

Highlights
► Distinct subsets of ssRNA, dsRNA and dsDNA viruses trigger RIG-I and MDA5 activation of IPS-1. ► RLR signaling is regulated through multiple protein modifications by host cellular factors. ► Viral proteins antagonize multiple components of the RLR signaling pathway to subvert immunity. ► RLR, NLR and Caspase crosstalk promote alternate recognition pathways to sense cytosolic infection.
Abstract
During virus infection, multiple immune signaling pathways are triggered, both within the host cell and bystander cells of an infected tissue. These pathways act in concert to mediate innate antiviral immunity and to initiate the inflammatory response against infection. The RIG-I-like receptor (RLR) family of pattern recognition receptors (PRRs) is a group of cytosolic RNA helicase proteins that can identify viral RNA as nonself via binding to pathogen associated molecular pattern (PAMP) motifs within RNA ligands that accumulate during virus infection. This interaction then leads to triggering of an innate antiviral response within the infected cells through RLR induction of downstream effector molecules such as type I interferon (IFN) and other pro-inflammatory cytokines that serve to induce antiviral and inflammatory gene expression within the local tissue. Cellular regulation of RLR signaling is a critical process that can direct the outcome of infection and is essential for governance of the overall immune response and avoidance of immune toxicity. Mechanisms of positive and negative regulation of RLR signaling have been identified that include signaling crosstalk between RLR pathways and nuclear oligomerization domain (NOD)-like receptor (NLR) pathways and Caspase networks. Furthermore, many viruses have evolved mechanisms to target these pathways to promote enhanced replication and spread within the host. These virus–host interactions therefore carry important consequences for host immunity and viral pathogenesis. Understanding the pivotal role of RLRs in immune regulation and signaling crosstalk in antiviral immunity may provide new insights into therapeutic strategies for the control of virus infection and immunity.
Introduction
The development of effective antiviral immunity requires robust and specific immune activation during acute virus infection. This process is dependent on the ability of the host cell to first sense the viral pathogen and then to signal within the infected cell, alerting neighboring bystander cells and adaptive immune cells that a viral infection is underway. This form of cellular communication requires that pattern recognition receptors (PRRs) work in concert to sense specific pathogen associated molecular patterns (PAMPs) expressed by the virus which are distinct from the host. Several PRRs have now been described to sense and distinguish viral PAMPs. The toll-like receptor (TLR) family of PRRs resides at the cell surface and in endosomal compartments and are poised to sense extracellular or actively engulfed pathogens but not those in the cytoplasm [1]. By contrast, several PRRs have been identified that recognize microbial products within the cytoplasm of infected cells. These include the nuclear oligomerization domain (NOD)-like receptors (NLRs), proinflammatory DNA-binding receptors such as AIM2 and DAI (DLM-1/ZBP1), and the RLRs [2, 3]. Although we now have a strong understanding of the signaling pathways which PRRs engage in response to viral infection, it is only in the past few years that we have began to understand the unique crosstalk that exists between these pathways. This review will present an overview of contemporary studies defining RLR function and their signaling crosstalk that programs the immune response to virus infection.
Structure of RLR family members
The RLR family members include retinoic acid inducible gene-I (RIG-I), melanoma differentiation gene-5 (MDA5) and laboratory of genetics and physiology-2 (LGP2). They contain a distinct DEX/DH box RNA helicase domain involved in RNA binding and ATPase function to drive a conformation change that initiates signaling activation [4]. In addition, RIG-I and MDA5 but not LGP2 contain two N-terminal Caspase activation and recruitment domains (CARDs) which facilitate their interactions with other CARD containing molecules. These CARD–CARD interactions promote RIG-I/MDA5 binding to interferon promoter stimulator-1 (IPS-1; also called MAVS/VISA/Cardif) through a CARD–CARD interaction, leading to IPS-1-dependent activation of interferon regulator factor (IRF)-3, IRF-7 and nuclear factor κB (NF-κB) [5]. These processes result in transcriptional activities that direct the expression of IFN and the induction of a variety of antiviral effector genes, including interferon-stimulated genes (ISGs), whose actions limit virus replication and spread [6]. Furthermore, the C-terminus of RIG-I and LGP2 has been described as a repressor domain (RD) that acts to keep these molecules in an inactive ‘closed’ confirmation in the absence of activating RNA [7].
Multiple RNA viruses are known to trigger RLR signaling through distinct RIG-I or MDA5 dependent responses (Figure 1 ). RIG-I has been shown to be involved in recognition of Paramyxoviruses, Newcastle disease virus (NDV), Sendai virus (SeV), and Respiratory syncytial virus (RSV) [8]; Rhabdoviruses, Vesicular Stomatitis virus (VSV) and Rabies virus [8, 9]; Orthomyxoviruses, Influenza A,B virus (IAV, IBV); Flaviviruses, Hepatitis C virus (HCV), and Japanese Encephalitis virus (JEV) [8, 10] and the Filovirus, Ebola virus [11]. By contrast, MDA5 was found to be responsible for the recognition of Picornaviruses such as EMCV, the Coronavirus, Murine Hepatitis virus and the Calicivirus, Murine Norovirus-1 as well as the dsRNA mimetic, poly(I:C) [8, 12]. Both RIG-I and MDA5 were shown to function in the recognition of the Flaviviruses, Dengue virus (DENV) and West Nile virus (WNV) as well as the dsRNA Reovirus, Rotavirus [10, 13, 14].
Figure 1.
