

Abstract
Objective
Pulmonary surfactant protein B (SP-B), an alveolar protein normally detectable at only very low concentrations in blood, circulates at higher levels among smokers and those with alveolar injury and inflammation. We hypothesized that SP-B may serve as a marker of the vascular effects of smoking, and would thus associate with subclinical measures of atherosclerosis.
Methods and Results
Plasma levels of SP-B were measured in 3,294 subjects, ages 30-65, enrolled in the Dallas Heart Study, a probability-based population sample of Dallas County adults. Coronary artery calcium (CAC) was measured by computed tomography and abdominal aortic plaque (AP) by magnetic resonance imaging. The cohort comprised 29% current and 17% former smokers. The overall prevalence of CAC was 22% and AP 39%. Median SP-B levels were 5-fold higher among current vs. never smokers (P<0.0001) and significantly correlated with estimated pack-years smoked (Spearman rho=0.35, P<0.0001). Increasing levels of SP-B also associated with other traditional cardiac risk factors and higher levels of inflammatory biomarkers. In univariable analyses, increasing SP-B quartiles associated with higher prevalence of both CAC and AP (Ptrend <0.0001 for each). In multivariable analyses adjusting for traditional cardiovascular risk factors, SP-B remained associated with AP (OR 1.87 for the 4th vs. 1st quartile, 95% CI 1.39 to 2.51; P<0.0001) but not CAC. An interaction was observed between SP-B, smoking status, and AP (Pinteraction=0.01), such that SP-B associated with AP in current smokers (adjusted OR 2.15 for the 4th vs. 1st quartile, 95% CI 1.26 to 3.67; P=0.005), but not in former or never smokers.
Conclusions
Circulating levels of SP-B increase with greater smoking burden and independently associate with abdominal aortic plaque among current smokers. Our findings support further investigation of the role of SP-B as a marker of the vascular effects of smoking.
Tobacco smoking promotes atherosclerosis and its complications via direct effects on the vasculature, as well as alterations in local inflammation, platelet function, and antithrombotic, prothrombotic, and fibrinolytic factors.1 While a causal role of smoking in atherosclerosis has been conclusively established, evidence to support a dose-response effect has been varied. While several epidemiologic studies have shown dose-dependent associations between tobacco smoke exposure and cardiovascular events such as myocardial infarction, stroke, and cardiovascular death, these effects attenuate quickly after smoking cessation,2 in contrast to the cumulative effects of smoking on other endpoints such as cancer. Moreover, studies evaluating dose-dependent effects on subclinical atherosclerosis phenotypes have been conflicting, with some studies suggesting a non-linear effect,3 and others suggesting a differential effect in peripheral vascular beds versus the coronary circulation.4-6 Several inflammatory biomarkers are increased in active smokers, and recent studies suggest a possible interaction between smoking, circulating levels of these inflammatory markers, and atherosclerosis phenotypes.7 However, evaluation of a more direct serological measure linking smoking-related toxicity and vascular disease in the general population may provide a means to assess individual risk for smoking-related vascular complications.
Pulmonary surfactant protein B (SP-B) is crucial to lung function.8 It is the oldest member of a family of proteins called saposins, which has been shown to have a significant impact on phospholipid organization.9 The primary role of SP-B is to maintain reduced surface tension, and to enhance the rate, adsorption, and spread of phospholipids during the respiratory cycle, thereby promoting formation and stability of the surfactant monolayer.10 SP-B is normally detectable in the systemic circulation, but only at very low levels.11 However, leakage of surfactant proteins into the circulation has been reported in several diseases that result in damage to the alveolar-capillary membrane, including acute respiratory distress syndrome (ARDS) and chronic heart failure.11, 12 Increased levels of SP-B in the circulation have also been reported in tobacco-smoke exposure due to alveolar inflammation and increased lung permeability.13 Given the reported associations of smoking exposure with higher plasma levels of SP-B, we hypothesized that SP-B may serve as a quantitative marker reflecting smoking exposure and its associated vascular risk. To evaluate this hypothesis, we explored the associations and interactions between smoking status, circulating SP-B and several atherosclerotic phenotypes in the Dallas Heart Study, a large multi-ethnic probability-based population cohort.
METHODS
Study Population
The Dallas Heart Study (DHS) is a population-based, probability sample of 6,101 Dallas County residents.14 Following an initial in-home visit for collection of survey data, body mass index and measurement of blood pressure, participants between the ages of 30 and 65 were invited to participate in a second visit where they provided in-home fasting blood and urine specimens. Those completing visit two were invited to a third visit at UT Southwestern Medical Center, where imaging studies including cardiac magnetic resonance imaging and electron beam computed tomography were performed. Demographics, blood pressure and body composition were similar between subjects completing visits 1 and 2, and laboratory data did not significantly differ between those completing visits 2 and 3.14 The study population for the present analyses includes 3,294 DHS subjects, ages 30-65, who underwent measurement of plasma levels of SP-B at Visit 2. A subset of the population underwent electron beam computed tomography (EBCT) to measure coronary artery calcium (CAC; n = 2519), and magnetic resonance imaging to measure aortic plaque prevalence (AP; n=2305).
Definition of Variables
Hypertension was defined as an average (5 repeated measurements) systolic blood pressure ≥140 mm Hg; or diastolic blood pressure ≥90 mm Hg; or use of anti-hypertensive medication. Diabetes was defined as a fasting blood glucose level of ≥126mg/dL; or a non-fasting blood glucose level of ≥200mg/dL; or prevalent medically treated diabetes. Hypercholesterolemia was defined as a fasting low-density lipoprotein (LDL) cholesterol ≥160mg/dL; a total cholesterol ≥240mg/dL; or use of a statin medication. Hypertriglyceridemia was defined as a fasting triglyceride concentration ≥200mg/dL. Low high-density lipoprotein (HDL) cholesterol was defined as HDL-C <40mg/dL in men and <50mg/dL in women. Race was self-reported.
Smoking was determined by self-report and categorized in several ways. Current smokers were defined as those who had smoked greater than 100 cigarettes in a lifetime and continued to smoke. Among current smokers, frequency of smoking was subcategorized as daily smoking, defined as smoking every day in the past 30 days, or intermittent smoking, defined as smoking some days in the past 30 days. Former smokers were those who had smoked greater than 100 cigarettes in a lifetime, but did not currently smoke. Never smokers were those who had smoked less than 100 cigarettes in a lifetime. Cumulative smoking exposure was quantified by pack-years among current smokers. Tobacco exposure was also evaluated for smoking cigars and pipes, or chewing tobacco.
