and glia were present (Figure 1C and D) albeit in varying proportions from tumor to tumor. For some tumors, cells with antigenic characteristics of both neurons and glia were present, as previously described 6, 7and shown in Figure 1.
Figure 1.
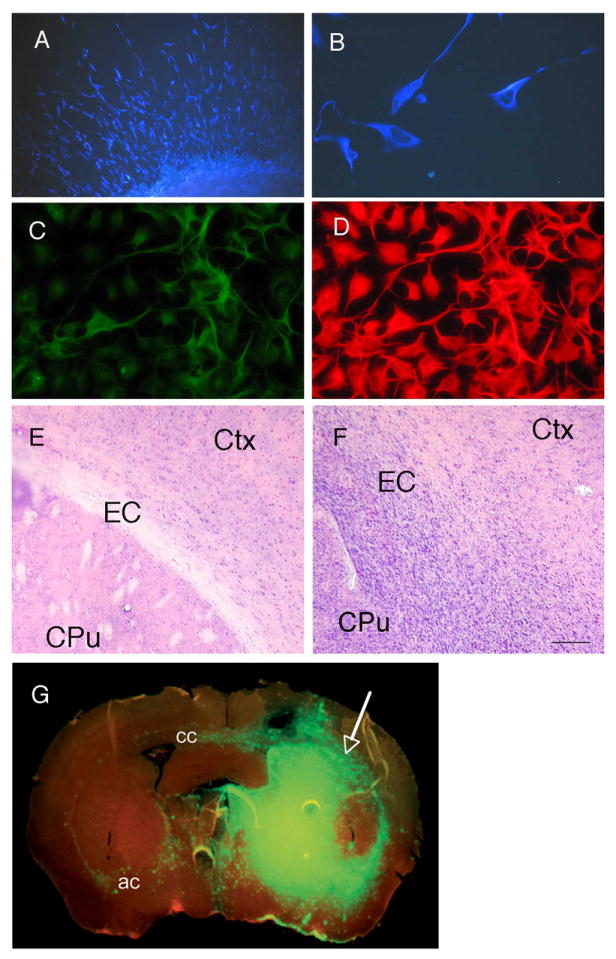
Human cultured glioblastoma cells. (A) & (B) Nestin staining (blue) of undifferentiated neurospheres. (C) TUJ-1 staining (green) of differentiated cells. (D) GFAP staining (red) of differentiated cells. (E) Coronal section through the striatum shows normal cellular composition in the hemisphere contralateral to the injection site for xenotransplant into the brain of immunodeficient mice (EC=external capsule, Ctx=Cortex, CPu= caudate putamen (striatum)). (F) Hemisphere of injection site with dense nuclei of infiltrating tumor cells. (G) Coronal section through the striatum shows EGFP expressing human glioblastoma cells forming a tumor mass (arrow) in the injected (right) hemisphere. The xenograft here exhibits a highly infiltrative character that is typical of high grade glioma. Major routes of infiltration from the injected hemisphere to the contralateral side are the corpus callosum (cc) and the anterior commissure (ac). Section was pre-treated with RNAse and counterstained with Ethidium Bromide (red). Typhoon Scanner image, at 10μm resolution. The scale bar in F represents 200 microns in A, 32 microns in B, 50 microns in C and D, 133 microns in E and F, and 800 microns in G.
Six out of the 8 intracranial neurosphere xenografts formed tumors in the mouse brain (Figure 1 E-G). These tumors infiltrated into the surrounding tissue in a manner that is characteristic of high grade glioma. All of the tumor-initiating neurosphere cultures originated from GBMs. Five originated from primary GBM, one from a recurrent GBM. The two xenografted neurosphere cultures that did not initiate tumors were also primary GBMs.
Associations between renewable neurosphere formation, tumorigenic capacity, and clinical outcome measures were examined using Cox proportional hazards models (Table 2). In the full population of glioma (n=32), renewable neurosphere formation was significantly associated with a 7.71 hazard of death (P=0.013) and a 5.83 hazard of progression (P=0.002). These significant associations persisted in the subpopulations of adult glioma (n=30) and primary glioma (n=22) (Table 2).
Table 2.
