

Abstract
This Letter describes the discovery and SAR of three novel series of mGluR5 noncompetitive antagonists/negative allosteric modulators (NAMs) not based on manipulation of an MPEP/MTEP chemotype identified by a functional HTS approach. This work demonstrates fundamentally new mGluR5 NAM chemotypes with submicromolar potencies, and further examples of a mode of pharmacology `switch' to provide PAMs with a non-MPEP scaffold.
Metabotropic glutamate receptors (mGluRs) are members of the G protein-coupled receptor (GPCR) family C, which is distinguished from other GPCR families by its large extracellular N-terminal agonist binding site. There are eight subtypes of mGluRs (Group I: mGluR1 and mGluR5; Group II: mGluR2 and mGluR3; Group III: mGluRs 4,6,7,8) based on their sequence homology, pharmacology and coupling to effector mechanisms.1 The mGluRs bind glutamate to modulate neurotransmitter release or postsynaptic excitatory neurotransmission, hence they modulate the strength of synaptic transmissions. Different from ionotropic glutamate receptors which mediate the fast excitatory neurotransmissions, metabotropic glutamate receptors play a more modulatory role and have been proposed as alternative targets for pharmacological interventions.1
Group I receptor mGluR5 has been implicated in a number of physiological processes in the central nervous system (CNS). The discovery of highly selective mGluR5 receptor antagonists MPEP (1) and MTEP (2) was a major breakthrough (Fig.1). MPEP (1) and MTEP (2) in preclinical models demonstrated that selective antagonism of mGluR5 has therapeutic potential for the treatment of pain, anxiety, depression, cocaine addiction and Fragile X Syndrome (FXS).2
Figure 1.
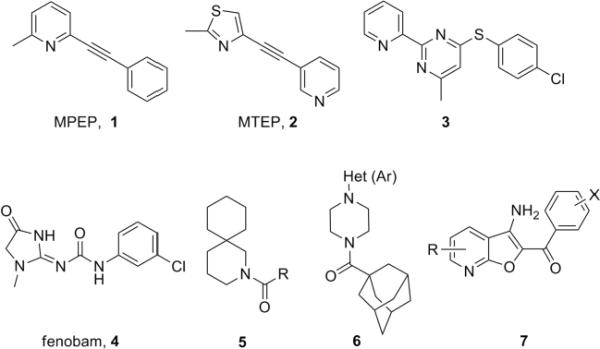
Reported mGluR5 non-competitive antagonists/negative allosteric modulators (NAMs).
A majority of reported non-competitive mGluR5 antagonists have employed the MPEP (1) and MTEP(2) chemotypes as a basis for ligand design.3–11 Only a few more diverse structures, such as thiopyrimidine (3) and fenobam (4), have been disclosed.12,13 Recently our group reported three novel non-MPEP chemotypes of mGluR5 antagonists (5, 6, and 7), which exemplified a dramatic departure from the MPEP scaffold.14
As a continuing effort to design and synthesize novel non-MPEP-based mGluR5 negative allosteric modulators (NAMs), in this letter, we describe the discovery and SAR of three novel mGluR5 NAM series represented by 8, 9 and 10 (Fig. 2). These novel leads were identified from a functional high-throughput mGluR5 antagonist screen of over 160,000 compounds (624 mGluR5 antagonists identified, 0.39% hit rate in the primary screen).14 Leads 8, 9 and 10 all possess submicromolar IC50s from the HTS DMSO stock solutions, good clogP values and low molecular weights, which are ideal properties for further SAR development.
Figure 2.
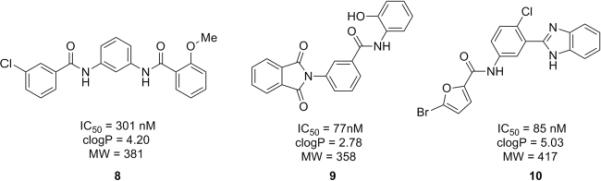
Novel non-MPEP mGluR5 non-competitive antagonists 8, 9 and 10 identified from a functional HTS campaign.