Measurement of SP-B and Other Biomarkers
Venous blood was collected in standard blood collection tubes containing citrate EDTA and samples were maintained at 4°C for ≤4 hours and then centrifuged (1,430g for 15 minutes) at 4°C. Plasma was then removed, aliquoated, and frozen at -80°C until assays were performed. SP-B was measured from thawed frozen plasma at Alere, Inc. (San Diego, CA) using a proprietary sandwich platform with minimum and maximum detection limits of 1ng/mL and 950ng/mL, respectively. The intra-assay coefficient of variation (CV) was 10% and inter-assay CV was 11%. Assays were performed by individuals blinded to all clinical data. Samples had been thawed once for aliquoting prior to biomarker measurement. Tumor necrosis factor-alpha 1 receptor (TNFR1A), myeloperoxidase (MPO) and matrix metalloproteinase-9 (MMP-9) were measured on a similar platform: minimum and maximum detection limits for TNFR1A were 0.094 and 100 ng/mL, for MPO were 3.7 and 250 ng/mL, and for MMP-9 were 1.0 and 730 ng/mL; intra-assay and inter-assay CVs for TNFR1A were 9% and 10%, for MPO were 6% and 7%, and for MMP-9 were 5% and 7%.
The following analytes were measured previously and the methods have been described: high-sensitivity C-reactive protein (hsCRP);15 N-terminal pro-B-type natriuretic peptide (NT-proBNP);16 monocyte chemoattractant protein (MCP-1);17 interleukin-18;18 osteoprotegerin;19 cystatin C;20 and soluble receptor for advanced glycation end products (sRAGE).21 The homeostasis model assessment of insulin resistance index (HOMA-IR) was calculated as described previously [fasting insulin (μIU/mL) X fasting glucose (mmol/L)/22.5].22
Atherosclerosis Assessment
EBCT scans were performed in duplicate to measure coronary artery calcium (CAC). The scans were scored using the Agatston method and the results of the two scans were averaged. Prevalent CAC was defined as >10 Agatston units, a data-derived threshold selected to maximize the signal to noise ratio, as previously described.23 To determine AP, six transverse slices of the infrarenal abdominal aorta were obtained using a 1.5 Tesla whole-body MRI system (Intera, Philips Medical Systems). Investigators blinded to all subject data used the Magnetic Resonance Analytic Software Systems (MASS) cardiac analysis software package (Version 4.2 beta, Medis Medical Imaging Systems Inc) to analyze the images, using a previously published definition of AP as a hyperintense signal volume that protruded ≥1mm from the endoluminal surface of the aortic wall.24
Death Events
All-cause and cardiovascular (CV) mortality were ascertained using National Death Index data through December 31, 2007 (median follow-up time 6.3 [5.9, 6.8] years). CV death was defined using ICD10 codes I00-I99.25 Death status for all participants analyzed was ascertained to the end of 2007 with no loss of follow-up.
Statistical Analysis
Demographic and clinic variables were compared across quartiles of SP-B levels using the χ2 trend test for categorical variables and the Jonckheere-Terpstra test for continuous variables. Correlations between selected biomarkers and SP-B were evaluated by Spearman rank correlation coefficients. Logistic regression was performed to assess associations between SP-B and prevalent CAC and AP in unadjusted models, as well as models adjusting for age, sex, race, diabetes, hypertension, hypertriglyceridemia, hypercholesterolemia, low HDL, BMI, and smoking exposure. Smoking exposure was entered into models as both a categorical variable (current, former, never) and as a continuous measure of pack-years smoked (never/former = 0 pack-years). Additional adjustments were made for cigar, pipe, and snuff tobacco exposure in sensitivity analyses. Due to the highly skewed distribution of SP-B, it was evaluated as both a categorical variable (in quartiles) and as a log transformed continuous variable. Cox proportional hazards models were used to assess the associations between SP-B and all-cause and CV death, with adjustment for the same covariates as described above. Proportional hazards assumptions were met for all Cox models. Tests for statistical interactions were performed between smoking (current, former, never), SP-B, and all atherosclerosis phenotypes and death endpoints. For all interaction tests with P<0.1, stratified analyses were performed in subgroups defined by smoking status. All two-sided P-values ≤ 0.05 were considered statistically significant, and no adjustments were made for multiple testing. All analyses were performed using SAS 9.2 (Cary, NC, USA) and all box-plot figures using GraphPad PRISM 5.01 (La Jolla, CA, USA).
RESULTS
Associations of SP-B with Atherosclerosis Risk Factors and Biomarkers
The overall median [IQR] level of SP-B was 2.67 ng/mL [1.00, 7.57]. Increasing quartiles of SP-B associated with male sex, older age, black race, hypertension, metabolic syndrome, and hypercholesterolemia (Table 1). SP-B did not associate with HDL cholesterol or diabetes. The strongest correlations between SP-B and the continuous variables tested include TNFR1A, MPO, age, MCP-1, cystatin C, osteoprotegerin, interleukin-18, and hsCRP (Table 2). SP-B did not correlate strongly with measures of adiposity (BMI) or insulin resistance (HOMA-IR) (Table 1).
Table 1.
Clinical and Biological Variables by Quartile of Pulmonary Surfactant Protein B
| Variable | Q1 (n=873) | Q2 (n=773) | Q3 (n=825) | Q4 (n=823) | p trend |
|---|---|---|---|---|---|
| SP-B, ng/mL | <1.0 | 1.0 – 2.6 | 2.67 – 7.57 | 7.63 – 829.72 | |
| Age, years | 39 [33, 47] | 43 [36, 51] | 45 [37, 54] | 47 [40, 54] | <0.0001 |
| Men | 341 (39%) | 331 (43%) | 402 (49%) | 381 (46%) | 0.000 |
| Black | 345 (40%) | 362 (47%) | 464 (56%) | 525 (64%) | <0.0001 |
| White | 294 (34%) | 237 (31%) | 251 (30%) | 192 (23%) | <0.0001 |
| Hispanic | 209 (24%) | 148 (19%) | 99 (12%) | 98 (12%) | <0.0001 |
| Hypertension | 212 (25%) | 212 (28%) | 323 (40%) | 355 (44%) | <0.0001 |
| Diabetes | 81 (9%) | 100 (13%) | 104 (13%) | 101 (12%) | 0.072 |
| Metabolic Syndrome | 225 (29%) | 255 (33%) | 293 (36%) | 300 (36%) | 0.001 |
| Hypercholesterolemia | 80 (9%) | 95 (12%) | 125 (15%) | 131 (16%) | <0.0001 |
| Low HDL | 356 (41%) | 316 (41%) | 336 (41%) | 359 (44%) | 0.273 |
| Current Smokers | 94 (11%) | 117 (15%) | 264 (32%) | 486 (59%) | <0.0001 |
| Former Smokers | 137 (16%) | 153 (20%) | 172 (21%) | 94 (12%) | 0.056 |
| Never Smokers | 641 (73%) | 502 (65%) | 389 (47%) | 240 (29%) | <0.0001 |
| Pack-Years (current smokers) | 2.9 [0.7, 8.0] | 6.9 [1.7, 19.8] | 13.1 [5.3, 24.0] | 17.5 [8.0, 29.0] | <0.0001 |
SP-B=Pulmonary Surfactant Protein B
Levels of continuous variables are reported as median [IQR] and categorical variables as percentages.
