

Abstract
This Letter describes the discovery and SAR of three novel series of mGluR5 non-competitive antagonists/negative allosteric modulators (NAMs) not based on manipulation of an MPEP/MTEP chemotype. This work demonstrates fundamentally new mGluR5 NAM chemotypes with submicromolar potencies, and the first example of a mode of pharmacology `switch' to provide PAMs with a non-MPEP scaffold.
Glutamate is the major excitatory transmitter in the central nervous system, exerting its effects through both ionotropic and metabotropic glutamate receptors. The metabotropic glutamate receptors (mGluRs) are members of the GPCR family C, characterized by a large extracellular amino-terminal agonist binding domain. To date, eight mGluRs have been cloned, sequenced and assigned to three groups (Group I: mGluR1 and mGluR5; Group II: mGluR2 and mGluR3; Group III: mGluRs 4,6,7,8) based on their sequence homology, pharmacology, and coupling to effector mechanisms.1 In preclinical models, studies with the non-competitive antagonists MPEP (1) and MTEP (2) have demonstrated that selective antagonism of mGluR5 has therapeutic potential for chronic disorders such as pain, anxiety, depression, cocaine addiction and Fragile X syndrome.2
The vast majority of reported non-competitive mGluR5 antagonists have been designed based on the MPEP (1) and MTEP (2) scaffolds.3,4 Many recent efforts have produced diverse heterobicylic analogs 3,5,6 along with other directed efforts to replace the acetylinic linker with amides 47 and heterocycles 5.8 Other reports describe homologated variants such as 69 and novel heterobiaryls such as 7.10 In terms of structural diversity, the thiopyrimidine 811 and fenobam 912 display the greatest departure from the MPEP chemotype; however, all of these scaffolds bear structural and topological similarities to MPEP and/or employed the MPEP/MTEP scaffolds as a basis for ligand design (Figure 1).3–10
Figure 1.

Reported mGluR5 non-competitive antagonists.
In an effort to make a dramatic departure from the MPEP chemotype, we conducted a functional high-throughput mGluR5 antagonist screen to identify novel, non-MPEP chemotypes. We screened a collection of 160,000 compounds and identified 624 mGluR5 antagonists in the primary screen (0.39% hit rate). Following hit verification and generation of full concentration-response-curves for all the primary hits, this effort produced 345 confirmed mGluR5 non-competitive antagonists. In this Letter, we describe the synthesis and SAR of three novel, non-MPEP mGluR5 non-competitive antagonists series 10, 11 and 12 identified from the functional HTS with submicromolar IC50s, low molecular weight and good clogP values (Figure 2).
Figure 2.

Novel, non-MPEP mGluR5 non-competitive antagonists 10, 11 and 12 identified from a functional HTS campaign.
Our attention first focused on lead 10, a furyl amide of a 2-azaspiro[5.5]undecane core. We employed an iterative parallel synthesis approach,13 and resynthesized 10 in the context of a 24-member library prepared by standard acylation (24 RCOCls) of commercial 2-azaspiro[5.5]undecane 13 to provide analogs 14, which were then purified to >98% by prep LCMS.14 As shown in Table 1, clear SAR was observed; however, upon resynthesis, lead 10 was a considerably weaker antagonist with an IC50 of 1.54 μM (Table 1). We have noted HTS DMSO stocks providing discrepancies with newly synthesized material on several occasions for various programs.15 While a thienyl analog 14a proved slightly more potent than 10, other aryl and heteroaryl congeners were far less potent or inactive. Cyclic alkyl moieties proved the most intriguing in this series, with the cyclohexyl congener 14g inactive, a cyclopentyl analog 14h weak (IC50 > 10 μM), a cyclobutyl variant 14i affording submicrolar inhibition (IC50 = 820 nM), and further contraction to a cyclopropyl derivative 14j provides inhibition comparable to cyclopentyl (IC50 > 10 μM). 14i was further evaluated and found to be selective for mGluR5 (>30 μM vs. mGluRs 1 (Group I), 2,3 (Group II) and 4,7,8 (Group III)) and displaced [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine with a Ki of 840 nM – a value in agreement with the IC50 (820 nM).
Table 1.
Structures and activities of analogs 14.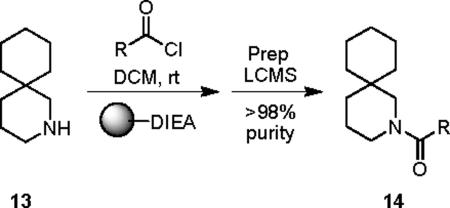
| Cmpd | R | mGluR5a IC50 (μM) | % Glu Maxb |
|---|---|---|---|
| 10 |
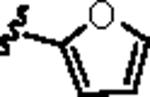
|
1.54 | 2.64 |
| 14a |
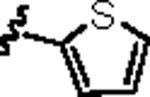
|
1.18 | 2.69 |
| 14b |
|
>30 | ND |
| 14c |
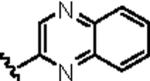
|
5.02 | 2.39 |
| 14d |
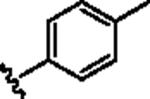
|
>10 | 56.2 |
| 14e |
|
5.24 | 4.23 |
| 14f |
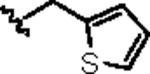
|
>10 | 52.1 |
| 14g |
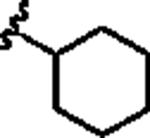
|
>30 | ND |
| 14h |
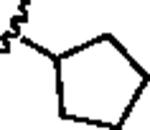
|
>10 | 26.1 |
| 14i |

