

Abstract
This Letter describes the first account of the synthesis and SAR, developed through an iterative analogue library approach, of analogues of the highly selective M1 allosteric agonist TBPB. With slight structural changes, mAChR selectivity was maintained, but the degree of partial M1 agonism varied considerably.
The muscarinic acetylcholine receptors (mAChRs) are members of the GPCR family A that mediate the metabotropic actions of the neurotransmitter acetylcholine (ACh).1–3 To date, five distinct subtypes of mAChRs (M1–M5) have been cloned and sequenced. M1, M3 and M5 activate phospholipase C and calcium through Gq whereas M2 and M4 block the action of adenylyl cyclase through Gi/o.1–3 mAChR-regulated cholinergic signalling plays a critical role in a wide variety of CNS and peripheral functions including memory and attention mechanisms, motor control, nociception, regulation of sleep wake cycles, cardiovascular function, renal and gastrointestinal function and many others.4–6 As a result, agents that can selectively modulate the activity of specific mAChRs have therapeutic potential in multiple pathological states.1–6 However, due to high sequence conservation within the orthosteric binding site of the five mAChR subtypes, historically it has been difficult to develop subtype selective ligands.1–6 The native orthosteric ligand for the mAChRs is acetylcholine 1 (ACh), and numerous synthetic congeners such as carbachol 2 (CCh) have been employed for assay development (Figure 1).1–6 Prototypical mAChR activators include the pan-mAChR agonists tascilidine 3 and oxotremorine 4 and the M1/M4 preferring agonist xanomeline 5.1–6 More recently, AF267B 6 and related AF congeners have been reported as M1 selective agonists.7 However, in our hands, AF267B 6 activates M1, M3 and M5, and is M3 preferring.8 While AC-42 7 (an ectopic agonist) and brucine 8 (an M1 positive allosteric modulator) lack M1 potency and clean ancillary pharmacology to probe the effects of selective M1 activation in vitro or in vivo, they validated that allosteric sites can confer M1 mAChR-subtype selectivity by exploiting allosteric binding sites.9,10
Figure 1.

Structures of representative orthosteric and allosteric mAChR activators.
In numerous Phase II and III clinical trails, pan-mAChR agonists were shown to improve cognitive performance in AD patients, nevertheless, the GI-and/or cardiovascular side effects, resulting from activation of peripheral mAChRs, were deemed intolerable and the trials were discontinued.1–10 Importantly, both 3 and AF102B, a congener of 6, promoted a reduction of Aβ42 in the cerebral spinal fluid of AD patients, suggesting that mAChR activation has the potential to be disease modifying as well as providing palliative cognitive therapy.1–10 More recent studies in 3xTg-AD mice with 6 further support a disease modifying role for mAChR activation.1–10 Interestingly, the M1/M4 preferring 5, in addition to improving cognitive performance, had robust therapeutic effects on the psychotic symptoms and behavioral disturbances associated with AD, and more recently demonstrated positive effects in a schizophrenia trial.11,12
We recently reported on TBPB 9, a potent, centrally active and highly selective M1 allosteric agonist, which displayed robust efficacy in several preclinical antipsychotic models as well as significant effects on the processing of amyloid precursor protein (APP) towards the non-amyloidogenic pathway and decreased Aβ production.8 However, TBPB 9 was an un-optimized screening lead with antagonist activity at D2 (IC50 = 2.6 μM).8,13 Despite an [18F]-fallypride micro-PET study that confirmed the antipsychotic activity observed with 9 was the result of selective M1 activation and not due to inhibition of D2, we hoped to diminish D2 activity through a lead optimization campaign.8 In this Letter, we describe the synthesis, SAR and pharmacological profile of the first analogues ever reported for this important M1 selective small molecule probe.