Table 2.
Spearman Rank Correlations of Biomarkers with Pulmonary Surfactant Protein B in Smokers and Non-Smokers
| Smokers | Non-Smokers | |||
|---|---|---|---|---|
| Variable | Coefficient | p-value | Coefficient | p-value |
| TNFR1A | 0.51 | <0.0001 | 0.60 | <0.0001 |
| MPO | 0.45 | <0.0001 | 0.41 | <0.0001 |
| Age | 0.24 | <0.0001 | 0.27 | <0.0001 |
| MCP-1 | 0.19 | <0.0001 | 0.24 | <0.0001 |
| CRP | 0.15 | <0.0001 | 0.10 | <0.0001 |
| Cystatin C | 0.13 | 0.0001 | 0.21 | <0.0001 |
| Osteoprotegerin | 0.12 | 0.0002 | 0.14 | <0.0001 |
| IL-18 | 0.09 | 0.04 | 0.08 | 0.001 |
| NT-proBNP | 0.09 | 0.009 | 0.09 | <0.0001 |
| Body Mass Index | -0.08 | 0.02 | 0.05 | 0.03 |
| eGFR | -0.07 | 0.05 | -0.13 | <0.0001 |
| HOMA-IR | -0.07 | 0.06 | 0.06 | 0.005 |
| LDL-C | 0.06 | 0.05 | 0.07 | 0.002 |
| HDL-C | -0.04 | 0.25 | -0.03 | 0.23 |
| MMP-9 | -0.02 | 0.65 | -0.08 | 0.000 |
| sRAGE | -0.02 | 0.46 | -0.03 | 0.19 |
| Pack-Years | 0.35 | <0.0001 | ||
TNFR1A=Tumor necrosis factor receptor 1 alpha; MPO=myeloperoxidase; IL-18=interleukin-18; CRP=C-reactive protein; eGFR=estimated glomerular filtration rate; sRAGE=soluble receptor for advanced glycation end products; MMP-9=matrix metalloproteinase-9; MCP-1=monocyte chemoattractant protein-1; HOMA-IR=homeostasis model assessment of insulin resistance index
Associations of SP-B and Tobacco Exposure
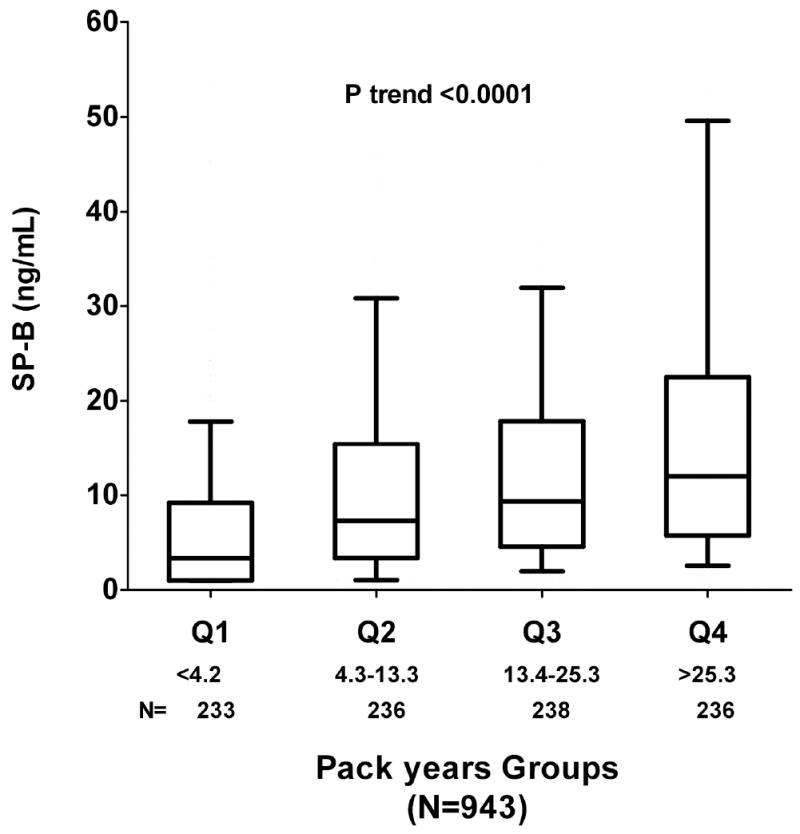
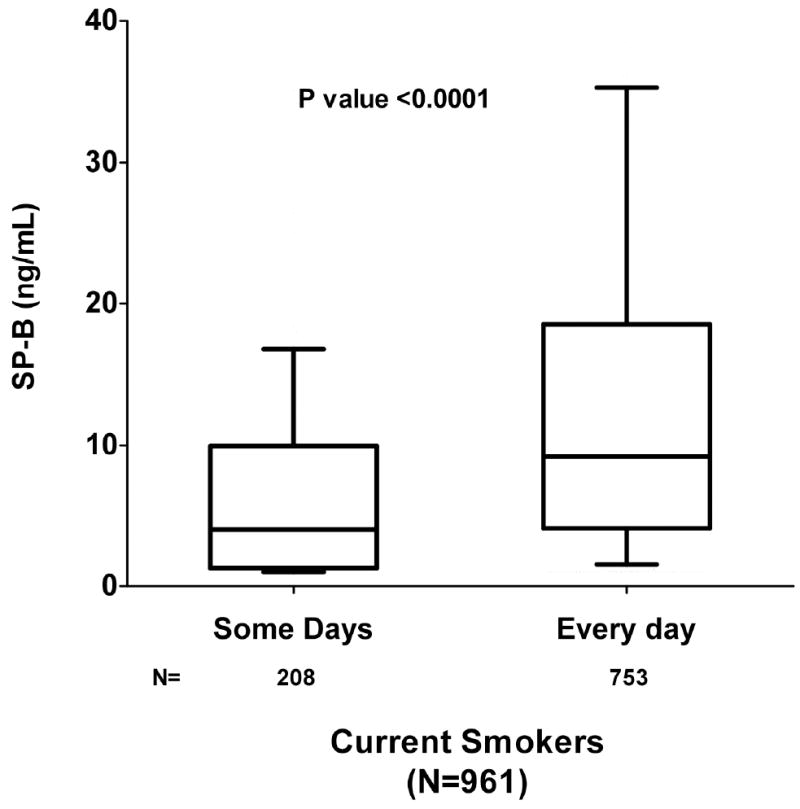
Median SP-B levels were markedly higher among current vs. never smokers (7.83 ng/mL vs. 1.64 ng/mL; P<0.0001) and significantly correlated with estimated pack-years smoked (Spearman rho=0.35, P<0.0001). SP-B levels were highest among current smokers, intermediate among former smokers, and lowest among never smokers (Ptrend <0.001; Figure 1a); among current smokers, SP-B exhibited a graded association with increasing tobacco exposure as measured by pack-years smoked (Ptrend <0.0001; Figure 1b). Moreover, among current smokers, higher levels of SP-B were found in subjects who smoked everyday in the past 30 days, as compared with those who only smoked intermittently over the same time period (P<0.0001; Figure 1c). Higher levels of SP-B were found in subjects who smoked cigars or pipes than those who did not smoke tobacco products (Table 3). In contrast, no difference in SP-B levels was found between subjects who chewed tobacco vs. those who did not (Table 3).