|
0.82 | 1.12 |
| 14j |
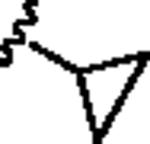
|
>10 | 40.6 |
IC50s are average of three determinations.
Determined at 30 μM test compound. ND, not determined.
Thus, 14i, possessing no aryl/heteroaryl features, represents a fundamentally new mGluR5 non-competitive antagonist chemotype that inhibits mGluR5 function by interaction with the MPEP allosteric binding site. Further libraries focused on other spirocyclic systems 15 as well as simple 3,3-dimethyl congeners 16 (Figure 3). Only analog 17 displayed activity (IC50 = 9.9 μM).
Figure 3.

Further analogs of novel mGluR5 antagonist 10/14.
Attention was then directed at lead 11, an adamantyl amide of 2-pyridinylpiperazine. Once again, we employed an iterative parallel synthesis approach,13 and resynthesized 11 in the context of a 12-member library prepared by standard acylation chemistry of 12 diverse aryl/heteroaryl piperazines 18 and adamantyl chloride 19 to deliver analogs 20. Upon resynthesis, lead 11 suffered a two-fold loss in potency (IC50 = 990 nM) relative to the HTS stock solution (IC50 = 414 nM). Solid SAR was noted for this series (Table 2). Moving the pyridine nitrogen from the 2-position (11) to the 3-position (20a), leads to a >10-fold loss in activity (IC50 > 10 μM), and the 4-pyridyl congener (20b) loses all mGluR5 inhibitory activity. A thiazole derivative 20c provided the most potent mGluR5 non-competitive antagonist in the series (IC50 = 540 nM). Functionalized aromatic analogs 20d–20f were generally weak to inactive with IC50s ranging from 2.3 μM to >10 μM. 20c was further evaluated and found to be selective for mGluR5 (>30 μM vs. mGluR 2,3 (Group II) and 4,7,8 (Group III) with modest activity at mGluR1 (IC50 = 2.3 μM)) and displaced [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine with a Ki of 440 nM – a value in accord with the IC50 (540 nM). Thus, 20c represents a fundamentally new mGluR5 non-competitive antagonist chemotype that inhibits mGluR5 function by interaction with the MPEP allosteric binding site.
Table 2.
Structures and activities of analogs 20.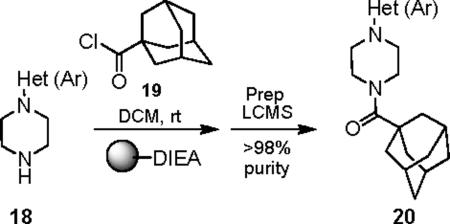
| Cmpd | R | mGluR5a IC50 (μM) | % Glu Maxb |
|---|---|---|---|
| 11 |
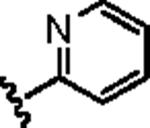
|
0.99 | 3.75 |
| 20a |
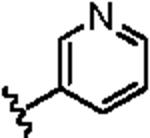
|
>10 | 30.2 |
| 20b |
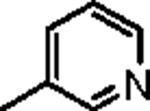
|
>30 | ND |
| 20c |
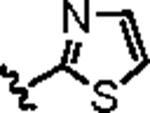
|
0.54 | 1.16 |
| 20d |
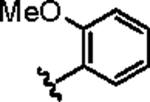
|
>10 | 52.0 |
| 20e |
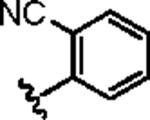
|
2.35 | 3.68 |
| 20f |
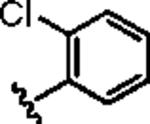
|
>30 | ND |
IC50s are average of three determinations.
Determined at 30 μM test compound. ND, not determined.
Further libraries focused on replacements for the adamantyl ring system 21–23, but only moderate micromolar antagonists were discovered (Figure 4).
Figure 4.