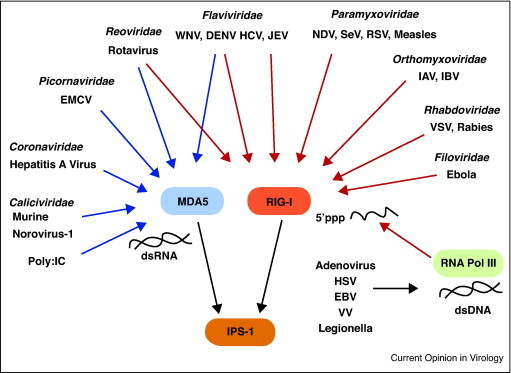
Requirements for RIG-I and MDA5 in recognition of distinct viral families. Interactions between viruses and RIG-I and MDA5 are delineated by arrows. Red arrows depict known requirements for RIG-I in sensing viruses while blue arrows denote known requirements for MDA5. Of note, members of the Flaviviridae and Reoviridae can trigger both MDA5 and RIG-I activation. The RNA polymerase III pathway for generation of RIG-I ligands and the DNA viruses and bacteria, Legionella, known to activate this pathway are also shown.
Recent work by Ablasser et al. [15••] and Chiu et al. [16••] now provides evidence that RLR signaling is critical to a broader range of pathogens. In these studies, the DNA mimetic poly (dA:dT) was found to trigger IFN responses in a RIG-I-dependent manner. This was driven by the host cell RNA polymerase III, which functioned to transcribe DNA to RNA ligands that were then recognized by RIG-I in the cytosol. Several DNA viruses have now been shown to activate this pathway including herpes-simplex virus-1, Adenovirus, Epstein-Barr virus and Vaccinia virus (VV) (Figure 1) [15••, 16••, 17, 18]. Surprisingly, the intracellular gram negative bacterium, Legionella pnuemophila, was also shown to activate type I IFN responses through RIG-I signaling [16••]. It is likely that other intracellular bacterium will be identified to trigger this pathway thus reflecting a broad role for RLR signaling in anti-microbial immunity.
The characterization of PAMP motifs that are recognized by RLRs to trigger innate immune signaling is ongoing. RIG-I was initially described to bind dsRNA [19] however, key findings have now identified that ssRNA containing a 5’tri-phosphate motif (5’ppp) was required for recognition by RIG-I [20, 21]. This interaction provides a way by which RIG-I can distinguish between host and viral RNAs as host mRNAs are capped at their 5’ ends while mature tRNA and rRNA lack 5’ppp and are covered as ribonucleoproteins respectively, thus preventing 5’ppp from recognition by RIG-I. This concept has been called into question by studies that showed that in addition to 5’ppp, RIG-I triggering requires some double stranded nature in the RNA, and that previous studies involving the 5’ppp contained double stranded hairpins that were introduced by the T7 polymerase in vitro [22]. An examination of 5’ppp RNA generated with non-hairpin coding polymerases is necessary to for definitive demonstration of this however. A study by Saito et al. [23] has also revealed a mechanism for self/non-self recognition by RIG-I. Here, it was shown that RIG-I engagement actually requires 5’ppp and specific sequences including uridine or adenosine-rich regions that are found within a PAMP motif of the Hepatitis C virus RNA and a number of other viruses recognized by RIG-I [23]. In the case of HCV, deletion of this region form the viral RNA abolished RIG-I binding despite the presence of 5’ppp. Thus, multiple motifs likely mark a viral RNA as non-self, including 5’ppp and specific sequence domains and secondary structures.
The RNA motifs which drive the activation of MDA5 are less clear. As mentioned above, the recognition of poly(I:C) by MDA5 but not RIG-I suggested that dsRNA was the ligand for MDA5. In line with these observations, recent studies suggested that higher order dsRNA ‘web’ structure which is produced during EMCV and VV virus infection were responsible for the activation of MDA5 [24]. However, the exact mechanism by which this higher order RNA would be engaged by MDA5 to trigger this response is still unclear.
RLR signaling cascade
RLR activation is thought to trigger the formation of an IPS-1 antiviral signaling complex or signalsome anchored at mitochondria-associated membranes, mitochondria, and peroxisomes. However, it is currently unclear how signaling at these distinct sites is orchestrated [25, 26•]. IPS-1 signaling leads to activation of Tank Binding Kinase-1 (TBK-1) and IκB kinase ɛ (IKKɛ) which then mediates downstream activation of IRFs and NF-κB as well as IFN and pro-inflammatory cytokine expression [12]. Several adaptors have been characterized to function in this complex including the tumor necrosis factor (TNF) associated factors (TRAFs), TRAF3 [27], and TRAF2/TRAF6 [28, 29], NEMO/Ikkγ [30], Fas-associated death domain (FADD), receptor interacting protein-1 (RIP1), TRADD and Caspase 8 and 10 [31]. These molecules participate in distinct signaling responses which drive the bifurcation of IRF and NF-κB; the findings of which have been reviewed in great detail elsewhere [12, 32] (Figure 2 ). In addition, to IPS-1, another adaptor molecule, stimulator of interferon genes (STING, also known as MITA) was shown to interact with RIG-I and IPS-1 and potentiate IRF/IFN activation [33, 34]. Further analysis has demonstrated that STING/MITA play crucial roles to IFN induction upon DNA stimulation however, the significance for STING/MITA in RLR signaling during RNA viral infection is still an area of contention.
Figure 2.