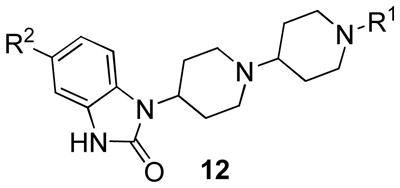
Lead optimization efforts employed an iterative parallel synthesis approach as shown in Scheme 1.14 Analogues 12 were prepared by a reductive amination sequence employing a functionalized piperidone 11 and 4-(2-keto-1- benzoimidazolinyl)piperidine or the 5-chloro congener and polymer-supported triacetoxy borohydride.16 In the first round of library synthesis, efforts first focused on commercially available piperidinones 11 (wherein R1 is either an alkyl or carbamate moiety) to determine the effect of modulating basicity and/or topology. Subsequent rounds of library synthesis utilized benzylic analogues of 11 generated by standard alkylation chemistry with 10 and substituted benzyl halides. Table 1 highlights representative SAR for this effort. Out of the initial 24-member library with simple aliphatic and carbamate derivatives of 11, only one retained M1 agonist activity. Compound 12a, an ethyl carbamate analogue (Figure 3) displayed improved M1 efficacy (EC50 = 2.1 nM, 65% CCh Max), but lost selectivity versus M2–M5 (EC50s = 11.9 nM, 113 nM, 9.1 nM and 34.3 nM, respectively) degenerating into a pan-mAChR partial agonist.
Scheme 1.
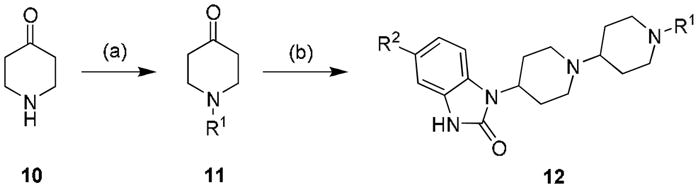
Reagents: (a) K2CO3, R1Br, DCM, 70–88%; (b) MP-B(OAc)3H, 4-(2-keto-1-benzoimidazolinyl)piperidine or 5-chloro-1-(4-piperidinyl)-2-benzimidazolone, DCM, rt, 24 hr, 85–95%. All compounds purified by mass-directed HPLC to analytical purity (>98%).15
Table 1.
Functional Activity of TBPB Analogues 12.
 | |||||
|---|---|---|---|---|---|
| Cmpd | R1 | R2 | M1 EC50 (nM)a | %CCh Maxa | D2 IC50 (μM)a |
| TBPB | 2-MeBn | H | 289 | 82 | 2.65 |
| 12a | CO2Et | H | 2.1 | 65 | >10 |
| 12b | 2-MeBn | Cl | 1,030 | 84 | 35%@10 μM |
| 12c | Bn | Cl | 356 | 74 | 35%@10 μM |
| 12d | 2-CF3Bn | H | 410 | 82 | 30%@10 μM |
| 12e | 2-CF3Bn | Cl | 1,500 | 83 | ND |
| 12f | 2-ClBn | H | 260 | 44 | 2.06 |
| 12g | 2-ClBn | Cl | 1,800 | 49 | ND |
| 12h | 2-NO2Bn | H | 120 | 48 | 1.13 |
| 12i | 2-NO2Bn | Cl | 1,100 | 42 | ND |
| 12j | 2-CNBn | H | 490 | 23 | 40%@10 μM |
| 12k | 2-CNBn | Cl | 1,600 | 27 | ND |
EC50s, % CCh maximum and IC50s are the mean of at least three independent determinations. All analogues in this Table are selective for M1 (>50 μM vs. M2–M5), except 12a. ND, not determined.
Figure 3.

Concentration response curves for TBPB analogue 12a at M1–M5. Data represent the mean±SEM of three independent determinations. M1 EC50 = 2.1 nM (82.6% CCh Max), M2 EC50 = 11.9 nM (80.6% CCh Max), M3 EC50 = 113 nM (24.1% CCh Max), M4 EC50 = 9.1 nM (96.9% CCh Max), M5 EC50 = 34.3 nM (50.8% CCh Max).
In the second round of analogue synthesis, we focused on alternative benzyl moieties for the distal piperidine ring of TBPB while maintaining the 4-(2-keto-1-benzoimidazolinyl)piperidine moiety, a well known GPCR privileged structure, with either hydrogen or a chlorine atom in the 5-position, the latter of which to block a site of oxidative metabolism we identified. This effort generated over 60 analogues, and the SAR proved to be rather ‘flat’. If the benzyl group possessed substituents in the 3- or 4-positions, all M1 agonism was lost (EC50 >20 μM). As shown in Table 1, it was possible to synthesize other analogues with selective M1 partial agonism (M1 EC50s 120 nM to 1.8 μM, >50 μM vs. M2–M5), but the degree of agonism varied widely (23–84%). Incorporation of a 5-Cl atom into TBPB to block metabolism results in 12b, a compound that maintains the same degree of agonism as TBPB, but loses ~5-fold in efficacy. Removal of the 2-Me group results in an analogue comparable to TBPB (12c). Attempts to replace the metabolically labile 2-methyl group with alternative chemical moieties (12d–12k) generally led to a significant diminution in M1 agonism (<50%), M1 efficacy, or both. However, the 2-nitrobenzyl analogue 12h proved more potent than TBPB (M1 EC50 = 120 nM), but the degree of agonism fell to 42% of the maximum CCh response. One exception to this was the 2-CF3 group, which provided an analogue with an M1 EC50 of 410 nM and 82% of the maximum CCh response.