Lead 8, an N,N′-(1,3-phenylene)dibenzamide, was first investigated and SAR explored through the synthesis of two small 24-membered libraries.15 The first library aimed at holding the 2-methoxybenzamide moiety constant, while varying the other amide portion of the molecule. As illustrated in Scheme 1, synthesis of key intermediate 13 was achieved by coupling of 3-nitroaniline 11 and 2-methoxybenzoyl chloride under mild basic conditions to provide 12, followed by hydrogenation with Raney nickel to deliver 13. We applied an iterative parallel synthesis strategy for this library preparation, as well as all the other libraries in this letter, and resynthesized 8 in the context of a 24-member library through standard acylation of 13 with 24 commercial acid chlorides. Excess reagents were scavenged by PS-isocyanate and PS-trisamine, and the final products were purified by mass-directed preparative LCMS to >98% purity.16
Scheme 1.
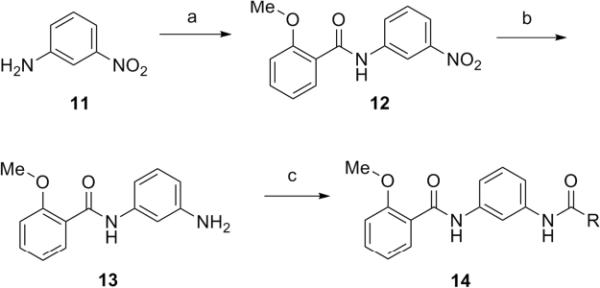
Reagents and conditions: (a) 2-methoxybenzoyl chloride, DIEA, DMAP, DMF, 64%, (b) Raney nickel, H2 (50 psi), EtOH/EtOAc (3:2), 94%, (c) (i) RCOCl, PS-DMAP, PS-DIEA, (ii)PS-isocyanate, PS-trisamine, 25–90%.
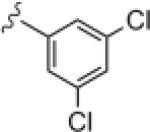
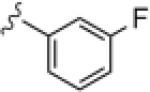
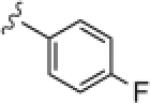
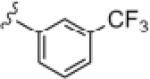
Upon resynthesis, lead 8 suffered a 3-fold loss in potency (IC50 = 880 nM) compared to the HTS stock solution (IC50 = 301 nM); however, clear SAR was still noted for this series (Table 1). Lead 8 was the most potent compound. Aromatic groups were the only active analogs; meta- substitution was favored (14e, 14g, 14h and 14i, potency Cl > Me > CN > F > CF3), any other substitution patterns in the aromatic ring were not tolerated (14a–14d, 14f and 14j). Introduction of other groups, such as heterocyclic derivative (14k), benzyl variant (14l), cyclic alkyl moieties (14n–14r) or acyclic alkyl derivatives (14s–14u), were all proven inactive.
Table 1.
Structures and activities of analogs 14.