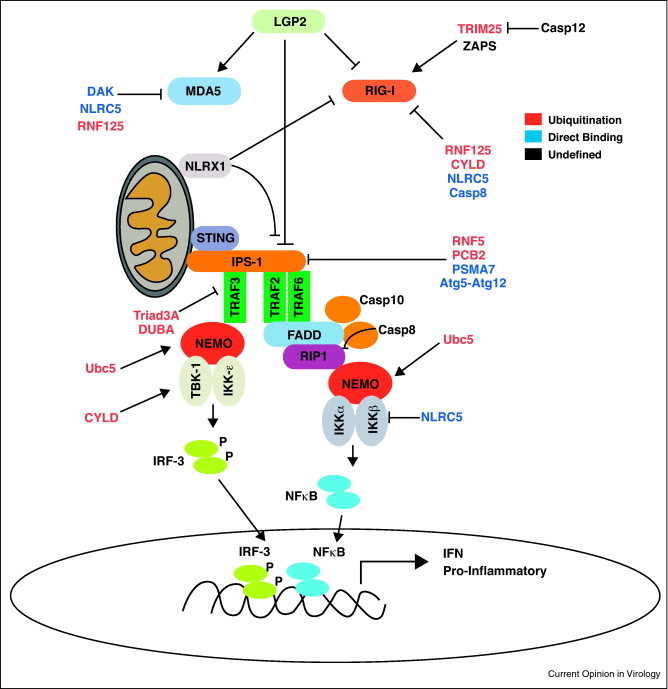
RLR signaling and cellular regulation. RIG-I and MDA5 signal through the IPS-1 signalsome located at mitochondria, mitochondrial membranes or peroxisomes. Signaling proceeds through multiple adaptors and leads to the bifurcation and activation of IRF-3 and NF-κB via the kinases TBK-1 and IKKɛ. Cellular factors known to contribute to regulation of multiple stages of RLR signaling are depicted in the figure. Factors involved in ubiquitin mediated regulation are denoted in red text, factors which require direct interaction with RLR signaling components are depicted in blue text and factors with unknown mechanisms of regulation are shown in black text.
Cellular regulation of RLR signaling
RLR signaling is tightly controlled. The first regulatory mechanism described was the identification of the RD of RIG-I and LGP2, which autoregulates RLR function through dynamic intramolecular interactions [7]. Multiple protein factors have also been identified to function in RLR regulation. LGP2 itself was found to act as both a positive and negative regulator of RLR signaling, wherein its over expression enhanced ISG induction to a variety of viruses including SeV and NDV [35, 36, 37], and LGP2−/− mice showed increased IFN responses and decreased susceptibility to VSV but not EMCV infection in vitro and in vivo [38]. By contrast, examination of an independently generated LGP2−/− mouse line or mice with mutations in the LGP2 ATP binding site (LGP2K30A/K30A) demonstrated decreased IFN responses and increased susceptibility to VSV, EMCV, SeV, JEV but not IAV [39]. These differences could be due in part to genetic background distinctions of mouse lines, as well as the nature of each targeting construct used to make the LGP2 null lines. A more complete examination of LGP2 in a cell/murine strain-specific manner is required for understanding the function of LGP2 in RLR signaling of immunity.
RLR ubiquitination
RIG-I is also regulated though interaction with and modification by the E3 ubiquitin ligase, tripartate motif 25 (TRIM25) which binds to RIG-I and mediates lysine 63 (K63) ubiquitin ligation at residue 172. This modification was critical to the ability of RIG-I to interact with IPS-1 and mediate downstream signaling [40]. In addition, the E2 ubiquitin-conjugating enzyme Ubc5 has also been shown to be involved in activation of RLR signaling. This event occurs downstream of RIG-I and IPS-1 and may function via conjugation of K63-ub to NEMO which enables recruitment of TBK-1 and IRF/NF-kB activation [41]. Neither TRIM25 nor Ubc5 were shown to ubiquitinate MDA5.
It should also be noted that ubiquitination of RLRs has also been shown to function in RLR negative regulation. The Ring Finger 125 (RNF125) E3 ligase, was shown to conjugate K48 ubiquitin to RIG-I and MDA5 promoting their proteosomal degradation [42]. Similar to this response, RNF5 directly interacts with IPS-1 and mediates K48-ub at positions K362 and K461 leading to degradation of IPS-1 during SeV infection [43]. In line with these observations, the E3 ligase, Triad3A was also shown to target TRAF3 for degradation via K48-ubiquitination [44]. These observations demonstrate that negative regulation by K48 ubiquitination occurs at multiple levels to control RLR signaling. A key push in understanding the mechanism of action by which these modifications operate should be a prime focus of future studies in this area.
In addition to ubiquitination, de-ubiquitinases (DUBs) play an important role in negative regulation of RLRs. For example, the DUB enzyme cylindromatosis (CYLD) was shown to directly bind RIG-I and mediate the removal of K63-ub and limit IFN induction [45]. In addition, CYLD was shown to de-ubiquitinate TBK-1 suggesting a broad regulatory role for DUB activity in regulating the effects of TRIM25 and Ubc5. In line with this idea, the de-ubiquitinating enzyme A (DUBA) has been shown to directly interact with TRAF3 and mediate the removal of K63-ub chains. The functional consequence of this response is the loss of interaction with TBK-1, alteration of the IPS-1 signalsome, and a block in signaling downstream of RLR signaling [46].