Cox Proportional Hazards Regressions
| Patient Population | Variable | Hazard of Patient Death | Hazard of Patient Tumor Progression |
|---|---|---|---|
| Glioma* (n=32) | Neurosphere Formation | 7.71, CI (1.54-38.5) P value 0.013 | 5.83, CI (1.91-17.8) P value 0.002 |
| Glioma (n=26) | Xenograft Tumor Formation | 3.31 (not significant), CI (0.588-18.6) P value 0.175 | 3.99, CI (1.23-12.9) P value 0.021 |
| Adult Glioma (Above 18) (n=30) | Neursophere Formation | 6.62, CI (1.33-32.9) P value 0.021 | 6.51, CI (1.82-23.2) P value 0.004 |
| Adult Glioma (Above 18) (n=24) | Xenograft Tumor Formation | 2.88 (not significant), CI (0.516-16.1) P value 0.228 | 4.06, CI (1.18-13.9) P value 0.026 |
| Primary Glioma (n=22) | Neurosphere Formation | 1.03E+16, CI (3.8e+15-2.79e+16) P value <0.001 | 5.28, CI (1.73-16.1) P value 0.004 |
| Primary Glioma (n=22) | Xenograft Tumor Formation | 4.6 (not significant), CI (0.393-53.7) P=0.224 | 3.62, CI (1.10-11.9) P value 0.034 |
| Primary Glioblastoma** (n=15) | Neurosphere Formation | 3.28E+15, CI (1.03e+15-1.04e+16) P value <0.001 | 3.16 (not significant), CI (0.439-22.7) P value 0.253 |
| Primary Glioblastoma (n=11) | Xenograft Tumor Formation | 1.41 (not significant), CI (0.126-15.8) P value 0.778 | 1.44 (not significant), CI (0.255-8.14) P value 0.670 |
CI denotes the 95% Confidence Interval
Mean Age 47. Tumors with Neurosphere Formation 14/32. 17 grade 4, 8 grade 3, 4 grade 2, 3 grade 1.
27 are primary tumors, 5 are recurrent.
Mean Age 58. All patients are adult. Primary GBM tumors with Neurosphere Formation 11/15.
To control for the effects of tumor grade on survival, a subsequent analysis was restricted to the subpopulation of grade 4, primary GBM patients (n=15). In this small subpopulation, renewable neurosphere formation was significantly associated with a hazard of death (HR>10, P<0.001) but not progression (Table 2). Table 2 reports the 95% confidence intervals of the hazard ratios. In glioma (n=26), tumorigenic capacity was significantly associated with a 3.99 hazard of progression (P=0.021). This significant association persisted in the subpopulations of adult glioma (n=24, P= 0.026) and primary glioma (n=22, P=0.034) but not in the group of GBM (n=11) (Table 2).
Figure 2 shows Kaplan-Meier curves displaying the proportion of patient survival as a function of days from surgery. These graphs represent fitted values from the Cox regression model and the inferences that follow are based on these fitted values. After two years, in the full cohort of glioma, the group with neurosphere formation had 31% survival, whereas the group without neurosphere formation had 86% survival. After two years, in the subpopulation of primary GBM, the group with neurosphere formation had 48% survival, whereas the group without neurosphere formation had 100% survival.
Figure 2.

Kaplan-Meier curves display the proportion of patients surviving as a function of time since surgery. (A) In the full cohort of glioma (n=32), the group with renewable neurosphere formation has a significantly higher hazard of death (Cox Regression, P=0.032). (B) In the subpopulation of primary glioblastoma (n=15), the group with renewable neurosphere formation has a significantly higher hazard of (Cox Regression, P<0.001).
Kaplan-Meier curves displaying the proportion of progression free survival as a function of days from surgery are shown in Figure 3A. After two years, in the full cohort of glioma, we found that the group with neurosphere formation (neurosphere=1) had 0% progression free survival, whereas the group without neurosphere formation (neurosphere=0) had 37% progression free survival. The median duration of progression for the neurosphere group was 370 days while the median duration of progression for the non-neurosphere group was 700 days. After two years, in a cohort of glioma (n=26), the group with tumorigenic capacity (tumor=1) had 2% progression free survival, whereas the group without tumorigenic capacity (tumor=0) had 35% progression free survival (Figure 3B). The median duration of progression for the tumorigenic group was 350 days (Figure 3B) while the median duration of progression for the non-neurosphere group was 620 days.
Figure 3.