Figure 1.

a: Pulmonary Surfactant Protein B in Current, Former, Never Smokers Levels of pulmonary surfactant protein B (SP-B) across never, former, and current smokers. Lower and upper borders of box indicate 10th and 90th percentile, respectively. Horizontal bar indicates median, and whiskers 5th and 95th percentiles. Median SP-B levels were markedly higher among current vs. never smokers (7.83ng/mL vs. 1.64ng/mL; P<0.0001).
b: Pulmonary Surfactant Protein B in Current Smokers by Pack Years Levels of pulmonary surfactant protein B (SP-B) in current smokers by quartiles of pack-years smoked. Lower and upper borders of box indicate 10th and 90th percentile, respectively. Horizontal bar indicates median, and whiskers 5th and 95th percentiles.
c: Pulmonary Surfactant Protein B in Current Smokers by Smoking Frequency Levels of Pulmonary surfactant protein B (SP-B) in current smokers who smoked intermittently or everyday in the past 30 days. Lower and upper borders of box indicate 10th and 90th percentile, respectively. Horizontal bar indicates median, and whiskers 5th and 95th percentiles.
Table 3.
Pulmonary Surfactant Protein B and Other Tobacco Use
| Current Use | No Current Use | P-value | |
|---|---|---|---|
| Cigars (n=118/3208) | 5.95 [2.07, 13.29] | 2.57 [1, 7.31] | <0.0001 |
| Pipes (n=13/3208) | 8.15 [4.86, 10.42] | 2.62 [1, 7.51] | 0.02 |
| Snuff (n=55/3208) | 2.38 [1.00, 6.40] | 2.65 [1, 7.55] | 0.39 |
Levels of SP-B reported as median [25th,75th percentile] by current tobacco use status.
Associations of SP-B with Atherosclerosis
The overall prevalence of CAC was 22% and AP 39%. CAC prevalence (Ptrend<0.0001) and AP prevalence (Ptrend<0.0001) increased across higher quartiles of SP-B. Similarly, when SP-B was analyzed as a log transformed continuous variable, univariable associations were observed with CAC and AP (P<0.0001 for each; Table 4). In multivariable models adjusting for traditional atherosclerosis risk factors, former and current smoking, and pack-years tobacco exposure, a graded association remained between higher SP-B quartiles and AP (OR 1.87 for the 4th vs. 1st quartile, 95% CI 1.39 to 2.51; P<0.0001Table 4). Similar results were observed when log SP-B was entered into the model as a continuous variable (P<0.0001; Table 4). These findings were not changed by further adjustment for cigar and pipe smoking. In contrast, after multivariable adjustment, associations of SP-B with CAC were attenuated and no longer significant (Table 4).
Table 4.
Overall Association of Pulmonary Surfactant Protein B with Atherosclerotic Phenotypes and Mortality
| Unadjusted | Adjusted | |||
|---|---|---|---|---|
| OR [95% CI] | P-value | OR [95% CI] | P-value | |
| CAC > 10 | N = 2485 | |||
| log SP-B* | 1.54 [1.41-1.69] | <0.0001 | 1.10 [0.97-1.25] | 0.15 |
| Q1 | 1.0 (referent) | 1.0 (referent) | ||
| Q2 | 1.62 [1.18-2.24] | 0.003 | 1.13 [0.78-1.65] | 0.52 |
| Q3 | 2.57 [1.90-3.46] | <0.0001 | 1.13 [0.78-1.62] | 0.52 |
| Q4 | 3.75 [2.80-5.03] | <0.0001 | 1.33 [0.91-1.94] | 0.14 |
| Former smoking | 1.27 [0.94-1.73] | 0.13 | ||
| Current Smoking | 2.63 [1.81-3.83] | <0.0001 | ||
| Pack-years* | 1.04 [0.93-1.16] | 0.54 | ||
| AP | N = 2296 | |||
| log SP-B* | 1.60 [1.47-1.75] | <0.0001 | 1.24 [1.11-1.38] | <0.0001 |
| Q1 | 1.0 (referent) | 1.0 (referent) | ||
| Q2 | 1.70 [1.31-2.19] | <0.0001 | 1.32 [1.00-1.74] | 0.05 |
| Q3 | 2.39 [1.87-3.06] | <0.0001 | 1.44 [1.09-1.89] | 0.01 |
| Q4 | 3.84 [3.01-4.92] | <0.0001 | 1.87 [1.39-2.51] | <0.0001 |
| Former Smoking | 1.28 [0.99-1.66] | 0.06 | ||
| Current Smoking | 1.36 [1.00-1.84] | 0.05 | ||
| Pack-years* | 1.21 [1.08-1.37] | 0.002 | ||
| HR [95% CI] | P-value | HR [95% CI] | P-value | |
| Overall Death | N = 2788 | |||
| log SP-B* | 1.57 [1.35-1.83] | <0.0001 | 1.20 [1.00-1.46] | 0.05 |
| Q1 | 1.0 (referent) | 1.0 (referent) | ||
| Q2 | 1.30 [0.62-2.73] | 0.49 | 0.94 [0.45-1.99] | 0.87 |
| Q3 | 3.57 [1.92-6.63] | <0.0001 | 1.70 [0.90-3.25] | 0.10 |
| Q4 | 4.60 [2.51-8.44] | <0.0001 | 1.85 [0.95-3.59] | 0.07 |
| Former Smoking | 1.84 [1.12-3.03[ | 0.02 | ||
| Current Smoking | 2.38 [1.37-4.14] | 0.002 | ||
| Pack-years* | 0.96 [0.82-1.12] | 0.60 | ||
| CV Death | N = 2788 | |||
| log SP-B* | 1.46 [1.15-1.85] | 0.002 | 1.16 [0.86-1.56] | 0.32 |
| Q1 | 1.0 (referent) | 1.0 (referent) | ||
| Q2 | 1.44 [0.54-3.87] | 0.47 | 1.08 [0.40-2.94] | 0.87 |
| Q3 | 2.61 [1.08-6.28] | 0.03 | 1.32 [0.53-3.31] | 0.56 |
| Q4 | 2.97 [1.25-7.07] | 0.01 | 1.30 [0.50-3.38] | 0.59 |
| Former Smoking | 1.17 [0.55-2.49] | 0.69 | ||
| Current Smoking | 1.49 [0.63-3.54] | 0.36 | ||
| Pack-years* | 0.98 [0.74-1.30] | 0.88 | ||
CAC=coronary artery calcium; AP=aortic plaque; SP-B=pulmonary surfactant protein B
Adjusted models include age, sex, ethnicity, hypertension, diabetes, hypercholesterolemia, low HDL-C, hypertriglyceridemia, BMI, smoking status (current, former, never smoker), and pack-years smoked.