Regulatory protein interactions
RLR signaling regulation also occurs through direct interaction with specific activating or repressor factors. The zinc finger antiviral protein shorter isoform (ZAPS) was found to directly interact with RIG-I to potentiate signaling actions to suppress virus infection [47]. By contrast, the proteosome molecule, PSMA7(α4) subunit was shown to bind directly to IPS-1 and limit its ability to translate downstream IFN signals. This regulation operates though a mechanism dependent on proteosomal degradation of IPS-1. In addition, since VSV infection triggers expression of PSMA7α4, this factor may act as a negative feedback loop stimulated by virus infection [48]. PCBP2 was also shown to interact with IPS-1 and mediate its ubiquitination and degradation by the E3 ligase AIP4 [49], while the autophagy conjugate Atg5–Atg12 was shown to regulate RLR signaling by interacting directly with both RIG-I and IPS-1 to limit downstream IFN production [50]. Specific regulation of MDA5 function by protein interactions is less well understood. However, regulation of MDA5 has been described to occur via the dihydroacetone kinase (DAK), in which overexpression of the molecule led to decreased IFN responses to MDA5 agonists. Further, DAK was found to interact with MDA5 but not RIG-I suggesting that the mechanism for action was at the level of preventing MDA5 activation and interaction with IPS-1 [33]. Further comparison of regulatory mechanism involved in RIG-I versus MDA5-specific signaling are required in order to define the processes of RLR regulation through protein interaction.
Caspases and RLR signaling control
The Caspase family of proteins function in the activation of apoptotic cascades as well as in triggering inflammasome activation through the processing and release of IL-1β, IL-18 and IL-33 [51]. Two recent studies have identified unique roles for Caspase family members in the regulation of RLR signaling. Caspase-8 has been shown to associate with the IPS-1 signalsome via interactions with FADD, RIP-1 and IPS-1 after dsRNA stimulation of cells [31]. Caspase-8 may function to suppress RLR signaling through two distinct mechanisms. First, Caspase-8 promotes a direct depletion of RIG-I, through a mechanism dependent on its ability to bind FADD but independent of its catalytic cleavage ability. Secondly, Caspase-8 cleaves RIP1 at the IPS-1 signalsome/RLR signaling complex which leads to loss of IFN induction [52•]. Both of these functions of Caspase-8 are thought to act to negatively regulate and shut down the RLR response at later times after initial RLR triggering during acute virus infection. However further interrogation into the mechanism by which RIP-1 cleavage mediates this regulation is required in order to fully understand how this process regulates the IPS-1 signalsome and RLR functions.
The Caspase family member, Caspase-12, has also been identified as a negative regulator of RLR signaling. Using a model of WNV infection, Caspase-12−/− mice showed increased mortality to viral infection. Further, cells from knockout mice failed to mediate TRIM25 ubiquitination of RIG-I and this correlated with decreased IFN responses and lack of protection against WNV [53]. The exact mechanism by which Caspase-12 regulates TRIM25 was not described in this study, therefore warranting further analyses. In addition, as only a small percentage of the human population expresses Caspase-12 its role in regulation of the RLRs in humans remains debatable. Despite this caveat, these studies identify the potential for cross-talk between RLRs and Caspase signaling pathways as a regulatory feature that governs the innate antiviral immune response. Since CARD–CARD interactions play an important role in both RLR signaling and Caspase responses, further interactions between such CARD proteins will likely be identified and revealed as regulatory interactions.
Viral antagonism of RLR signaling
Pathogenic viruses have evolved mechanisms to target and disrupt RLR signaling programs in order to escape from the immune response to infection. Numerous viral proteins have now been described as RLR antagonists to block RLR recognition of viral RNA, target and bind RLRs, and modulate or disrupt downstream signaling components of the RLR pathway (Figure 3 ).
Figure 3.
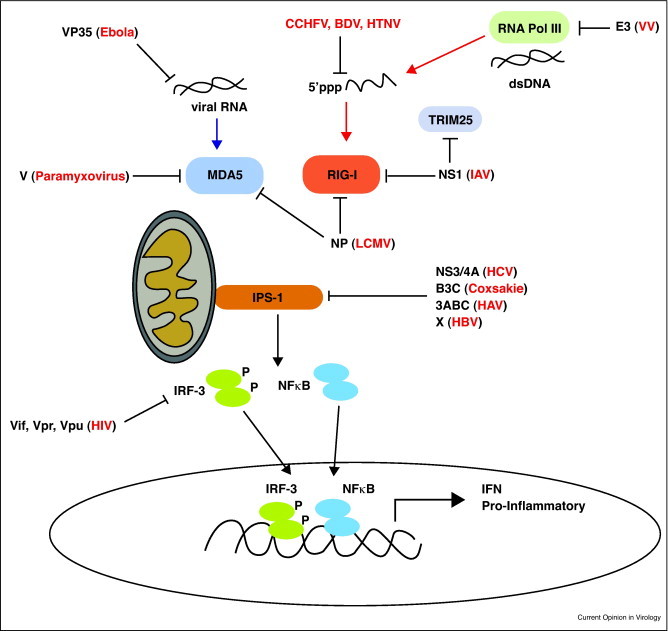
Viral regulation of RLR signaling. The RLR signalsome is shown including the RNA polymerase/RIG-I pathway for indirect recognition of cytosolic foreign DNA. Viral proteins known to block RLR signaling are shown with lines associating the specific factors they target. viruses or viral genus are denoted in parenthesis next to their specific proteins. Abbreviations: CCHFV (Crimean Congo Hemorrhagic Fever virus), BDV (Borna Disease virus), HTNV (Haantan virus).