Kaplan-Meier curves display the proportion of patients surviving without tumor progression as a function of time since surgery. (A) In the full cohort of glioma (n=32), the group with renewable neurosphere formation has a significantly higher hazard of progression (Cox Regression, P=0.002). (B) In the population of glioma (n=26), the group with tumorigenic capacity in culture has a significantly higher hazard of progression (P<0.021).
The maximum observed percentage of Ki67 measurement values was significantly associated with neurosphere formation in the full population of gliomas (T-Test, P<0.001) but not in the subpopulation of GBM. Ki67 levels were also significantly associated with tumorigenic capacity in the full population of gliomas (T-Test, P=0.0351) but not within GBM. In addition, Ki67 levels were associated with both patient survival (HR=1.04, p=0.05) and progression (HR=1.03, p=0.017). Within the subpopulation of glioblastoma patients, Ki67 was not associated with either clinical outcome. Multivariate Cox regression models demonstrate that neurosphere formation status remained a significant predictor of patient survival (HR=17, P=0.049) and progression (HR=4.7, P=0.034) even after adjusting for Ki67 staining. In contrast, Ki67 staining was not a significant predictor of clinical outcome in the multivariate Cox model after adjusting for neurosphere formation. Therefore, neurosphere formation status was independent of Ki67 and a more robust predictor of clinical outcome.
To determine the most robust prognostic variables, we carried out multivariate cox proportional hazards regressions, adjusting for tumor grade and patient age within the full population, and for patient age and ki67 within the GBM subpopulation. Within the full population of glioma, grade was the most robust, independent predictor of survival (OR 3.25, P=0.018) (Table 3). Neurosphere formation was not associated with survival after adjusting for grade and age. However, within the full population of glioma samples, even after adjusting for grade and age, neurosphere formation remained a significant predictor of tumor progression (OR 3.90, P=0.024) (Table 3). This demonstrates that neurosphere formation is an independent predictor of tumor progression. The other two explanatory variables were also demonstrated to be independent predictors of glioma tumor progression; tumor grade, OR=2.14 (P=0.020), and patient age, OR =0.972 (P=0.049) (Table 3). Within the subpopulation of primary glioblastoma, neurosphere formation was not associated to progression or survival after adjusting for age and Ki67.
Table 3.
Multivariate Cox Proportional Hazards Regressions
| Patient Population | Independent Variable | Controlled Variables | Hazard of Patient Death | Hazard of Patient Tumor Progression |
|---|---|---|---|---|
| Glioma* (n=32) | Neurosphere Formation | Grade, Age | 3.66 (not significant), CI (0.592-22.6) P value 0.163 | 3.90, CI (1.20-12.7) P value 0.024 |
| Glioma* (n=32) | Neurosphere Formation | Age | 6.38, CI(1.13-36.1) P value 0.036 | 5.35, CI(1.82-15.8) P value 0.002 |
| Glioma (n=30) | Neurosphere Formation | Ki67 | 17.0, CI(1.01-286) P value <0.049 | 4.71, CI (1.12-19.7) P value 0.034 |
| Glioma* (n=32) | Grade | Neurosphere Formation, Age | 3.25, CI (1.22-8.67) P value 0.018 | 2.14, CI (1.13-4.05) P value 0.020 |
| Glioma (n=26) | Grade | Xenograft Tumor Formation, Age | 5.83, CI (1.65-20.6) P value 0.006 | 2.12, CI (1.19-3.76) P value 0.010 |
| Glioma* (n=32) | Age | Neurosphere Formation, Grade | 0.993 CI (not significant) (0.958-1.03) P value 0.717 | 0.972, CI (0.945-0.999) P value 0.049 |
| Glioma (n=26) | Age | Grade, Xenograft Tumor Formation | 0.979 (not significant), CI (0.930-1.03) P=0.422 | 0.98 (not significant), CI (0.955-1.00) P value 0.113 |
| Glioma (n=26) | Xenograft Tumor Formation | Grade, Age | 1.12 (not significant), CI (0.185-6.77) P value 0.903 | 2.25 (not significant), CI (0.504-10.1) P value 0.287 |
CI denotes the 95% Confidence Interval
Mean Age 47. Tumors with Neurosphere Formation 14/32. 17 grade4, 8 grade3, 4 grade2, 3 grade1.
27 are primary tumors, 5 are recurrent.