With respect to D2 inhibition, 12d maintained an mAChR profile comparable to TBPB, but D2 inhibition was greatly diminished (~30%@10 μM), indicating that 12d might prove to be a more useful small molecule tool to probe M1 than TBPB. The only analogue more potent than TBPB, 12h, also displayed increased D2 inhibition (IC50 = 1.13 μM); however, a dual M1 allosteric agonist/D2 antagonist may prove to be an excellent profile for a novel schizophrenia treatment.
We then decided to evaluate the effect of a small structural modification and targeted fluorine substituted 4-(2-keto-1-benzoimidazolinyl)piperidines, prepared by modification of the Merck route (Scheme 2).17
Scheme 2.

Reagents: (a) Ethyl 4-aminopiperidine, Na2CO3, KI, cyclohexanol, μw, 180°C, 10 min, 70–90%; (b) Zn, 1N HCl, MeOH; (c) tripshosgene, Et3N, THF, rt, 2 h; (d) 10% NaOH, μw, 130°C, 30 min, 50–60% from 14. All compounds purified were by mass-directed HPLC to analytical purity (>98%).15
Beginning with commercial fluorine-substituted ortho-fluoro-nitroaromatic compounds 13 were heated under microwave irradiation with ethyl 4-amino-1-piperdinecarboxylate to deliver the SNAr products 14 in yields ranging from 70–90%. A zinc mediated nitro reduction afforded 15 which was then treated with triphosgene to provide the benzimidazolone 16. Removal of the ethyl carbamate was accomplished under another microwave-assisted protocol to deliver the 4-, 5- and 6-fluoro-1-(piperdin-4-yl)-1-H-benzo[d]imidazol-2(3H)-ones 17 in 50–60% yield from 14.
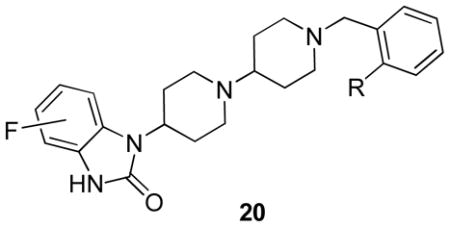
Analogues 20 were prepared by a reductive amination sequence employing either 2-methyl benzyl piperidone 18 or 2-trifluoromethyl benzyl piperidone 19 and the 4-, 5- or 6-fluoro-1-(piperdin-4-yl)-1-H-benzo[d]imidazol-2(3H)-ones 17. As shown in Table 2, TBPB analogues 20 containing a fluorine atom were uniformly less active than the unsubstituted parents, TBPB and 12d. However, analogues such as 20b and 20c possessed sub-micromolar EC50s and were fully efficacious (850 nM, 85% CCh max and 760 nM, 84% CCh max, respectively); moreover, they afforded no significant D2 inhibition. Based on these data, we prepared additional 4-(2-keto-1-benzoimidazolinyl)piperidines and TBPB analogues with alternative substitutions (Br, CN, CF3), but most were found to be either inactive, or partial agonists with <20% efficacy, or they were no longer selective for M1. After hundreds of analogues had been synthesized and evaluated, we found that this was a rare instance, from our experience, where the original screening lead, TBPB, could not be significantly improved upon. However, we have encountered ‘flat’ SAR with other allosteric ligands for class C GPCRs such as mGluR5.18–20
Table 2.
Functional Activity of TBPB Analogues 20.
 | |||||
|---|---|---|---|---|---|
| Cmpd | R | F | M1 EC50 (μM)a | %CCh Maxa | D2 IC50 (μM)a |
| 20a | CH3 | 4-F | 1.06 | 73 | >10 |
| 20b | CH3 | 5-F | 0.85 | 85 | 40%@10 μM |
| 20c | CH3 | 6-F | 0.76 | 84 | >10 |
| 20d | CF3 | 4-F | 1.71 | 75 | >10 |
| 20e | CF3 | 5-F | 1.04 | 80 | 2.69 |
| 20f | CF3 | 6-F | >10 | ND | >10 |
EC50s, % CCh maximum and IC50s are the mean of at least three independent determinations. All analogues in this Table are selective for M1 (>50 μM vs. M2–M5). ND, not determined.