Several viruses have found ways to prevent recognition of their viral RNA in the cytoplasm by the RLRs. The Ebola virus VP35 protein has been shown to sequester dsRNA in the cytosol and prevent viral RNA recognition by RIG-I [11]. In addition, Picornaviruses utilize their Vpg protein to cap viral RNA [8]. This mechanism may serve to block the 5’ppp RNA motif associated with RIG-I activation thus preventing RIG-I recognition and signaling. One interesting mechanism of direct subversion was found to occur in the Haantan virus, Crimean-Congo Hemorrhagic Fever virus and Borna Disease virus which modify their viral RNA to remove the 5’ppp motif thus remaining hidden from recognition by RIG-I [54].
Viral targeting of RLR signaling components has also been described. In particular, the cleavage and inactivation of IPS-1 by multiple viruses has been shown as a mechanism for RLR antagonism. The HCV protease, NS3/4A cleaves IPS-1, removing it from intracellular membranes and preventing RLR signaling of IFN induction, resulting in increased cell permissiveness to HCV [55, 56]. In addition, the Coxsackie virus protease B3C was recently shown to cleave IPS-1 and block downstream signaling responses, while the Hepatitis A virus protease precursor, 3ABC, was shown to cleave IPS-1 [57, 58]. Other viruses have been shown to block RLR signaling by sequestering signaling components or targeting their degradation through host machinery. The V proteins of several Paramyxoviruses have been shown to directly bind MDA5 and block its downstream signaling actions [59, 60]. Further, the NS1 protein of IAV was also shown to directly bind and sequester RIG-I from its downstream signaling complex [61]. Importantly, NS1 was also shown to directly bind TRIM25 and effectively block K63-ub of RIG-I [62•]. This activity is of interest as it demonstrates the ability of viruses to not only target the RLR signaling pathway but a key pathway involved in RLR signaling and metabolism as well. More recently, the Arenavirus family member, LCMV, was also shown to directly interact with RIG-I and MDA5 through its nuclear protein (NP). Here, NP functioned to modulate the late phase of IFN production both in vitro and in vivo; however, the mechanism of this regulation and interaction has yet to be elucidated [63].
The identification RNA polymerase III products as RLR agonists has identified DNA viruses as a new subset of viruses likely recognized, albeit indirect, by RLRs. Multiple DNA viruses have now been identified to modulate RLR signaling in order to escape its antiviral actions. The Hepadna virus, Hepatitis B virus X protein can bind IPS-1 and target it for degradation via K63 ubiquitination [64]. The VV virus protein E3 was also shown to bind RNA polymerase III generated RNAs and may act to prevent their recognition by RIG-I [18]. The protease of HIV, a lentivirus, was also recently shown to sequester RIG-I and limit ISG induction, thus linking RIG-I signaling to recognition of HIV-derived nucleic acid products [65]. However, as HIV RNA is known to be capped and polyadenylated it is unclear as to the actual viral RNA species which could actively trigger the RLR pathway during HIV infection. In addition, to blockade of RLR signaling by HIV, recent studies from our lab and others have demonstrated HIV directly depletes IRF-3 to block ISG induction downstream of TLR, RLR and other signaling pathways [66, 67]. This activity has been linked to the viral proteins Vpr, Vif [67] and now Vpu (Doehle et al. submitted) and appears a common mechanism employed by multiple viruses [68]. These observations reflect a broad theme among pathogenic viruses to suppress innate immune signaling at multiple levels, thus supporting the complex viral replication cycle.
Cross talk between the RLR and NLR signaling platforms
NLRs play important roles in the induction of pro-inflammatory cytokines, antimicrobial genes, and inflammasome activation. For example, the NLR, NOD2 is known to signal through the adaptor RIP2 to trigger NF-κB upon activation by the bacterial cell wall component, muramyl-dipeptide (MDP) [69]. NLRs, NLRC4, NLRP1 and NLPR3 are likewise involved in the activation of inflammasome cleavage of IL-1β, IL-18 and IL-33 [51]. Interestingly, two recent reports have identified unique links between RLRs and NLRs in these signaling responses. First, Sabbah et al. [70••] demonstrated that NOD2 could respond to viral infection and mediate the activation of IRF-3 and that this was mediated through direct interactions between NOD2 and IPS-1. This response occurred through the recognition of ssRNA derived from respiratory syncytial virus (RSV), a novel ligand for NOD2, which triggered the activation of IRF-3 and IFN expression. This study indicates for the first time, a model in which a bacterial trigger of NOD2 drives a RIP2-dependent activation of NF-κB genes while viral infection triggers an NOD2/IPS-1 driven IRF-3 response, thus defining host-pathogen interactions of polymicrobial infection. It is unclear whether NOD2 acts as a direct sensor for ssRNA or simply associates with RNA binding factors. One intriguing possibility is that NOD2 may interact with RIG-I or MDA5 which then sense the ssRNA, thus triggering IPS-1 dependent signaling (Figure 4 ). Polymicrobial infection presents complexities of host interactions that are likely to engage a variety of response pathways, including RLRs, NLRs, and others. Future studies are necessary to define these interactions and their role in polymicrobial infection and immunity.
Figure 4.