Discussion
The current study provides evidence of an inverse correlation between clinical outcome and both neurosphere growth and tumorigenic capacity of cultured human glioma. Even within the most malignant tumors of grade 4, GBM patients, and despite the relatively small sample size used, renewable neurosphere formation distinguished a subpopulation with a highly significant increased hazard of death. This study demonstrates that the ability to form multi-passaged neurospheres reflects the clinical severity of malignant gliomas and lends credence to this culture system as an in vitro model of brain tumors.
Only GBM patients who underwent full treatment (both chemotherapy and radiation therapy) were included in this study. Although it may have introduced a selection bias into our cohort, this criterion was used because the course of treatment is expected to affect clinical outcome and therefore, to compare those patients who underwent full therapy with those that did not would have compromised the uniformity of our target population.
The use of the neurosphere culture system as a model for brain tumors has been supported by the capacity of neurosphere-generated cells to form tumors with many features of the parent tumor as shown here and previously 5, 9, 10. As all of the xenografted samples had first been passaged in vitro as neurosphere cultures, tumorigenic capacity is not an independent variable from neurosphere growth, but it is still an important validation of the relevance of the method used. Neurosphere cultures have a capacity to form xenografted tumors that is superior to unselected patient-derived cultures grown in serum 9. In vivo xenograft formation is a defining characteristic of tumor-initiating cells. However, a potential concern with xenografting is that this tumorigenic capacity could be an artifact of the culture system or of an in vivo environment that does not replicate that of the human brain. Our study provides evidence that neurosphere formation and tumorigenic capacity indicate clinical features of the parent tumor.
It is important to note that we did not see a clear relationship between the ability to form xenograft tumors and clinical outcome within the GBM population. This is not entirely surprising, since our study did not examine tumorigenesis in a quantitative fashion but rather asked a simple “yes/no” question. The sensitivity of our assays may have been limited by small sample size. Further study on a larger cohort will be needed to resolve these issues. Another important caveat is that, while current and previous studies demonstrate the tumorigenicity of neurosphere cultures, they do not prove that neurosphere-derived tumors are the single best xenograft model. Another means to reliably and repeatedly obtain tumors that reproduce the molecular and pathological features of the parent tumor is through the serial subcutaneous transplantation of tumor tissue in immunosuppressed mice13. It will be interesting, in the future, to directly compare the tumors derived from these different methods of amplification and determine whether one model or the other provides a more accurate portrayal of the patient tumor and its behavior during the course of therapy.
A key question is whether the ability to form neurospheres reflects the number or aggressiveness of cancer stem cells within the tumor prior to resection. Previous work has suggested that the expression levels of genes and proteins associated with brain tumor stem cells, such as nestin 14 and CD133 15 within brain tumors are correlated with clinical severity. Phillips et al. 16 demonstrated that subclasses of gliomas with gene signatures highly associated with neural stem cells are associated with a poor prognosis. Furthermore, in oligodendrogliomas, neurosphere formation is greater in the subset of tumors that have a poor prognosis17. Thus, there is at least some correlation between stem cell-associated features and clinical outcome in brain tumors. Our data provide further links between the biological capacity of brain tumor stem cells and patient outcome. It is likely that our cultures are the product, to at least some degree, of the propagation of brain tumor stem cells. They are passagable for long periods of time and remain tumorigenic. Thus, the correlation between neurosphere formation and clinical severity is consistent with, but does not prove the hypothesis that poor prognosis is associated with brain tumor stem cell activity. Clonal studies accompanied by xenografts with appropriately small numbers of cells will be needed to test this hypothesis.
For this study, we used a phenotypic selection—the ability to grow in neurosphere media—for neural stem-like cells, rather than utilize cell sorting prior to culturing. Although studies have demonstrated that brain tumor stem cells isolated from many malignant gliomas express CD133 on their surface 8, 18, there are now reports of tumors where this is not the case 19. Furthermore, in our experience, cell sorting negatively influences our ability to produce long term, viable cultures from primary brain tumors (not shown). While the methods we used have the drawback of resulting in starting material that may not be highly enriched in brain tumor stem cells, we 6, 20 and others 9, 21 have found them useful in capturing a highly significant self-renewing and tumor-initiating cell population or populations. On the other hand, it must be noted that our inability to grow long-term sphere cultures from certain patients does not confirm the absence of cancer stem cells in those tumors. It is possible that cancer stem cells from certain tumors have different growth requirements than the conditions used here.