In summary, we have identified a novel series of allosteric partial to full agonists with high selectivity for M1 versus M2–M5 based on a (1-(1′-substituted)-1,4′-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one scaffold. SAR was ‘flat’ within the TBPB series, with subtle changes resulting in pan-mAChR agonism, loss of M1 efficacy, significant decreases in the degree of partial agonism or undesirable ancillary pharmacology. For instance, 12h afforded an over 2-fold improvement in M1 EC50, relative to TBPB, but the degree of agonism diminished to 48% of CCh max. This was a rare example in which the screening lead TBPB could not be optimized, but possessed a profile that enabled both in vitro and in vivo studies to be conducted. Moreover, this study further exemplifies the challenges in the development of allosteric ligands for GPCRs. Further refinements to the TBPB scaffold are in progress and will be reported in due course.
Figure 2.

Concentration response curves for TBPB (9) at M1–M5. Data represent the mean±SEM of three independent determinations. M1 EC50 = 289 nM, 82% CCh Max, M2–M5 EC50 >50 μM.
Scheme 3.
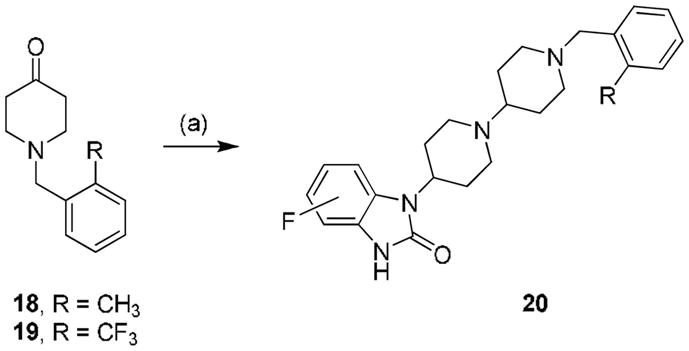
Reagents: (a) MP-B(OAc)3H, 4-, 5- and 6-fluoro-1-(piperdin-4-yl)-1-H-benzo[d]imidazol-2(3H)-ones 17, 50–90%. All compounds purified were by mass-directed HPLC to analytical purity (>98%).15
Acknowledgments
The authors thank the NIH and NIMH for support of our programs (1RO1MH082867-01). The authors specifically acknowledge the support of a Vanderbilt Institute of Chemical Biology Pilot Project Grant, the Alzheimer’s Association (IIRG-07-57131). T.M.B. acknowledges an ITTD (T90-DA022873) pre-doctoral training grant and A.E.B. is supported by a National Research Service Award (1FM32 MH079678-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]; (b) Bonner TI, Young AC, Buckley NJ, Brann MR. Neuron. 1988;1:403–410. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 2.Felder CC, Bymaster FP, Ward J, DeLapp N. J Med Chem. 2000;43:4333–4353. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- 3.Bymaster FP, McKinzie DL, Felder CC, Wess J. Neurochem Res. 2003;28:437–442. doi: 10.1023/a:1022844517200. [DOI] [PubMed] [Google Scholar]
- 4.Eglen RM, Choppin A, Dillon MP, Hedge S. Curr Opin Chem Biol. 1999;3:426–432. doi: 10.1016/S1367-5931(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 5.Birdsall NJM, Nathanson NM, Schwarz RD. TRENDS Pharm Sci. 2001;22:215–219. [Google Scholar]
- 6.(a) Eglen RA, Choppin A, Watson N. TRENDS Pharm Sci. 2001;22:409–414. doi: 10.1016/s0165-6147(00)01737-5. [DOI] [PubMed] [Google Scholar]; (b) Tarsy D, Simon DK. N Engl J Med. 2006;335:818–829. doi: 10.1056/NEJMra055549. [DOI] [PubMed] [Google Scholar]
- 7.(a) Fisher A. Jpn J Pharmacol. 2000;84:101–112. doi: 10.1254/jjp.84.101. [DOI] [PubMed] [Google Scholar]; (b) Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 8.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane A, Bridges TM, Kennedy JP, Bradley SR, Peterson T, Baldwin RM, Kessler R, Deutch A, Levey AI, Lindsley CW, Conn PJ. J Neurosci. doi: 10.1523/JNEUROSCI.1850-08.2008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmead CJ, Fry VAH, Forbes IT, Branch CL, Christopoulos A, Wood MD, Hedron HJ. Mol Pharm. 2006;69:236–246. doi: 10.1124/mol.105.017814. [DOI] [PubMed] [Google Scholar]