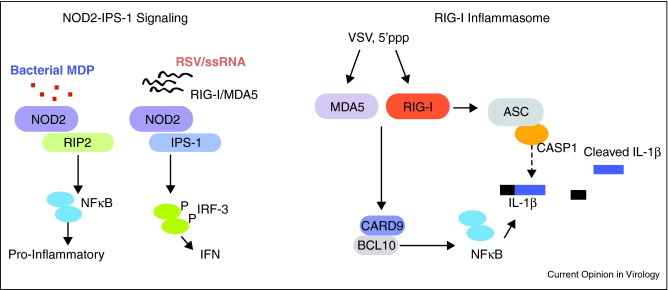
RLR–NLR interactions in signaling against pathogens. The NOD2 signaling pathway for recognition of bacterial and viral PAMPs is shown on the left. NOD2 is triggered by bacterial MDP to activate NF-κB while ssRNA from RSV leads to activation of IRF-3 signaling. The RIG-I inflammasome is depicted on the right. Both RIG-I and MDA5 can interact with CARD9/BCL10 to activate NF-κB. This leads to induction of pro-IL-1β. RIG-I but not MDA5 can directly associate with ASC bifurcating NLR mediated activation of Caspase-1. This leads to cleavage of IL-1β and secretion from the cell.
Work by Poeck and Ruland [71••] has now shown that RIG-I has the capacity to directly trigger the inflammasome and secretion of IL-1β through two distinct interactions. First, the authors reveal a novel interaction between RIG-I and the CARD9-BCL10, factors know to be involved in triggering of NF-κB activation. Interestingly, RIG-I and MDA5 could directly interact with CARD9 to drive this response during acute VSV infection or 5’ppp RNA stimulation. In addition, RIG-I alone was shown to interact with ASC to promote Caspase-1 mediated cleavage of pro-IL-1β independent of the known NLR, NLRP3 (Figure 4). These observations demonstrate unique signaling pathway for RIG-I that occurs independently of IPS-1 within a novel inflammasome pathway. A highly rigorous examination of this response is necessary to truly place it in the context of anti-viral signaling and innate immune programing.
NLRs have also been identified as negative regulators of RLR signaling. The NLR family member NLRX1 localizes to the mitochondria and was shown to block RIG-I signaling in a mechanism that required its binding to IPS-1. Further, siRNA knockdown of NLRX1 led to enhanced IFN and NF-kB activation to Sindbis virus infection [72]. NLRP5 was also recently described to limit multiple pro-inflammatory pathways including NF-κB and IFN response pathways, either through modulation of IKK phosphorylation or direct binding to RIG-I and MDA5 [73, 74, 75]. The ability of NLRs to act both as positive and negative regulators of RLR signaling suggests an important new avenue in understanding PRR cross-regulation.
Concluding remarks
The RLR pathway plays a critical role in the induction of both IFN and pro-inflammatory responses to viral infection. Our initial understanding of the complexity of this pathway has recently been expanded by key findings in the mechanisms that trigger activation and regulation of RLRs and their crosstalk with other innate immune signaling programs. Moreover, a role for RLR signaling crosstalk has been revealed as paramount in controlling polymicrobial infections involving RLRs, NLRs, and TLRs as specific PRRs that induce the immune response to viral and bacterial infections. Consideration of RLR signaling regulation, ligand and protein interactions, and signaling crosstalk will be important for future vaccine and immune adjuvant strategies aimed at suppressing microbial infection and controlling virus replication and spread.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
The authors would like to thank Brian Doehle, Stacy Horner and Maggie Brassil for helpful discussions and reading of the manuscript. Work in the Gale Laboratory is supported by NIH grants, the State of Washington, and the Burroughs Wellcome Fund.
References
- 1.Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 2.Wilkins C., Gale M., Jr. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yanai H., Savitsky D., Tamura T., Taniguchi T. Regulation of the cytosolic DNA-sensing system in innate immunity: a current view. Curr Opin Immunol. 2009;21:17–22. doi: 10.1016/j.coi.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Saito T., Gale M., Jr. Principles of intracellular viral recognition. Curr Opin Immunol. 2007;19:17–23. doi: 10.1016/j.coi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Hiscott J., Lin R., Nakhaei P., Paz S. MasterCARD: a priceless link to innate immunity. Trends Mol Med. 2006;12:53–56. doi: 10.1016/j.molmed.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Loo Y.M., Gale M., Jr. Viral regulation and evasion of the host response. Curr Top Microbiol Immunol. 2007;316:295–313. doi: 10.1007/978-3-540-71329-6_14. [DOI] [PubMed] [Google Scholar]
- 7.Saito T., Hirai R., Loo Y.M., Owen D., Johnson C.L., Sinha S.C., Akira S., Fujita T., Gale M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 9.Faul E.J., Wanjalla C.N., Suthar M.S., Gale M., Wirblich C., Schnell M.J. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 2010;6:e1001016. doi: 10.1371/journal.ppat.1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loo Y.M., Fornek J., Crochet N., Bajwa G., Perwitasari O., Martinez-Sobrido L., Akira S., Gill M.A., Garcia-Sastre A., Katze M.G. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardenas W.B., Loo Y.M., Gale M., Jr., Hartman A.L., Kimberlin C.R., Martinez-Sobrido L., Saphire E.O., Basler C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoneyama M., Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 13.Broquet A.H., Hirata Y., McAllister C.S., Kagnoff M.F. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol. 2011;186:1618–1626. doi: 10.4049/jimmunol.1002862. [DOI] [PubMed] [Google Scholar]
- 14.Sen A., Pruijssers A.J., Dermody T.S., Garcia-Sastre A., Greenberg H.B. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J Virol. 2011;85:3717–3732. doi: 10.1128/JVI.02634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Ablasser A., Bauernfeind F., Hartmann G., Latz E., Fitzgerald K.A., Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]; See annotation for reference [16••].