OR per 1 SD increment in SP-B
Odds ratio for CAC and AP derived from logistic regression models. Hazard ratios for all-cause death (n=125) and CV death (n=52) derived from Cox-proportional hazards models.
Current smoking was significantly associated with both CAC and AP in unadjusted and adjusted analyses (Table 4). Number of pack-years smoked was significantly associated with AP but not CAC in analyses adjusted for smoking status, SP-B levels, and traditional risk factors. (Table 4).
Interaction between SP-B, Smoking, and Aortic Plaque
A significant interaction was observed between SP-B quartile, smoking status, and AP (Pinteraction=0.01). In subgroups defined as current, former or never smokers, SP-B was associated with AP after adjustment for traditional risk factors and pack-years in current smokers (adjusted OR 2.15 in 4th quartile, 95% CI 1.26 to 3.67; P=0.005) (Figure 2). In contrast, no significant association was seen between SP-B and AP in former or never smokers (Figure 2). Among current smokers, levels of SP-B were significantly higher among those with compared to without prevalent AP (10.9 ng/mL [4.5, 19.1] vs. 6.2 ng/mL [2.2, 12.9]; p<0.0001), a difference that persisted after adjustment for self-reported pack year exposure (p<0.0001). Adjustment for inflammatory markers TNFR1A, hsCRP, cystatin C, and interleukin-18 separately did not significantly attenuate the interaction between SP-B, smoking status, and AP (Pinteraction≤0.03 for SP-B quartile x smoking with additional inflammatory markers).
Figure 2. Pulmonary Surfactant Protein B with Prevalent Aortic Plaque, Stratified by Smoking Status.

Association of pulmonary surfactant protein B (SP-B) with prevalent abdominal aortic plaque (AP) after adjustment for traditional risk factors in current, former, and never smokers (total n=2296).
Associations of SP-B with All-Cause and CVD Mortality
In univariable analysis, SP-B was significantly associated with overall mortality and CVD mortality (Table 4). This association was attenuated for both overall and CVD mortality when adjusted for traditional risk factors (Table 4). There was a significant interaction between log SP-B and smoking for overall mortality (P=0.05), but stratified analyses by smoking status did not reveal any significant associations of SP-B with mortality among smokers in adjusted models (data not shown). Although current smoking and former smoking were significantly associated with overall mortality adjusted for traditional risk factors, there was no association between pack-years smoked and overall mortality in unadjusted or adjusted analyses (Table 4).
DISCUSSION
In a large, multi-ethnic probability-based population study, we report novel associations between circulating levels of a pulmonary surfactant protein, SP-B, and cardiovascular risk factors, inflammatory biomarkers, and atherosclerotic phenotypes. We found that increasing levels of SP-B correlated remarkably with multiple measures of self-reported smoking exposure, including current smoking status, cumulative smoking burden, and frequency of smoking. Moreover, higher levels of SP-B were found in those who smoked cigars or pipes, but not in those who chewed tobacco, further supporting the relationship between tobacco inhalation and higher circulating levels of SP-B.
Circulating plasma levels of SP-B associated with both prevalent coronary and peripheral atherosclerosis phenotypes, but after adjustment for smoking exposure and other risk factors, an independent association was observed only for abdominal aortic atherosclerosis. Moreover, this association was modified by smoking status, such that SP-B was independently associated with abdominal aortic plaque among current smokers but not among former or never smokers. This observation is concordant with the clinical observation of a stronger association of smoking with peripheral arterial disease than coronary artery disease.4 We found that compared with smokers without aortic plaque, those with aortic plaque had higher SP-B levels even after adjusting for self-reported pack-year exposure, supporting the concept that biomarkers of alveolar toxicity may help to quantify smoking dose-exposure and help to explain variability in the adverse vascular effects of smoking.
The associations of smoking dose-exposure with atherosclerosis phenotypes and mortality were mirrored consistently by the associations of SP-B levels with these endpoints. For example, current smoking independently associated with both coronary and aortic atherosclerosis in our study sample; however, among current smokers, a dose-response association with pack-years smoked was seen only with aortic atherosclerosis, consistent with prior reports.4-6, 26-28 In a parallel fashion, SP-B was independently associated with aortic but not coronary atherosclerosis. In contrast, while current smoking independently associated with all-cause mortality, pack-years smoked did not, mirroring the absence of an association of SP-B with all-cause mortality.
It is interesting to note that considerable variability in SP-B levels was observed among individuals reporting similar tobacco exposures. This variability may have resulted from inaccurate self-reporting, differences in the type of cigarettes smoked, the mode of smoking, degree of inhalation, and “downstream” factors such as the ability of the lungs to clear toxins and defend against injury to the alveolar-capillary membrane. Importantly, renal clearance does not appear to play a role in circulating SP-B levels, consistent with prior reports.29
Biology of SP-B
Mature SP-B is a hydrophobic 79-amino acid peptide of low molecular mass (~18 kD) that associates as a thiol-dependent homo-dimer, and so far has only been detected in the lumen of the alveolus, in alveolar type II cells, and in alveolar macrophages.10 SP-B is normally associated with complexes of surfactant phospholipids, likely making it too large to breach the alveolar-capillary membrane.30 However, labeling studies of alveolar type II cells have shown that some of the protein is secreted into the alveolus as a hydrophilic, monomeric proprotein and processing intermediate with molecular weights of ~45kD and ~25kD, respectively.31 Since these forms are considerably smaller, they can more readily breach the alveolar-capillary membrane.