| Cmpd | R | mGluR5a IC50 (μM) | % Glu Max |
|---|---|---|---|
| 8 |
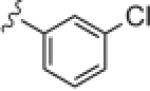
|
0.88 | 2.6 |
| 14a |
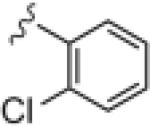
|
>30 | ND |
| 14b |
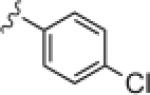
|
>30 | ND |
| 14c |
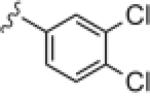
|
>30 | ND |
| 14d |
|
>30 | ND |
| 14e |
|
6.48 | 25 8 |
| 14f |
|
>30 | ND |
| 14g |
|
10.60 | 26.1 |
| 14h |
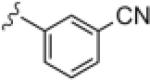
|
1.80 | 3.22 |
| 14i |
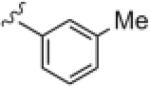
|
1.35 | 3.82 |
| 14j |
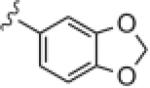
|
>30 | ND |
| 14k |
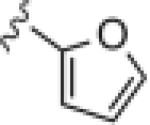
|
>30 | ND |
| 14l |
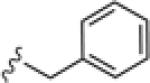
|
>30 | ND |
| 14m |
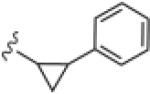
|
>30 | ND |
| 14n |
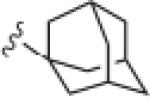
|
>30 | ND |
| 14o |
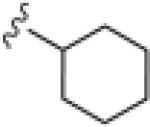
|
>30 | ND |
| 14p |
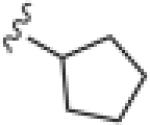
|
>30 | ND |
| 14q |
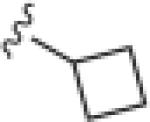
|
>30 | ND |
| 14r |
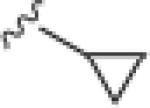
|
>30 | ND |
| 14s |
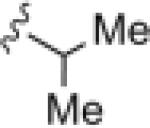
|
>30 | ND |
| 14t |
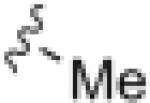
|
>30 | ND |
| 14u |
|
>30 | ND |
IC50s are average of three determinations. ND. not determined.
A second library was prepared in similar fashion with the goal of holding the 3-chloro group constant, while changing the other amide moiety. As shown in Scheme 2, synthesis of key intermediate 16 was achieved by coupling of 3-nitroaniline 11 and 3-chorobenzoyl chloride under mild basic conditions to provide 15. A subsequent Raney nickel-catalyzed hydrogenation at atmospheric pressure delivers 16. A 24-member library 17 was synthesized through standard acylation of 16 with 24 readily available acid chlorides.14 Excess reagents were scavenged by PS-isocyanate and PS-trisamine, and the final products were purified by mass-directed preparative LCMS with purity >98%.16
Scheme 2.
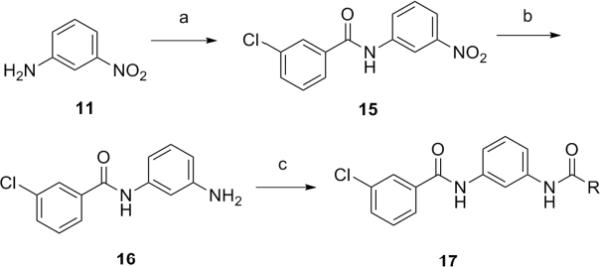
Reagents and conditions: (a) 3-chlorobenzoyl chloride, DIEA, DMAP, DMF, 89%, (b) Raney nickel, H2 (1.0 atm), EtOH/EtOAc (3:2), 89%, (c) (i) RCOCl, PS-DMAP, PS-DIEA, (ii)PS-isocyanate, PS-trisamine, 28–92%.
Generally (Table 2), aromatic groups were not well tolerated (17b–17i), with exception of the 2-chlorophenyl analog 17a (IC50=2.62 μM) which proved to be the most potent NAM in this series, along with the cylcopropyl amide 17q. Benzyl analog 17k, phenylcyclopropane derivative 17l and adamantyl compound 17m proved inactive. Interestingly, 6- and 5-member cyclic alkyl moieties were inactive (17n and 17o). However, a cyclobutyl amide provided a NAM of modest potency (17p, IC50 = 5.27 μM), and further contraction to the cyclopropyl derivative 17q led to improved inhibition (IC50 = 2.23 μM). Acyclic alkyl analogs 17r and 17s both demonstrated weak inhibition, while the ether variant 17t showed no activity. Most surprising from this library (Fig. 3) was the identification of two weak, but unexpected mGluR5 positive allosteric modulators (PAMs) 17u (EC50 = 1.87 μM, 49% Glu Max) and 17v (EC50 = 5.54 μM, 56% Glu Max), both regiosiomeric pyridine amide analogs.14,17,18 While weak, 17u and 17v represent a fundamentally new mGluR5 PAM chemotype, worthy of further optimization.17–22
Table 2.