- 16••.Chiu Y.H., Macmillan J.B., Chen Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; These studies identify a pathway for cytosolic DNA recognition via indirect activation of RIG-I. Using different systems, the authors of these studies independently demonstrated that the host RNA polymerase III transcribes synthetic or microbial derived DNA into dsRNA that stimulated RIG-I activation. In addition, these studies identified distinct DNA viruses and bacteria that can activate this response suggesting a broader scope for RLR in recognition of cellular pathogens.
- 17.Melchjorsen J., Rintahaka J., Soby S., Horan K.A., Poltajainen A., Ostergaard L., Paludan S.R., Matikainen S. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J Virol. 2010;84:11350–11358. doi: 10.1128/JVI.01106-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valentine R., Smith G.L. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J Gen Virol. 2010;91:2221–2229. doi: 10.1099/vir.0.021998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 20.Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. 5’-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 21.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt A., Schwerd T., Hamm W., Hellmuth J.C., Cui S., Wenzel M., Hoffmann F.S., Michallet M.C., Besch R., Hopfner K.P. 5’-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A. 2009;106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito T., Owen D.M., Jiang F., Marcotrigiano J., Gale M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pichlmair A., Schulz O., Tan C.P., Rehwinkel J., Kato H., Takeuchi O., Akira S., Way M., Schiavo G., Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 26•.Dixit E., Boulant S., Zhang Y., Lee A.S., Odendall C., Shum B., Hacohen N., Chen Z.J., Whelan S.P., Fransen M. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; IPS-1 has been thought to mediate its signaling responses from anchored positions at the mitochondrial. This study identifies peroxisomes as another platform in which the IPS-1 signalsome is anchored. Further this data suggests that differential IPS-1 localization may prime distinct IFN and ISG responses.
- 27.Saha S.K., Pietras E.M., He J.Q., Kang J.R., Liu S.Y., Oganesyan G., Shahangian A., Zarnegar B., Shiba T.L., Wang Y. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikkelsen S.S., Jensen S.B., Chiliveru S., Melchjorsen J., Julkunen I., Gaestel M., Arthur J.S., Flavell R.A., Ghosh S., Paludan S.R. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J Biol Chem. 2009;284:10774–10782. doi: 10.1074/jbc.M807272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida R., Takaesu G., Yoshida H., Okamoto F., Yoshioka T., Choi Y., Akira S., Kawai T., Yoshimura A., Kobayashi T. TRAF6 and MEKK1 play a pivotal role in the RIG-I-like helicase antiviral pathway. J Biol Chem. 2008;283:36211–36220. doi: 10.1074/jbc.M806576200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao T., Yang L., Sun Q., Arguello M., Ballard D.W., Hiscott J., Lin R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K., Kawai T., Kumar H., Sato S., Yonehara S., Akira S. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol. 2006;176:4520–4524. doi: 10.4049/jimmunol.176.8.4520. [DOI] [PubMed] [Google Scholar]
- 32.Nakhaei P., Genin P., Civas A., Hiscott J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009;21:215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Diao F., Li S., Tian Y., Zhang M., Xu L.G., Zhang Y., Wang R.P., Chen D., Zhai Z., Zhong B. Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc Natl Acad Sci U S A. 2007;104:11706–11711. doi: 10.1073/pnas.0700544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong B., Yang Y., Li S., Wang Y.Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Komuro A., Horvath C.M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006;80:12332–12342. doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothenfusser S., Goutagny N., DiPerna G., Gong M., Monks B.G., Schoenemeyer A., Yamamoto M., Akira S., Fitzgerald K.A. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 37.Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K., Foy E., Loo Y.M., Gale M., Jr., Akira S. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 38.Venkataraman T., Valdes M., Elsby R., Kakuta S., Caceres G., Saijo S., Iwakura Y., Barber G.N. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- 39.Satoh T., Kato H., Kumagai Y., Yoneyama M., Sato S., Matsushita K., Tsujimura T., Fujita T., Akira S., Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gack M.U., Shin Y.C., Joo C.H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 41.Zeng W., Xu M., Liu S., Sun L., Chen Z.J. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell. 2009;36:315–325. doi: 10.1016/j.molcel.2009.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arimoto K., Takahashi H., Hishiki T., Konishi H., Fujita T., Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong B., Zhang Y., Tan B., Liu T.T., Wang Y.Y., Shu H.B. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J Immunol. 2010;184:6249–6255. doi: 10.4049/jimmunol.0903748. [DOI] [PubMed] [Google Scholar]
- 44.Nakhaei P., Mesplede T., Solis M., Sun Q., Zhao T., Yang L., Chuang T.H., Ware C.F., Lin R., Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedman C.S., O’Donnell M.A., Legarda-Addison D., Ng A., Cardenas W.B., Yount J.S., Moran T.M., Basler C.F., Komuro A., Horvath C.M. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kayagaki N., Phung Q., Chan S., Chaudhari R., Quan C., O’Rourke K.M., Eby M., Pietras E., Cheng G., Bazan J.F. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 47.Hayakawa S., Shiratori S., Yamato H., Kameyama T., Kitatsuji C., Kashigi F., Goto S., Kameoka S., Fujikura D., Yamada T. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat Immunol. 2011;12:37–44. doi: 10.1038/ni.1963. [DOI] [PubMed] [Google Scholar]
- 48.Jia Y., Song T., Wei C., Ni C., Zheng Z., Xu Q., Ma H., Li L., Zhang Y., He X. Negative regulation of MAVS-mediated innate immune response by PSMA7. J Immunol. 2009;183:4241–4248. doi: 10.4049/jimmunol.0901646. [DOI] [PubMed] [Google Scholar]
- 49.You F., Sun H., Zhou X., Sun W., Liang S., Zhai Z., Jiang Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009;10:1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 50.Jounai N., Takeshita F., Kobiyama K., Sawano A., Miyawaki A., Xin K.Q., Ishii K.J., Kawai T., Akira S., Suzuki K. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanneganti T.D. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52•.Rajput A., Kovalenko A., Bogdanov K., Yang S.H., Kang T.B., Kim J.C., Du J., Wallach D. RIG-I RNA, helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34:340–351. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]; This work identifies a new interaction between Caspase family members and the RLR pathway. Here the authors show that regulation of both RIG-I and RIP1 is mediated through interactions with Caspase-8 and this acts as a mechanism to down modulate IFN responses after primary activation. This study highlights the unique crosstalk between Caspases and regulation of the RLR pathway.