The route by which proteins leak through the alveolar-capillary membrane into the circulation is not completely understood. Doyle and coworkers measured levels of surfactant proteins in patients with acute respiratory failure, and postulated that the alveolar-capillary membrane acts as a sieve. With respiratory disorders that cause inflammation and damage to the membrane, a dynamic alteration occurs to the radius of the pores, allowing proteins that are normally restricted by their size to cross the membrane and enter the circulation.11 A large concentration gradient of >1500:1 of SP-B between the alveoli and plasma supports the notion that SP-B may flow naturally down its concentration gradient into the circulation during states that alter the permeability of the alveolar-capillary membrane.29
Is SP-B a Biomarker of the Cumulative Vascular Effects of Tobacco Smoke?
Although evidence supports a dose-response effect of smoking on cardiovascular events, demonstration of a linear and cumulative effect on subclinical atherosclerosis has not been consistent. The dose-response effect seen with tobacco exposure differs among vascular beds, with weaker dose-response effects seen in the coronary arteries than the aorta and peripheral arteries.5, 6, 27, 28 The toxic effects of smoking may vary among individuals with similar apparent tobacco exposures due to multiple factors including inter-individual variability in delivery of toxins to lung parenchyma, pulmonary defense mechanisms to clear inhaled toxins, nicotine pharmacokinetics, environmental factors, and tissue-specific responses to the vascular changes induced by inhaled tobacco smoke.
Multiple studies have investigated the role of biomarkers in assessing the variable toxicity resulting from inhaled tobacco smoke.32 Blood and urine cotinine levels, a nicotine metabolite, have been shown to correlate with both active and passive smoking exposure and are associated with CVD mortality and atherosclerosis.33-37 In addition, expired air carbon monoxide levels have also been shown to correlate with smoking exposure and CV disease.37 However, the relationship between levels of these markers and self-reported pack-years is not linear.38 Furthermore, there is marked variation in levels of cotinine and carbon monoxide at similar levels of pack-year exposure, indicating variable pharmacokinetics and supporting the notion that smoking may have variable down-stream effects.39, 40 Though nicotine metabolites may be associated with smoking toxicity, they are not a direct measure of smoking-related tissue toxicity as is SP-B. Studies of non-specific systemic inflammatory markers such as CRP, fibrinogen, and leukocyte count have also shown increased levels in smokers7 and several studies have suggested interactions between smoking, biomarker levels, and CV disease.41
Biomarkers of direct lung toxicity have been limited mostly to assessment of broncho-alveolar lavage fluid or expired air. The pulmonary surfactant proteins (SP-A, -B, -D) are detectable at low levels in the circulation and, except for SP-D, are specific to lung tissue, making them attractive blood-based biomarkers for lung-related smoking toxicity.29 Circulating levels of SP-B have been shown to correlate with tobacco exposure,13 as well as with increased pulmonary microvascular pressures in conditions such as acute respiratory failure and heart failure.12 Our novel findings of significant correlations between SP-B and several inflammatory biomarkers that have previously been associated with the development and progression of atherosclerosis, including TNF-alpha receptor-1A,42, 43 MPO,44 osteoprotegerin,19 MCP-117, and Cystatin C,45, 46 support a link between alveolar injury from smoking and inflammation and atherosclerosis. In contrast, no association was seen between SP-B and CRP, MMP-9, or MCP-1, suggesting that SP-B may reflect specific inflammatory pathways related to the vascular effects of smoking.
Study Limitations
Several study limitations must be noted. First, our cross sectional associations should not be interpreted to suggest a causal relationship between SP-B, smoking, and aortic atherosclerosis. Indeed, we believe it is more likely that SP-B is a marker than a mediator of tobacco-smoke related vascular risk. Cumulative pack-year exposure was not available for former smokers. Circulating SP-B reflects several pro-protein and processing intermediates of varying molecular weights,29 and the high-throughput methodology for determining plasma levels used in this study did not allow characterization of these different moieties or direct biological activity of the measured analyte. Other reported markers of smoking exposure and smoking-related toxicity were not measured, precluding direct comparison of biomarkers of smoking toxicity. Non-fatal CV events were not available at the time of analysis.
Conclusions
Circulating levels of SP-B are increased in proportion to the burden of inhaled tobacco exposure in the general population. Higher levels of SP-B independently associate with aortic atherosclerosis and correlate with a dose-response effect in smokers, suggesting that SP-B may be a useful marker of the dose-dependent vascular effects of smoking. These findings require confirmation in other study samples and should prompt further investigation into the role this protein may play as a marker of the interaction between inhaled toxins and the development and progression of atherosclerosis.
Acknowledgments
none
Sources of Funding: Grant support for the Dallas Heart Study was provided by the Donald W. Reynolds Foundation at UT Southwestern Medical Center, Dallas, TX, and by USPHS GCRC grant #M01-RR00633 from NIH/NCRR-CR. This publication was supported in part by the North and Central Texas Clinical and Translational Science Initiative (NIH UL1 RR024982). Assay measurements for SP-B were provided by Alere, Inc. (San Diego, CA).
Disclosures: Dr. de Lemos has received grant support from Roche Diagnostics and Alere, Inc (formerly Biosite), and consulting income from Johnson and Johnson and Tethys Biomedical. Dr. McGuire has received grant support from Alere and consulting income from Tethys Biomedical.