Structures and activities of analogs 17.
| Cmpd | R | mGluR5a IC50 (μM) | % Glu Max |
|---|---|---|---|
| 17a |
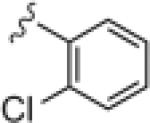
|
2.62 | 8.78 |
| 17b |
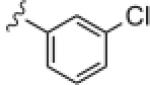
|
>30 | ND |
| 17c |
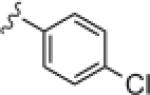
|
>30 | ND |
| 17d |
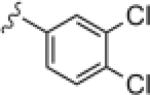
|
>30 | ND |
| 17e |
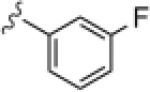
|
>30 | ND |
| 17f |
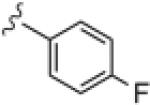
|
>30 | ND |
| 17g |
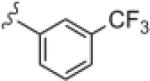
|
>30 | ND |
| 17h |
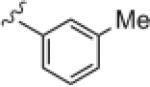
|
>30 | ND |
| 17i |
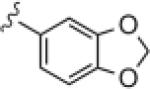
|
>30 | ND |
| 17j |
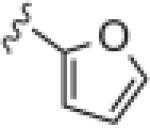
|
4.51 | 49.3 |
| 17k |
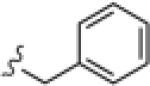
|
>30 | ND |
| 17l |
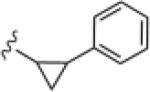
|
>30 | ND |
| 17m |
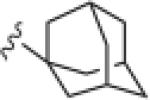
|
>30 | ND |
| 17n |
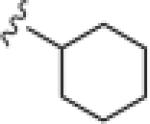
|
>30 | ND |
| 17o |
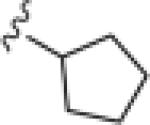
|
>30 | ND |
| 17p |

|
5.27 | 44.0 |
| 17q |
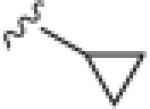
|
2.23 | 32.3 |
| 17r |
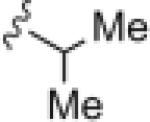
|
5.82 | 26.9 |
| 17s |
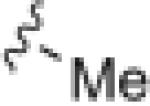
|
7.97 | 39.6 |
| 17t |
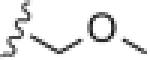
|
>30 | ND |
IC50s are average of three determinations. ND. not determined.
Figure 3.
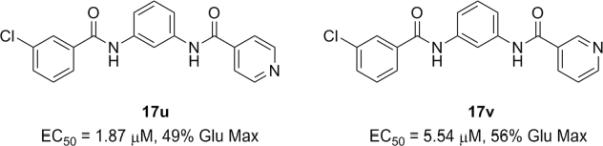
Novel non-MPEP mGluR5 PAMs 17u and 17v.
From this N,N′-(1,3-phenylene)dibenzamide series, 8 was further evaluated and found to displace [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine, a radioligand for the MPEP allosteric binding site on mGluR5, with a Ki value of 1.1 μM, comparable to the IC50 value (880 nM). Thus 8 represents a new mGluR5 non-competitive antagonist chemotype, inhibiting mGluR5 function by interaction with MPEP allosteric binding site. Future libraries will focus on SAR studies of the central phenyl core modifications.
Our attention was then directed to lead 9, 3-(phthalimidyl)-N-(2-hydroxyphenyl)benzamide. Lead 9 possesses very similar structure to CPPHA,20,21 the only known mGluR5 allosteric ligand, a PAM, that binds at an allosteric site distinct from the MPEP binding site (Fig. 4). We resynthesized 9 in the context of a 22-member library. Preparation of the library was straightforward as indicated in Scheme 3. Condensation between readily available methyl 3-aminobenzoate 18 and phthalic anhydride 19 in acetic acid afforded methyl 3-(N-phthalimidyl)benzoate 20. Saponification of 20 using lithium hydroxide gave benzoic acid 21. Scaffold 21 was then coupled to 22 different amines employing HOBt/EDC, to deliver the 3-(N-phthalimidyl) benzamide library 22. The final products were purified by mass-directed preparative LCMS to >98% purity.16
Figure 4.