- 10.Lazareno S, Gharagozloo P, Kuonen D, Popham A, Birdsall NJ. Mol Pharm. 1998;53:573–589. doi: 10.1124/mol.53.3.573. [DOI] [PubMed] [Google Scholar]
- 11.Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Arch Neurol. 1997;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- 12.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am J Psychiatry. 2008 Jul 1; doi: 10.1176/appi.ajp.2008.06091591. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 13.Kinney GG. Neuropsychopharmacology. 2006;31(supplement 1):S26. [Google Scholar]
- 14.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J Comb Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 15.Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J Comb Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 16.Experimental details: Synthesis of N-benzyl piperidinones. Each of sixty-four glass reaction vials containing 3 mL of CH2Cl2 and 0.1 mL of MeOH were loaded with piperidone hydrochloride (25 mg, 0.185 mmol, 1.0 equivalents) and K2CO3 (51 mg, 0.370 mmol, 2.0 equivalents). Then, one of sixty-four functionalized benzyl bromides (0.185 mmol, 1.0 equivalents) was added to each reaction tube. The reactions were stirred overnight at room temperature, partitioned between CH2Cl2 and H2O, and the organics were concentrated on a heat-air block. Purification by preparative LCMS was performed to afford N-benzyl piperidinones products for subsequent reductive amination.Reductive Amination. Each of sixty-four glass reaction vials containing 3 mL of CH2Cl2 were loaded with MP-B(OAc)3H (142 mg, 0.345 mmol, 2.42 mmol/g, 3.0 equivalents) and 4-(2-keto-1-benzoimidazolinyl)piperidine (25 mg, 0.115 mmol, 1.0 equivalents) or 5-chloro-1-(4-piperidinyl)-2-benzimidazolone (29 mg, 0.115 mmol, 1.0 equivalents). Then, one of sixty-four tertiary amine hydrates (0.115 mmol, 1.0 equivalents) from the previous alkylation reactions was added to each vial, and the reactions were stirred overnight at room temperature. The next day, the solutions were filtered and washed with CH2Cl2 (3 × 3 mL), then dried before purification by mass-directed preparative HPLC.TBPB, 1-(1′-2-methylbenzyl)-1,4′-bipiperidin-4-yl)-1Hbenzo[d]imidazol-2(3H)-one (9). To a stirred solution of 4-piperidone monohydrate HCl (2.00 grams, 13.02 mmol) in dimethylformamide (100 mL) was added 2-methylbenzyl bromide (2.44 grams, 13.02 mmol) and K2CO3 (3.60 grams, 26.04 mmol, 2.0 equivalents) at room temperature. The reaction was then monitored by analytical LCMS, and once judged complete, the reaction was separated with water and EtOAc. The organics were next washed with a saturated solution of NaCl and dried over MgSO4 before being concentrated in vacuo to afford 2.59 grams (90%) of of the hydrate form of the piperidone 1-(2-methylbenzyl)piperidin-4,4-diol as a colorless oil. Analytical LCMS (J-Sphere80-C18, 3.0 × 50.0 mm, 4.1 min gradient, 5%[0.05% TFA/CH3CN]:95%[0.05% TFA/H2O]:1.64 min, >99% (214 nm, 254 nm and ELSD) M+1 peak m/e 204.1; 1H NMR (400 MHz, DMSO-d6) δ 7.28 (m, 1H), 7.16 (m, 3H), 3.53 (s, 2H), 2.67 (t, J = 6.4 Hz, 4H), 2.36 (s, 3H), 2.32 (t, J = 6.4 Hz, 4H). 13C NMR (100 MHz, DMSO-d6) δ 137.0, 136.4, 130.1, 129.3, 127.0, 125.4, 58.7, 52.4, 40.6, 18.8; HRMS calc’d for C13H20NO2[M+H]; 222.1494 found 222.1488. 