Further analogs of novel mGluR5 antagonist 11/20.
Finally, we initiated an optimization campaign on HTS lead 12, (3-amino-4,6-dimethylfuro[2,3-b]pyridine-2-yl)(4-fluorophenyl)methanone. We again employed an iterative parallel synthesis approach,13 and resynthesized 12 in the context of a multi-dimensional library prepared according to Scheme 1. Alkylation of a diverse collection of commercial 2-hydroxy-4-methylnicotinonitriles 24 with functionalized α-bromophenyl ketones provides a mixture of O- and N-alkylated products, where upon 25 is easily isolated by column chromatography. Exposure of 25 to K2CO3 in DMF at 100 °C under microwave irradiation delivers analogs 26 of HTS lead 12. Upon resynthesis, lead 12 (IC50 = 150 nM, 1.39 % Glu Max) was found to possess comparable potency to the HTS stock. 12 was further evaluated and found to be selective for mGluR5 (>30 μM vs. mGluRs 1 (Group I), 2,3 (Group II) and 4,7,8 (Group III)) and displaced [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine with a Ki of 410 nM – a value comparable to the IC50 (150 nM). Thus, 12 represents a fundamentally new mGluR5 non-competitive antagonist chemotype that inhibits mGluR5 function by interaction with the MPEP allosteric binding site. Unlike 10 and 11, SAR for this potent series of mGluR5 non-competitive antagonists was extremely shallow. Out of 36 analogs, only three analogs 27–29 possessed inhibitory activity, and three analogs displayed weak mGluR5 PAM activity 30–32 (Figure 5). This was the first example of this mode of pharmacology switch within a non-MPEP scaffold, and 30–32 represent another novel mGluR5 PAM scaffold.16–21 Thus, a functional HTS approach, coupled with iterative parallel synthesis, identified and developed three novel series of potent and selective mGluR5 non-competitive antagonists represented by 12, 14i and 20c that bind at the MPEP allosteric site, but share little or no structural or topological similarities to MPEP (Figure 6).
Scheme 1.
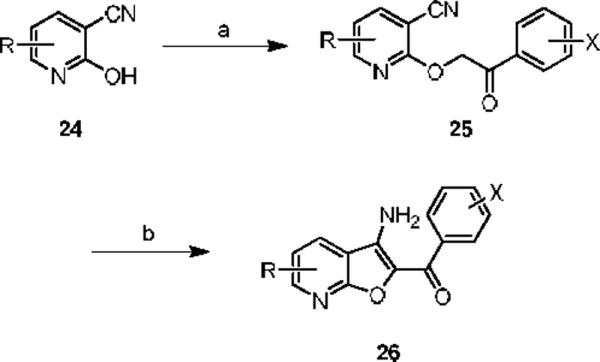
Reagents and conditions: (a) (i) NaH, DMF, (ii) 2-bromobenzophenones, (b) K2CO3, DMF, 100 °C, mw, 20 min, 18–56%.
Figure 5.

mGluR5 non-competitive antagonists 27–29 and mGluR5 PAMs 30–32 identified by optimization of HTS lead 12.
Figure 6.