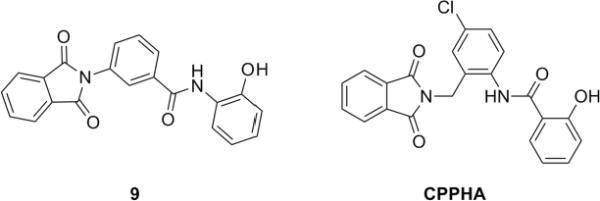
Structural similarity of 9 and non-MPEP binding mGluR5 PAM, CPPHA.
Scheme 3.
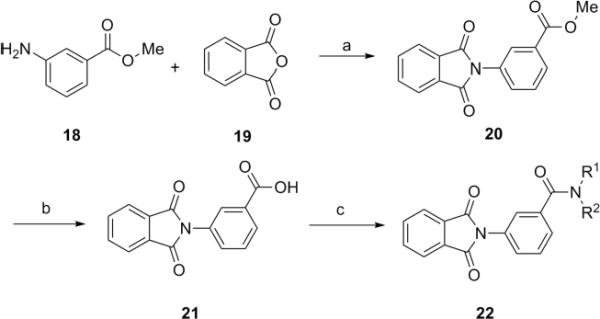
Reagents and conditions: (a) HOAc, 100 °C, overnight, 100%, (b) LiOH, THF/MeOH/H2O (4:1:1), 91%, (c) HOBt, EDC, DIEA, DMF, 35–90%.
Upon resynthesis, lead 9 suffered a five-fold loss in potency (IC50 = 417 nM) compared to the HTS stock solution (IC50 = 77 nM) (Table 3). The discrepancies between the resynthesized compounds and HTS DMSO stocks have been noted many times in our various programs previously.14,17 Lead 9 was still the most potent compound in this series. In general, functionalized aromatic and heteroaromatic analogs were more active (9, 22a–22h), and alkyl derivatives were inactive (22i, 22j, IC50 >μM). Moving the hydroxyl group of 9 from the 2-position to 3-position (22a), led to a 19-fold loss in potency (IC50 = 7.79 μM). 3,5-Dimethoxyl substrate 22b showed moderate inhibitory activity (IC50 = 3.58 μM), while 2-methoxyl analog 22c lost all the activity. 3-Chloro derivative 22d displayed improved potency (IC50 = 1.78 μM) compared to 3-hydroxyl analog 22a. 3-Pyridyl compound 22e proved inactive, however, its 2-pyridyl congener 22f showed reasonable activity (IC50 = 1.26 μM) as a partial antagonist.23 2-Flouro derivative 22g was favored over 4-flouro analog 22h, giving good potency (IC50 = 1.05 μM) as a partial antagonist as well.24 Considering the structural similarity of this series with the non-MPEP binding PAM CPPHA,20,21 we were hoping that this series would also interact with this distinct allosteric binding site and provide the first NAM tools to study the CPPHA binding site. However, further evaluation of 9 showed the displacement of [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine with a Ki of 319 nM – a value comparable to the IC50 (420 nM), which indicated its interaction with MPEP-binding site. Our continuing efforts will focus on modifications of both the phthalimide moiety and the central phenyl ring of the scaffold.
Table 3.