One question raised by experiments with tumor–derived neurospheres is whether by examining sphere formation, we are actually examining and recapitulating the previously identified process of anchorage-independent growth. It has long been known that anchorage-independent growth is correlated with tumor aggressiveness and tumorigenicity 22-26 in certain tumor types, although we have not encountered studies of low passage, patient-derived glioma samples. We do not feel that the results of the current study are merely a reflection of the anchorage-independent growth properties of cultured gliomas. Instead, the neurosphere cultures being studied are most likely a result of selection for a highly significant population of cancer stem cells and their progeny. The cells that are propagating have the capacity to both divide in a seemingly self-renewing manner and give rise to progeny with multilineage differentiation potential—the hallmarks of stem cells. It is likely that this selection occurs as a result of the serum-free medium containing supplements and growth factors, rather than simply anchorage independence. We do not routinely observed significant sphere formation in media that omits EGF and bFGF 6. Furthermore, cells grown in serum not only do not form neurospheres in uncoated plates, but they also do not form tumors after xenotransplantation. Finally, anchorage-independence is not a requirement for passaging neurosphere cultures with tumorigenic capacity under our conditions as at least one of our lines reported in this study grew as attached cells, and we have grown very similar cells on a variety of substrates (not shown).
Multivariate Cox regression analysis in the full glioma population shows that neurosphere propagation was a significant, prognostic factor for tumor progression that is independent of age and grade (Table 3). This no longer holds when considering survival time as the clinical outcome. Specifically, neurosphere formation is no longer significant when grade is added as covariate in a multivariate Cox model. The following reasons may explain why neurosphere is no longer a significant predictor of survival after adjusting for grade. First, neurosphere formation may capture the same information as grade. This is possible since the Spearman correlation between neurosphere status and grade is significant (r=0.60, p<0.001). Second, the sample size may be too small to establish the predictive capacity of neurosphere formation within each tumor grade.
Yet, we cannot say that grade is a confounder in our association between neurosphere formation and survival. Grade and neurosphere formation are not different exposures but rather different diagnostic criteria. While grade is a more significant predictor of survival than neurosphere formation, neurosphere formation is still significantly correlated with survival. Only when the association is adjusted for grade does the association disappear. The correlation between neurosphere formation and survival remains significant when adjusted for age alone. Why does the adjustment for grade obviate the correlation between neurosphere formation and survival? This may be the result of a small population size. Alternatively, this may be a true reflection that grade is more predictive of patient survival than neurosphere formation. If one assumes that the neurosphere assay is an assessment of cancer stem cell content, then one might hypothesize that our results suggest that cancer stem cell content is more predictive of tumor progression than it is of patient survival. These results make intuitive sense as the cancer stem cell hypothesis posits that cancer stem cells are directly and primarily responsible for tumor recurrence while patient survival may be dependent on multiple factors including the patient’s physical response to recurrence, overall health, and response to radiation and chemotherapy treatment. In other words, tumor progression may be a direct consequence of stem cell content while patient survival may be dependent on other factors as well. Further research will be needed to determine the validity of this hypothesis.
In contrast to survival, neurosphere formation had more robust associations with clinical outcome than did routine Ki67 measurement values. In multivariate analysis, neurosphere formation remained a significant predictor of clinical outcome while Ki67 did not. Within the subpopulation of GBM, neurosphere formation remained a significant predictor of clinical outcome while Ki67 measurement values did not. These latter findings are consistent with a previous study that found no association between Ki67 staining and clinical outcome of GBM 27. They also demonstrate that the ability to form neurospheres is not a simple product of the overall proliferative capacity of the brain tumor at the time of resection.
Conclusion
In conclusion, the current study demonstrates that neurosphere culture provides a unique model to investigate the role of cancer stem cells in glioma progression and severity. Here, we show that this model reflects the severity of the original patient tumor, independent of grade, age, and overall cell proliferation. While the lengthy duration of this assay and the technical expertise required to perform it may preclude direct clinical application, these results exemplify how neurosphere culture serves as a clinically relevant model for the study of malignant glioma. Furthermore, this study suggests that the ability to propagate brain tumor stem cells in vitro is associated with clinical outcome.