1-(2-Methylbenzyl)piperidin-4,4-diol (2.00 grams, 9.04 mmol) was then brought up in a stirred solution of CH2Cl2 (100 mL) to which 4-(2-keto-1-benzimidazolinyl)piperidine (2.16 grams, 9.95 mmol, 1.1 equivalents) was added at room temperature. Then, MP-triacetoxyborohydride (13.57 grams, 27.132 mmol [2.0 mmol/gram loading], 3.0 equivalents) was added and the reaction was monitored by analytical LCMS. Once judged complete, the reaction was filtered and the filtrate was purified by mass-directed preparative HPLC to afford 2.92 grams (80%) of title compound as a pure white solid. Analytical LCMS (J-Sphere80-C18, 3.0 × 50.0 mm, 4.1 min gradient, 5%[0.05% TFA/CH3CN]:95%[0.05% TFA/H2O]: 2.34 min, >99% (214 nm, 254 nm and ELSD) M+1 peak m/e 405.2; 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 9.97 (br s, 1H), 7.31 (d. J = 4.4 hz, 1H), 7.00 (m, 3H), 4.57 (m, 1H), 4.12 (br d, J = 11.6 Hz, 2H), 4.04 (q, J = 7.2 Hz, 2H), 3.59 (br d, J = 11.2 Hz, 2H), 3.47 (m, 1H), 3.24 (m, 2H), 2.82 (m, 2H), 2.66 (m, 2H), 2.05 (m, 2H), 1.94 (br d, J = 12.4 Hz, 2H), 1.59 (m, 2H), 1.19 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 154.4, 153.6, 128.7, 128.4, 120.9, 120.3, 109.1, 108.6, 62.5, 60.9, 48.1, 46.6, 42.1, 25.8, 14.5; HRMS calc’d for C25H33N4O[M+H]; 405.2654 found 405.2654; Ethyl 4-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-1,4′-bipiperidine-1′-carboxylate (12a). To a stirred solution of 4-(2-keto-1-benzimidazolinyl)piperidine (50 mg, 0.230 mmol) in CHCl2 (3 mL) was added ethyl 4-oxo-1-piperidinecarboxylate (43.3 mg, 0.253 mmol, 1.1 equivalents) and MP-B(OAc)3H (285 mg, 0.691 mmol, 2.42 mmol/g, 3.0 equivalents). The reaction was then monitored by analytical LCMS, and once judged complete, the reaction was filtered and concentrated. Purification by mass-directed preparative HPLC afforded 32.50 mg (38%) of title compound as a beige-white solid. Analytical LC/MS (J-Sphere80-C18, 3.0 × 50.0 mm, 4.1 min gradient, 5%[0.05% TFA/CH3CN]:95%[0.05% TFA/H2O]: 2.05 min, >99% (214 nm, 254 nm and ELSD) M+1 peak m/e 373.2; 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 9.97 (br s, 1H), 7.31 (d. J = 4.4 hz, 1H), 7.00 (m, 3H), 4.57 (m, 1H), 4.12 (br d, J = 11.6 Hz, 2H), 4.04 (q, J = 7.2 Hz, 2H), 3.59 (br d, J = 11.2 Hz, 2H), 3.47 (m, 1H), 3.24 (m, 2H), 2.82 (m, 2H), 2.66 (m, 2H), 2.05 (m, 2H), 1.94 (br d, J = 12.4 Hz, 2H), 1.59 (m, 2H), 1.19 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 154.4, 153.6, 128.7, 128.4, 120.9, 120.3, 109.1, 108.6, 62.5, 60.9, 48.1, 46.6, 42.1, 25.8, 14.5; HRMS calc’d for C20H29N4O3[M+H]; 373.2240 found 373.2235.
- 17.Burgey CS, Stump CA, Nguyen DM, Deng JZ, Quigley AG, Norton BR, Bell IM, Mosser SD, Salvatore CA, Rutledge RZ, Kane SA, Koblan KS, Vacca JP, Graham SL, Williams TM. Bioorg Med Chem Lett. 2006;16:5052–5056. doi: 10.1016/j.bmcl.2006.07.044. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien JA, Lemaire W, Chen TB, Chang RSL, Jacobson MA, Ha SN, Lindsley CW, Sur C, Pettibone DJ, Conn J, Wiliams DL. Mol Pharmacol. 2003;64(3):731–738. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Z, Wisnoski DD, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg Med Chem Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 20.Sharma S, Rodriguez A, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]