Structures and activities of analog 22.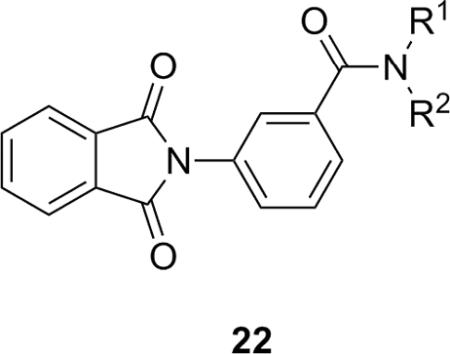
| Cmpd | R | mGluR5a IC50 (μM) | % Glu Max |
|---|---|---|---|
| 9 |
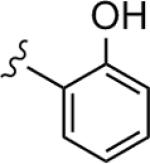
|
0.42 | 1.59 |
| 22a |
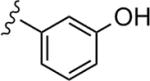
|
7.79 | 48.1 |
| 22b |
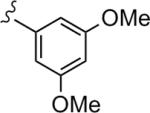
|
3.58 | 76.2 |
| 22c |
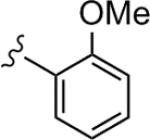
|
>30 | 86 |
| 22d |
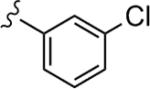
|
1.78 | 26.4 |
| 22e |
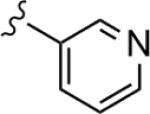
|
>30 | 86 |
| 22f |
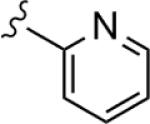
|
1.26 | 59.5 |
| 22g |
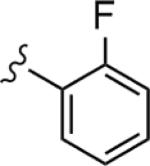
|
1.05 | 49.9 |
| 22h |
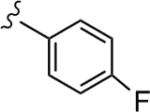
|
>30 | 73 |
| 22i |
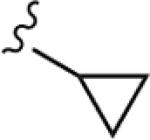
|
>30 | 68 |
| 22j |
|
>30 | 71 |
IC50s are average of three determinations.
Lastly, our attention was directed at lead optimization of 10, N-(3-(1H-benzo[d]imidazol-2-yl)-4-chlorophenyl)-5-bromofuran-2-carboxamide. Once again, we resynthesized 10 in the context of a small library of analogs.15 Surprisingly, upon resynthesis, lead 10 was totally inactive (IC50 >30μM) and the majority of analogs displayed only poor activity at best (IC50s 10–30μM). This effort produced three interesting compounds (Fig. 5) 23 wherein the chlorine of the central phenyl ring was replaced with hydrogen, 24 and 25 where the benzimidazole is replaced with a benzoxazole. In these examples, 23 was a weak mGluR5 NAM (IC50 = 3.5μM, 34% Glu Max), 25 was a full mGluR5 NAM (IC50 = 2.1 μM, 2.3% Glu Max) while 24 was a modestly potent mGluR5 PAM (EC50 = 2.2 μM, 68% Glu Max) – a very interesting `switch' in the mode of mGluR5 pharmacology.17,18 Unlike 8 and 9, which afforded comparable IC50s/Kis, 24 displaced [3H]3-methoxy-5-(2-pyridinylethynyl)pyridine with a Ki >10-fold less than the EC50. These data are consistent with the hypothesis that the PAM activity of 24 is mediated by interaction with the MPEP site.25,26 Previous studies suggest that mGluR5 PAMs acting at this site display strong cooperativity with orthosteric agonists so that the mGluR5 PAM potencies are significantly greater than their affinities. However, based on these data, it is also possible that 24 does not act solely through interaction with the MPEP site, but potentially at the CPPHA site or a potentially third allosteric site on mGluR5. Studies are underway to address this question. Importantly, 8, 9, 23, 24 and 25 were selective versus mGluRs 1, 2, 3, 4, 7 and 8.
Figure 5.
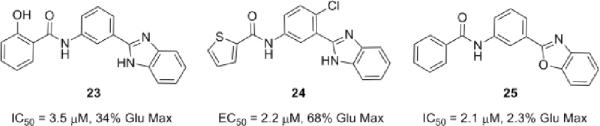
Non-MPEP Chemotype NAMs 23 and 25 as well as PAM 24, a non-MPEP site ligand, from a library of analogs of 10.