References
- 1.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 2.Lightwood JM, Glantz SA. Short-term economic and health benefits of smoking cessation: Myocardial infarction and stroke. Circulation. 1997;96:1089–1096. doi: 10.1161/01.cir.96.4.1089. [DOI] [PubMed] [Google Scholar]
- 3.Price JF, Mowbray PI, Lee AJ, Rumley A, Lowe GD, Fowkes FG. Relationship between smoking and cardiovascular risk factors in the development of peripheral arterial disease and coronary artery disease: Edinburgh artery study. Eur Heart J. 1999;20:344–353. doi: 10.1053/euhj.1998.1194. [DOI] [PubMed] [Google Scholar]
- 4.Leng GC, Lee AJ, Fowkes FG, Lowe GD, Housley E. The relationship between cigarette smoking and cardiovascular risk factors in peripheral arterial disease compared with ischaemic heart disease. The edinburgh artery study. Eur Heart J. 1995;16:1542–1548. doi: 10.1093/oxfordjournals.eurheartj.a060775. [DOI] [PubMed] [Google Scholar]
- 5.Solberg LA, Strong JP. Risk factors and atherosclerotic lesions. A review of autopsy studies. Arteriosclerosis. 1983;3:187–198. doi: 10.1161/01.atv.3.3.187. [DOI] [PubMed] [Google Scholar]
- 6.Strong JP, Richards ML. Cigarette smoking and atherosclerosis in autopsied men. Atherosclerosis. 1976;23:451–476. doi: 10.1016/0021-9150(76)90007-1. [DOI] [PubMed] [Google Scholar]
- 7.Levitzky YS, Guo CY, Rong J, Larson MG, Walter RE, Keaney JF, Jr, Sutherland PA, Vasan A, Lipinska I, Evans JC, Benjamin EJ. Relation of smoking status to a panel of inflammatory markers: The framingham offspring. Atherosclerosis. 2008;201:217–224. doi: 10.1016/j.atherosclerosis.2007.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frey SL, Pocivavsek L, Waring AJ, Walther FJ, Hernandez-Juviel JM, Ruchala P, Lee KY. Functional importance of the nh2-terminal insertion sequence of lung surfactant protein b. Am J Physiol Lung Cell Mol Physiol. 2010;298:L335–347. doi: 10.1152/ajplung.00190.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaccaro AM, Salvioli R, Tatti M, Ciaffoni F. Saposins and their interaction with lipids. Neurochem Res. 1999;24:307–314. doi: 10.1023/a:1022530508763. [DOI] [PubMed] [Google Scholar]
- 10.Pryhuber GS. Regulation and function of pulmonary surfactant protein b. Mol Genet Metab. 1998;64:217–228. doi: 10.1006/mgme.1998.2722. [DOI] [PubMed] [Google Scholar]
- 11.Doyle IR, Bersten AD, Nicholas TE. Surfactant proteins-a and -b are elevated in plasma of patients with acute respiratory failure. Am J Respir Crit Care Med. 1997;156:1217–1229. doi: 10.1164/ajrccm.156.4.9603061. [DOI] [PubMed] [Google Scholar]
- 12.De Pasquale CG, Arnolda LF, Doyle IR, Aylward PE, Chew DP, Bersten AD. Plasma surfactant protein-b: A novel biomarker in chronic heart failure. Circulation. 2004;110:1091–1096. doi: 10.1161/01.CIR.0000140260.73611.FA. [DOI] [PubMed] [Google Scholar]
- 13.Robin M, Dong P, Hermans C, Bernard A, Bersten AD, Doyle IR. Serum levels of cc16, sp-a and sp-b reflect tobacco-smoke exposure in asymptomatic subjects. Eur Respir J. 2002;20:1152–1161. doi: 10.1183/09031936.02.02042001. [DOI] [PubMed] [Google Scholar]
- 14.Victor RG, Haley RW, Willett DL, Peshock RM, Vaeth PC, Leonard D, Basit M, Cooper RS, Iannacchione VG, Visscher WA, Staab JM, Hobbs HH. The dallas heart study: A population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004;93:1473–1480. doi: 10.1016/j.amjcard.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 15.Khera A, McGuire DK, Murphy SA, Stanek HG, Das SR, Vongpatanasin W, Wians FH, Jr, Grundy SM, de Lemos JA. Race and gender differences in c-reactive protein levels. J Am Coll Cardiol. 2005;46:464–469. doi: 10.1016/j.jacc.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 16.Abdullah SM, Khera A, Das SR, Stanek HG, Canham RM, Chung AK, Morrow DA, Drazner MH, McGuire DK, de Lemos JA. Relation of coronary atherosclerosis determined by electron beam computed tomography and plasma levels of n-terminal pro-brain natriuretic peptide in a multiethnic population-based sample (the dallas heart study) Am J Cardiol. 2005;96:1284–1289. doi: 10.1016/j.amjcard.2005.06.073. [DOI] [PubMed] [Google Scholar]
- 17.Deo R, Khera A, McGuire DK, Murphy SA, Meo Neto Jde P, Morrow DA, de Lemos JA. Association among plasma levels of monocyte chemoattractant protein-1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J Am Coll Cardiol. 2004;44:1812–1818. doi: 10.1016/j.jacc.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 18.Zirlik A, Abdullah SM, Gerdes N, MacFarlane L, Schonbeck U, Khera A, McGuire DK, Vega GL, Grundy S, Libby P, de Lemos JA. Interleukin-18, the metabolic syndrome, and subclinical atherosclerosis: Results from the dallas heart study. Arterioscler Thromb Vasc Biol. 2007;27:2043–2049. doi: 10.1161/ATVBAHA.107.149484. [DOI] [PubMed] [Google Scholar]
- 19.Abedin M, Omland T, Ueland T, Khera A, Aukrust P, Murphy SA, Jain T, Gruntmanis U, McGuire DK, de Lemos JA. Relation of osteoprotegerin to coronary calcium and aortic plaque (from the dallas heart study) Am J Cardiol. 2007;99:513–518. doi: 10.1016/j.amjcard.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 20.Patel PC, Ayers CR, Murphy SA, Peshock R, Khera A, de Lemos JA, Balko JA, Gupta S, Mammen PP, Drazner MH, Markham DW. Association of cystatin c with left ventricular structure and function: The dallas heart study. Circ Heart Fail. 2009;2:98–104. doi: 10.1161/CIRCHEARTFAILURE.108.807271. [DOI] [PubMed] [Google Scholar]
- 21.Lindsey JB, de Lemos JA, Cipollone F, Ayers CR, Rohatgi A, Morrow DA, Khera A, McGuire DK. Association between circulating soluble receptor for advanced glycation end products and atherosclerosis: Observations from the dallas heart study. Diabetes Care. 2009;32:1218–1220. doi: 10.2337/dc09-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 23.Jain T, Peshock R, McGuire DK, Willett D, Yu Z, Vega GL, Guerra R, Hobbs HH, Grundy SM. African americans and caucasians have a similar prevalence of coronary calcium in the dallas heart study. J Am Coll Cardiol. 2004;44:1011–1017. doi: 10.1016/j.jacc.2004.05.069. [DOI] [PubMed] [Google Scholar]