Novel, non-MPEP mGluR5 non-competitive antagonists 12, 14i and 20c that bind at the MPEP allosteric site, but have little or no structural and topological similarity to MPEP.
In summary, we have identified three novel, non-MPEP series of selective non-competitive mGluR5 antagonists with IC50s ranging from 150 nM to 820 nM for the most potent ligands. These novel mGluR5 ligands bear little or no structural or topological similarity to MPEP and represent fundamentally new mGluR5 antagonist chemotypes. Within series 12, chemical optimization was able to provide both a potent mGluR5 antagonist 12 (IC50 = 150 nM) and 30–32, weak mGluR5 PAMs (EC50s of 6.1 to 7.6 μM). This represents the first example of switching modes of pharmacology in a non-MPEP series of mGluR5 ligands. Further studies in this arena are in progress and will be reported in due course.
Acknowledgement
The authors thank NIDA (DA023947-01) and Seaside Therapeutics (VUMC33842) for support of our programs in the development of mGluR5 non-competitive antagonists and partial antagonists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1).a) Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]; b) Conn PJ, Pin J-P. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2).a) Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]; b) Lea PM, IV, Faden AI. CNS Drug Reviews. 2006;12(2):149–166. doi: 10.1111/j.1527-3458.2006.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Alagille D, Baldwin RM, Roth BL, Wroblewski JT, Grajkowska E, Tamagnan GD. Bioorg. Med. Chem. Lett. 2005;15:945–949. doi: 10.1016/j.bmcl.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 4).Roppe JR, Wang B, Huang D, Tehrani L, Kamenecka T, Schweiger EJ, Anderson JJ, Brodkin J, Jiang X, Cramer M, Chung J, Reyes-Manalo G, Munoz B, Cosford NDP. Bioorg. Med. Chem. Lett. 2004;14:3993–3996. doi: 10.1016/j.bmcl.2004.05.037. [DOI] [PubMed] [Google Scholar]
- 5).Newman AH, Kulkarni SS. Bioorg. Med. Chem. Lett. 2007;17:2987–2991. doi: 10.1016/j.bmcl.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Galatis P, Yamagata K, Wendt JA, Connolly CJ, Mickelson JW, Milbank JBJ, Bove SE, Knauer CS, Brooker RM, Augelli-Szafran CE, Schwartz RD, Kinsora JJ, Kilgore KS. Bioorg. Med. Chem. Lett. 2007;17:6525–6528. doi: 10.1016/j.bmcl.2007.09.083. [DOI] [PubMed] [Google Scholar]
- 7).Newman AH, Kulkarni SS. Bioorg. Med. Chem. Lett. 2007;17:2074–2079. doi: 10.1016/j.bmcl.2006.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Eastman B, Chen C, Smith ND, Poon S, Chung J, Reyes-Manalo G, Cosford NDP, Munoz B. Bioorg. Med. Chem. Lett. 2004;14:5485–5488. doi: 10.1016/j.bmcl.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 9).Bach P, Nilsson K, Svensson T, Baur U, Hammerland LG, Peterson A, Wallberg A, Osterland K, Karis D, Boije M, Wensbo D. Bioorg. Med. Chem. Lett. 2006;16:4788–4791. doi: 10.1016/j.bmcl.2006.06.078. [DOI] [PubMed] [Google Scholar]
- 10).Micheli F, Bertani B, Bozzoli A, Crippa L, Cavanni P, Fabio RD, Donati D, Marzorati P, Merlo G, Paio A, Pergunni L, Zarantonello P. Bioorg. Med. Chem. Lett. 2008;18:1804–1809. doi: 10.1016/j.bmcl.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 11).Hammerland LG, Johansson M, Mattson JP, Minidis ABE, Nilsson K, Peterson A, Wensbo D, Wallberg A, Osterlund K. Bioorg. Med. Chem. Lett. 2006;16:2467–2469. doi: 10.1016/j.bmcl.2006.01.100. [DOI] [PubMed] [Google Scholar]
- 12).Porter RHP, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters J-U, Prinseen E, Wichmann J, Vieira E, Muhlemann A, Gatti S, Mutel V, Malherbe P. J. Pharm. Exp. Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 13).Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J.Comb. Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 14).Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J. Comb.Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 15).Lewis LM, Sheffler D, Williams R, Bridges TA, Kennedy JP, Brogan JT, Mulder MJ, Williams L, Nalywajko NT, Niswender C, Weaver CD, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:885–891. doi: 10.1016/j.bmcl.2007.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).O'Brien JA, Lemaire W, Chen T-B, Chang RSL, Jacobson MA, Ha SN, Lindsley CW, Sur C, Pettibone DJ, Conn J, Wiliams DL. Mol. Pharmacol. 2003;64(3):731–738. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 17).Sharma S, Rodriguez A, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Williams DL, Jr., Lindsley CW. Curr. Topics in Med. Chem. 2005;5:825–836. doi: 10.2174/1568026054750290. [DOI] [PubMed] [Google Scholar]
- 19).Zhao Z, Wisnoski DD, O'Brien JA, Lemiare W, Williams DL, Jr., Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg. Med. Chem. Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 20).Lindsley CW, Wisnoski DD, Leister WH, O'Brien JA, Lemiare W, Williams DL, Jr., Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. J. Med. Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 21).a) Le Poul E, Bessis AS, Lutgens R, Bonnet B, Rocher JP, Epping Jordan M, Mutel V. 5th International Metabotropic Glutamate Receptors Meeting; Taormina, Italy: 2005. [Google Scholar]; b) Epping-Jordan M, Le Poul M, Rocher JP. Innov. Pharm. Technol. 2007;24:24–26. [Google Scholar]; c) Bessis A-S, Bonnet B, Le Poul E, Rocher J-P, Epping-Jordan M. WO 044797. 2005 [Google Scholar]; d) Bugada P, Gagliardi S, Le Poul E, Mutel V, Palombi G, Rocher J-P. WO 6123249. 2006 [Google Scholar]; e) Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Wantuch C, Popiolek M, Day M, Khawaja X, Smith D, Olsen M, Kouranova E, Gilbert A, Lai M, Pausch MH, Pruthi F, Pulcicchio C, Brandon NJ, Comery TA, Beyer CE, Logue S, Rosenzweig-Lipson S, Marquis KL. J. Pharmacol. Exp. Therapeut. 2008;327:827–839. doi: 10.1124/jpet.108.136580. [DOI] [PubMed] [Google Scholar]; f) Engers DW, Rodriguez AL, Oluwatola O, Hammond AS, Venable DF, Williams R, Sulikowski GA, Conn PJ, Lindsley CW. Chem Med Chem. doi: 10.1002/cmdc.200800357. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]