In summary, our HTS campaign identified several novel non-competitive mGluR5 antagonists based on 8 and 9 with little or no structural and topological similarity to MPEP. An iterative parallel library synthesis strategy helped to rapidly develop SAR for these series. IC50s of analogs of 8 and 9 ranged from 420 nM to 880 nM for the most potent mGluR5 NAMs and, despite divergence from the MPEP chemotype, binding experiments indicated these NAMs bound to the MPEP site. Optimization efforts also identified fundamentally new mGluR5 PAM chemotypes, represented by 17u and 17v, as well as new mGluR5 partial antagonists 22f and 22g. While lead 10 was inactive upon resynthesis, both NAMs 23 and 25 and PAM 24 were identified within an analog library, and PAM 24 displayed only weak displacement of [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine, suggesting 24 may not act solely through interaction with the MPEP site. These data further highlight the complexities and subtle modifications that can alter modes of mGluR5 pharmacology for MPEP site ligands. Further refinements in this arena are in progress and will be reported in due course.
Acknowledgement
The authors thank NIDA (DA023947-01) and Seaside Therapeutics (VUMC33842) for support of our programs in the development of mGluR5 non-competitive antagonists and partial antagonists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1).a) Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]; b) Conn PJ, Pin J-P. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2).a) Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]; b) Lea PM, IV, Faden AI. CNS Drug Reviews. 2006;12(2):149–166. doi: 10.1111/j.1527-3458.2006.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Alagille D, Baldwin RM, Roth BL, Wroblewski JT, Grajkowska E, Tamagnan GD. Bioorg. Med. Chem. Lett. 2005;15:945–949. doi: 10.1016/j.bmcl.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 4).Roppe JR, Wang B, Huang D, Tehrani L, Kamenecka T, Schweiger EJ, Anderson JJ, Brodkin J, Jiang X, Cramer M, Chung J, Reyes-Manalo G, Munoz B, Cosford NDP. Bioorg. Med. Chem. Lett. 2004;14:3993–3996. doi: 10.1016/j.bmcl.2004.05.037. [DOI] [PubMed] [Google Scholar]
- 5).Newman AH, Kulkarni SS. Bioorg. Med. Chem. Lett. 2007;17:2987–2991. doi: 10.1016/j.bmcl.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Galatis P, Yamagata K, Wendt JA, Connolly CJ, Mickelson JW, Milbank JBJ, Bove SE, Knauer CS, Brooker RM, Augelli-Szafran CE, Schwartz RD, Kinsora JJ, Kilgore KS. Bioorg. Med. Chem. Lett. 2007;17:6525–6528. doi: 10.1016/j.bmcl.2007.09.083. [DOI] [PubMed] [Google Scholar]
- 7).Newman AH, Kulkarni SS. Bioorg. Med. Chem. Lett. 2007;17:2074–2079. doi: 10.1016/j.bmcl.2006.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Eastman B, Chen C, Smith ND, Poon S, Chung J, Reyes-Manalo G, Cosford NDP, Munoz B. Bioorg. Med. Chem. Lett. 2004;14:5485–5488. doi: 10.1016/j.bmcl.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 9).Bach P, Nilsson K, Svensson T, Baur U, Hammerland LG, Peterson A, Wallberg A, Osterland K, Karis D, Boije M, Wensbo D. Bioorg. Med. Chem. Lett. 2006;16:4788–4791. doi: 10.1016/j.bmcl.2006.06.078. [DOI] [PubMed] [Google Scholar]
- 10).Micheli F, Bertani B, Bozzoli A, Crippa L, Cavanni P, Fabio RD, Donati D, Marzorati P, Merlo G, Paio A, Pergunni L, Zarantonello P. Bioorg. Med. Chem. Lett. 2008;18:1804–1809. doi: 10.1016/j.bmcl.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 11).Kulkarni SS, Zhao M-F, Cao J, Deschamps JR, Rodriguez AL, Conn PJ, Newman AH. J. Med. Chem. 2009;52:3563–3575. doi: 10.1021/jm900172f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Hammerland LG, Johansson M, Mattson JP, Minidis ABE, Nilsson K, Peterson A, Wensbo D, Wallberg A, Osterlund K. Bioorg. Med. Chem. Lett. 2006;16:2467–2469. doi: 10.1016/j.bmcl.2006.01.100. [DOI] [PubMed] [Google Scholar]