- 24.Jaffer FA, O’Donnell CJ, Larson MG, Chan SK, Kissinger KV, Kupka MJ, Salton C, Botnar RM, Levy D, Manning WJ. Age and sex distribution of subclinical aortic atherosclerosis: A magnetic resonance imaging examination of the framingham heart study. Arterioscler Thromb Vasc Biol. 2002;22:849–854. doi: 10.1161/01.atv.0000012662.29622.00. [DOI] [PubMed] [Google Scholar]
- 25.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: A report from the american heart association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 26.He Y, Jiang Y, Wang J, Fan L, Li X, Hu FB. Prevalence of peripheral arterial disease and its association with smoking in a population-based study in beijing, china. J Vasc Surg. 2006;44:333–338. doi: 10.1016/j.jvs.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 27.Sackett DL, Gibson RW, Bross ID, Pickren JW. Relation between aortic atherosclerosis and the use of cigarettes and alcohol. An autopsy study. N Engl J Med. 1968;279:1413–1420. doi: 10.1056/NEJM196812262792602. [DOI] [PubMed] [Google Scholar]
- 28.Auerbach O, Garfinkel L. Atherosclerosis and aneurysm of aorta in relation to smoking habits and age. Chest. 1980;78:805–809. doi: 10.1378/chest.78.6.805. [DOI] [PubMed] [Google Scholar]
- 29.Doyle IR, Nicholas TE, Bersten AD. Partitioning lung and plasma proteins: Circulating surfactant proteins as biomarkers of alveolocapillary permeability. Clin Exp Pharmacol Physiol. 1999;26:185–197. doi: 10.1046/j.1440-1681.1999.03015.x. [DOI] [PubMed] [Google Scholar]
- 30.Longo ML, Waring A, Zasadzinski JA. Lipid bilayer surface association of lung surfactant protein sp-b, amphipathic segment detected by flow immunofluorescence. Biophys J. 1992;63:760–773. doi: 10.1016/S0006-3495(92)81643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weaver TE, Whitsett JA. Processing of hydrophobic pulmonary surfactant protein b in rat type ii cells. Am J Physiol. 1989;257:L100–108. doi: 10.1152/ajplung.1989.257.2.L100. [DOI] [PubMed] [Google Scholar]
- 32.Leone A. Biochemical markers of cardiovascular damage from tobacco smoke. Curr Pharm Des. 2005;11:2199–2208. doi: 10.2174/1381612054367391. [DOI] [PubMed] [Google Scholar]
- 33.Hamer M, Stamatakis E, Kivimaki M, Lowe GD, Batty GD. Objectively measured secondhand smoke exposure and risk of cardiovascular disease: What is the mediating role of inflammatory and hemostatic factors? J Am Coll Cardiol. 2010;56:18–23. doi: 10.1016/j.jacc.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whincup PH, Gilg JA, Emberson JR, Jarvis MJ, Feyerabend C, Bryant A, Walker M, Cook DG. Passive smoking and risk of coronary heart disease and stroke: Prospective study with cotinine measurement. BMJ. 2004;329:200–205. doi: 10.1136/bmj.38146.427188.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallo V, Neasham D, Airoldi L, Ferrari P, Jenab M, Boffetta P, Overvad K, Tjonneland A, Clavel-Chapelon F, Boeing H, Pala V, Palli D, Panico S, Tumino R, Arriola L, Lund E, Bueno-De-Mesquita B, Peeters PH, Melander O, Hallmans G, Riboli E, Saracci R, Vineis P. Secondhand smoke, cotinine levels, and risk of circulatory mortality in a large cohort study of never-smokers. Epidemiology. 2010;21:207–214. doi: 10.1097/EDE.0b013e3181c9fdad. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal S. The association of active and passive smoking with peripheral arterial disease: Results from nhanes 1999-2004. Angiology. 2009;60:335–345. doi: 10.1177/0003319708330526. [DOI] [PubMed] [Google Scholar]
- 37.Woodward M, Moohan M, Tunstall-Pedoe H. Self-reported smoking, cigarette yields and inhalation biochemistry related to the incidence of coronary heart disease: Results from the scottish heart health study. J Epidemiol Biostat. 1999;4:285–295. [PubMed] [Google Scholar]
- 38.Joseph AM, Hecht SS, Murphy SE, Carmella SG, Le CT, Zhang Y, Han S, Hatsukami DK. Relationships between cigarette consumption and biomarkers of tobacco toxin exposure. Cancer Epidemiol Biomarkers Prev. 2005;14:2963–2968. doi: 10.1158/1055-9965.EPI-04-0768. [DOI] [PubMed] [Google Scholar]
- 39.Gan WQ, Cohen SB, Man SF, Sin DD. Sex-related differences in serum cotinine concentrations in daily cigarette smokers. Nicotine Tob Res. 2008;10:1293–1300. doi: 10.1080/14622200802239132. [DOI] [PubMed] [Google Scholar]
- 40.Chahine R, Abchee A, Zalloua P. Nicotine metabolism in healthy smokers and patients with cardiovascular diseases. Mol Cell Biochem. 2005;280:241–244. doi: 10.1007/s11010-005-8840-9. [DOI] [PubMed] [Google Scholar]
- 41.Thakore AH, Guo CY, Larson MG, Corey D, Wang TJ, Vasan RS, D’Agostino RB, Sr, Lipinska I, Keaney JF, Jr, Benjamin EJ, O’Donnell CJ. Association of multiple inflammatory markers with carotid intimal medial thickness and stenosis (from the framingham heart study) Am J Cardiol. 2007;99:1598–1602. doi: 10.1016/j.amjcard.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 42.Jovinge S, Hamsten A, Tornvall P, Proudler A, Bavenholm P, Ericsson CG, Godsland I, de Faire U, Nilsson J. Evidence for a role of tumor necrosis factor alpha in disturbances of triglyceride and glucose metabolism predisposing to coronary heart disease. Metabolism. 1998;47:113–118. doi: 10.1016/s0026-0495(98)90203-7. [DOI] [PubMed] [Google Scholar]
- 43.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein e knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 44.Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 45.Ix JH, Shlipak MG, Chertow GM, Whooley MA. Association of cystatin c with mortality, cardiovascular events, and incident heart failure among persons with coronary heart disease: Data from the heart and soul study. Circulation. 2007;115:173–179. doi: 10.1161/CIRCULATIONAHA.106.644286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luc G, Bard JM, Lesueur C, Arveiler D, Evans A, Amouyel P, Ferrieres J, Juhan-Vague I, Fruchart JC, Ducimetiere P. Plasma cystatin-c and development of coronary heart disease: The prime study. Atherosclerosis. 2006;185:375–380. doi: 10.1016/j.atherosclerosis.2005.06.017. [DOI] [PubMed] [Google Scholar]