Acknowledgments
Financial Support: The Dr. Miriam and Sheldon Adelson Program in Neural Repair Research (HIK, PVM, KV), California Institute for Regenerative Medicine (BA), NIH NINDS NS052563 (HIK) and NS050151 (PVM) and the Ziering Family Foundation (TFC).
Abbreviations
- TTP
time to progression (progression free survival time)
- TTS
time to survival (overall survival time)
- GBM
glioblastoma multiforme
- bFGF
basic fibroblast growth factor
- EGF
epidermal growth factor
- EGFP
enhanced green fluorescent protein
- CI
confidence interval
- HR
hazard ratio
Footnotes
Dan R Laks: Conception and design, data analysis and interpretation, manuscript writing.
Michael Masterman-Smith: Collection and/or assembly of data, manuscript writing.
Koppany Visnyei: Collection and/or assembly of data.
Brigitte Angenieux: Collection and/or assembly of data.
Nicholas M. Orozco: Collection and/or assembly of data.
Ian Foran: Collection and/or assembly of data.
William H. Yong: Collection and/or assembly of data.
Harry V. Vinters: Provision of study material, collection and/or assembly of data.
Linda M. Liau: Provision of study material or patients, collection and/or assembly of data.
Jorge A. Lazareff: Provision of study material or patients, collection and/or assembly of data.
Paul S. Mischel: Provision of study material or patients, collection and/or assembly of data, conception and design, manuscript writing.
Timothy F. Cloughesy: Provision of study material or patients, collection and/or assembly of data, conception and design, manuscript writing.
Steve Horvath: Data analysis and interpretation, manuscript witing.
Harley I. Kornblum: Conception and design, manuscript writing, final approval of manuscript, financial support.
There are no potential conflicts of interest.
References
- 1.Buckner JC, Brown PD, O’Neill BP, et al. Central nervous system tumors. Mayo Clin Proc. 2007;82:1271–1286. doi: 10.4065/82.10.1271. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 5.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 6.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 8.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 9.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 11.Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. discussion 226-219. [DOI] [PubMed] [Google Scholar]
- 12.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 13.Shu Q, Wong KK, Su JM, et al. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells. 2008;26:1414–1424. doi: 10.1634/stemcells.2007-1009. [DOI] [PubMed] [Google Scholar]
- 14.Mangiola A, Lama G, Giannitelli C, et al. Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme: possible prognostic implications. Clin Cancer Res. 2007;13:6970–6977. doi: 10.1158/1078-0432.CCR-07-1229. [DOI] [PubMed] [Google Scholar]
- 15.Zeppernick F, Ahmadi R, Campos B, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 16.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 17.Beier D, Wischhusen J, Dietmaier W, et al. CD133 expression and cancer stem cells predict prognosis in high-grade oligodendroglial tumors. Brain Pathol. 2008;18:370–377. doi: 10.1111/j.1750-3639.2008.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SK, Clarke ID, Hide T, et al. Cancer stem cells in nervous system tumors. Oncogene. 2004;23:7267–7273. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 19.Joo KM, Kim SY, Jin X, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Laboratory investigation; a journal of technical methods and pathology. 2008;88:808–815. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 20.Nakano I, Masterman-Smith M, Saigusa K, et al. Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J Neurosci Res. 2008;86:48–60. doi: 10.1002/jnr.21471. [DOI] [PubMed] [Google Scholar]
- 21.Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 22.Cifone MA, Fidler IJ. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc Natl Acad Sci U S A. 1980;77:1039–1043. doi: 10.1073/pnas.77.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3:355–359. doi: 10.1016/0092-8674(74)90050-6. [DOI] [PubMed] [Google Scholar]
- 24.Shin SI, Freedman VH, Risser R, et al. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci U S A. 1975;72:4435–4439. doi: 10.1073/pnas.72.11.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colburn NH, Bruegge WF, Bates JR, et al. Correlation of anchorage-independent growth with tumorigenicity of chemically transformed mouse epidermal cells. Cancer Res. 1978;38:624–634. [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 27.Bouvier-Labit C, Chinot O, Ochi C, et al. Prognostic significance of Ki67, p53 and epidermal growth factor receptor immunostaining in human glioblastomas. Neuropathology and applied neurobiology. 1998;24:381–388. doi: 10.1046/j.1365-2990.1998.00137.x. [DOI] [PubMed] [Google Scholar]