- 13).Porter RHP, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters J-U, Prinseen E, Wichmann J, Vieira E, Muhlemann A, Gatti S, Mutel V, Malherbe P. J. Pharm. Exp. Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 14).Rodriguez AL, Williams R, Zhou Y, Lindsley SR, Le U, Grier MD, Weaver CD, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2009;19:3209–3213. doi: 10.1016/j.bmcl.2009.04.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) mGluR5 in vitro Functional Assay. HEK293A cells expressing rat mGluR5 receptor were plated (BD Falcon Poly-D-Lysine Cellware) at 50,000 cells/well in assay media (DMEM, 20 mM HEPES, 10% dialyzed FBS, and 1 mM sodium pyruvate). The plates were incubated overnight at 37°C in 5% CO2. Media was removed and assay buffer (Hanks Balanced Salt Solution, 20 mM HEPES, 2.5 mM Probenecid, pH 7.4) containing 4.0 μM Fluo4-AM dye (Invitrogen) was added. Cells were incubated for 45 minutes (37°C, 5% CO2) to allow for dye loading. Dye was removed, 20 μL assay buffer was added and the cell plate was allowed to incubate for 10 minutes. After incubation in assay buffer, cell plates were loaded into Flexstation II (Molecular Devices Corp). Test compound in assay buffer was added 19 seconds into the assay and subsequently, asubmaximal (250 nM) or nearly maximal (1.25 μM) amount of glutamate was added 109 seconds into the assay for potentiators and antagonists respectively. Controls included compound vehicle (0.2% DMSO) plus assay buffer, ECmax (100 μM glutamate), and submaximal or nearly maximal concentrations of glutamate. Compounds were tested in concentrations ranging from 46 pM to 100 μM. Fold shifts were determined using the same functional assay by varying the amount of glutamate in the presence of either a fixed concentration of compound (10 μM) or vehicle. A concentration response curve was generated using glutamate concentrations ranging from 4.6 nM to 10 μM. Controls included compound vehicle (0.2% DMSO) plus assay buffer, ECmax (100 μM glutamate), and glutamate concentration response curve. Assays were performed in triplicate on three different days. Concentration response curves were generated using GraphPad Prism 4.0
- 15).Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J.Comb. Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 16).Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J. Comb.Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 17).Sharma S, Rodriguez A, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Sharma S, Heiman JU, Rook JM, Jones CK, Conn PJ, Lindsley CW. J. Med. Chem. WEB ASAP. [Google Scholar]
- 19).O'Brien JA, Lemaire W, Chen T-B, Chang RSL, Jacobson MA, Ha SN, Lindsley CW, Sur C, Pettibone DJ, Conn J, Wiliams DL. Mol. Pharmacol. 2003;64(3):731–738. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 20).O'Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Sur C, Duggan ME, Pettibone DJ, Conn J, Williams DL. J. Pharmacol. Exp. Therapeut. 2004;309(2):568–579. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 21).Zhao Z, Wisnoski DD, O'Brien JA, Lemiare W, Williams DL, Jr., Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg. Med. Chem.Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 22).Lindsley CW, Wisnoski DD, Leister WH, O'Brien JA, Lemiare W, Williams DL, Jr., Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. J. Med. Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 23).Engers DW, Rodriguez AL, Oluwatola O, Hammond AS, Venable DF, Williams R, Sulikowski GA, Conn PJ, Lindsley CW. ChemMedChem. 2009;4:505–511. doi: 10.1002/cmdc.200800357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Rodriguez AL, Nong Yi., Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. Mol. Pharm. 2005;68:793–1802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- 25).de Paulis T, Hemstapat K, Chen Y, Zhang Y, Saleh S, Alagille D, Baldwin RM, Tamagnan GD, Conn PJ. J. Med. Chem. 2006;49:3332–3344. doi: 10.1021/jm051252j. [DOI] [PubMed] [Google Scholar]
- 26).Chen Y, Goudet C, Pin JP, Conn PJ. Mol. Pharm. 2008;73:909–918. doi: 10.1124/mol.107.040097. [DOI] [PubMed] [Google Scholar]