- 53.Wang P., Arjona A., Zhang Y., Sultana H., Dai J., Yang L., LeBlanc P.M., Doiron K., Saleh M., Fikrig E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat Immunol. 2010;11:912–919. doi: 10.1038/ni.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Habjan M., Andersson I., Klingstrom J., Schumann M., Martin A., Zimmermann P., Wagner V., Pichlmair A., Schneider U., Muhlberger E. Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One. 2008;3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loo Y.M., Owen D.M., Li K., Erickson A.K., Johnson C.L., Fish P.M., Carney D.S., Wang T., Ishida H., Yoneyama M. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 57.Mukherjee A., Morosky S.A., Delorme-Axford E., Dybdahl-Sissoko N., Oberste M.S., Wang T., Coyne C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y., Liang Y., Qu L., Chen Z., Yi M., Li K., Lemon S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andrejeva J., Childs K.S., Young D.F., Carlos T.S., Stock N., Goodbourn S., Randall R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Childs K., Stock N., Ross C., Andrejeva J., Hilton L., Skinner M., Randall R., Goodbourn S. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 61.Mibayashi M., Martinez-Sobrido L., Loo Y.M., Cardenas W.B., Gale M., Jr., Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62•.Gack M.U., Albrecht R.A., Urano T., Inn K.S., Huang I.C., Carnero E., Farzan M., Inoue S., Jung J.U., Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work identifies a unique mechanism for viral antagonism of the RLR pathway. Here the NS1 protein of IAV was shown to directly interact with TRIM25, a positive regulator of RIG-I, to block the induction of IFN responses. This highlights the ability of viruses to target not only antiviral signaling cascades as a mechanism for evasion, but also the pathways which regulate them.
- 63.Zhou S., Cerny A.M., Zacharia A., Fitzgerald K.A., Kurt-Jones E.A., Finberg R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J Virol. 2010;84:9452–9462. doi: 10.1128/JVI.00155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei C., Ni C., Song T., Liu Y., Yang X., Zheng Z., Jia Y., Yuan Y., Guan K., Xu Y. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. 2010;185:1158–1168. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 65.Solis M., Nakhaei P., Jalalirad M., Lacoste J., Douville R., Arguello M., Zhao T., Laughrea M., Wainberg M.A., Hiscott J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2011;85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doehle B.P., Hladik F., McNevin J.P., McElrath M.J., Gale M., Jr. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. 2009;83:10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okumura A., Alce T., Lubyova B., Ezelle H., Strebel K., Pitha P.M. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373:85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park J.H., Kim Y.G., McDonald C., Kanneganti T.D., Hasegawa M., Body-Malapel M., Inohara N., Nunez G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- 70••.Sabbah A., Chang T.H., Harnack R., Frohlich V., Tominaga K., Dube P.H., Xiang Y., Bose S. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a model of RSV infection, this study identified a novel role for NOD2 in activating IFN responses during viral infection. Furthermore this demonstrated a previously unappreciated role by which NOD2 recognizes cytosolic ssRNA promoting an interaction with IPS-1 and induction of IFN. This work highlights the crosstalk between cellular PRRs in recognition of viral infection.
- 71••.Poeck H., Bscheider M., Gross O., Finger K., Roth S., Rebsamen M., Hannesschlager N., Schlee M., Rothenfusser S., Barchet W. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]; This study identifies a novel pathway for inflammasome activation and demonstrates the first identification of RIG-I mediate immune activation independent of IPS-1. Using either VSV or 5’tri-phosphate RNA treatment, the authors show that RIG-I triggers IL-1β expression as well as cleavage through distinct direct interactions with the CARD9/BCL10 and ASC/Caspase-1 complexes.
- 72.Moore C.B., Bergstralh D.T., Duncan J.A., Lei Y., Morrison T.E., Zimmermann A.G., Accavitti-Loper M.A., Madden V.J., Sun L., Ye Z. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 73.Benko S., Magalhaes J.G., Philpott D.J., Girardin S.E. NLRC5 limits the activation of inflammatory pathways. J Immunol. 2010;185:1681–1691. doi: 10.4049/jimmunol.0903900. [DOI] [PubMed] [Google Scholar]
- 74.Cui J., Zhu L., Xia X., Wang H.Y., Legras X., Hong J., Ji J., Shen P., Zheng S., Chen Z.J. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141:483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neerincx A., Lautz K., Menning M., Kremmer E., Zigrino P., Hosel M., Buning H., Schwarzenbacher R., Kufer T.A. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem. 2010;285:26223–26232. doi: 10.1074/jbc.M110.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]