

Abstract
The control of mitochondria energy conversion by cytosolic processes is reviewed. The nature of the cytosolic and mitochondrial potential energy homeostasis over wide ranges of energy utilization is reviewed and the consequences of this homeostasis in the control network are discussed. An analysis of the major candidate cytosolic signaling molecules ADP, Pi and Ca2+ are reviewed based on the magnitude and source of the cytosolic concentration changes as well as the potential targets of action within the mitochondrial energy conversion system. Based on this analysis, Ca2+ is the best candidate as a cytosolic signaling molecule for this process based on its ability to act as both a feed-forward and feed-back indicator of ATP hydrolysis and numerous targets within the matrix to provide a balanced activation of ATP production. These targets include numerous dehydrogenases and the F1-F0-ATPase. Pi is also a good candidate since it is an early signal of a mismatch between cytosolic ATP production and ATP synthesis in the presence of creatine kinase and has multiple targets within oxidative phosphorylation including NADH generation, electron flux in the cytochrome chain and a substrate for the F1-F0-ATPase. The mechanism of the coordinated activation of oxidative phosphorylation by these signaling molecules in discussed in light of the recent discoveries of extensive protein phosphorylation sites and other post-translational modifications. From this review it is clear that the control network associated with the maintenance of the cytosolic potential energy homeostasis is extremely complex with multiple pathways orchestrated to balance the sinks and sources in this system. New tools are needed to image and monitor metabolites within subcellular compartments to resolve many of these issues as well as the functional characterization of the numerous matrix post-translational events being discovered along with the enzymatic processes generating and removing these protein modifications.
Introduction
It is believed that the presence of the mitochondrion within eukaryotic cells is the result of a symbiotic relationship between early eukaryotic cells and some early forms of bacteria. The current role of the mitochondria evolved to include numerous novel processes in the cell such as specialized biochemical synthesis1, lipid processing 2, reactive oxygen species signaling3, regulation of apoptosis4 and energy conversion via oxidative phosphorylation and substrate level phosphorylation.
The earliest studies of the relationship between cardiac work and mitochondrial ATP production, or respiration, demonstrated that mitochondrial ATP production is closely coupled to ATP hydrolysis at the myofibrils and active ion transport events associated with contraction. Since the extraction of oxygen is high and essentially constant, there is also a close coupling between tissue blood flow, mitochondrial respiration and work. These relationships are illustrated in Figure 1 from the study of Khouri et al 5 in the exercising awake dog. In these studies on canines working on a treadmill, a linear relationship between work, oxygen consumption and coronary blood flow was observed. Duncker and Bache 6 recently reviewed the relative contributions of heart rate (∼60%), contractility (20%) and end-diastolic volume (Starling)(20%) to the increase in myocardial work linearly driving myocardial oxygen consumption Though this phenomenon has been replicated over the years, the cellular mechanisms for these control processes remain unknown. In this review I will focus on acute regulatory mechanisms, the reader is referred to a recent review by Devin and Rigoulet on long term modifications of mitochondrial function7.
Figure 1.

Effect of cardiac work on coronary blood flow and oxygen consumption in awake canines. Exercise was conducted on an 11° incline treadmill with rates approaching 16km/min. A) Represents the oxygen usage as a function of coronary flow over the exercise range. B) Represents the relationship between coronary oxygen utilization from using the left main or circumflex artery flow values and left ventricular work. C) Represents the oxygen extraction values as a function of running speed. All data adapted from Khouri et al 5.
The initial models proposed that a simple feedback model of ATP metabolism products, that include ADP, Pi, and adenosine, would regulate the mitochondria via cytosolic ADP and Pi concentrations8-10, while adenosine would provide a feedback vasodilator to control flow 11. This was an extremely logical model since it provided the multifaceted control of respiration and blood flow via the breakdown of one molecule, ATP, intimately linked to the utilization of potential energy for cardiac work. However, in the late 1980's through 1990's non-invasive methods of monitoring high energy phosphate metabolites using nuclear magnetic resonance spectroscopy (NMRS) revealed that ATP and its hydrolysis products, ADP and Pi, did not change with large workloads when averaged over the heart wall 12-20. Evaluation of earlier classical biochemical “grind and find” experiments also confirmed this notion that changes in the concentration of ATP and its hydrolysis products were difficult to demonstrate with alterations in workload 21-23. Recent studies by Zhuo et al in a large work transition model in the pig confirms this notion24. This “metabolic homeostasis” or maintenance of the free energy and concentration of key energy metabolites in the face of large perturbation of energy flux suggested that the simple feedback model of mitochondrial ATP production and blood flow may not be correct25-28. Possibly a more specific term for this process would be a potential energy homeostasis since it is generally being referred to the cytosolic potential energy associated with ATP. At the extremes of cardiac output in the anesthetized animal in vivo, with minimal venous return, alterations in the hydrolysis products of ATP, ADP and Pi, can be detected suggesting limits to the regulatory pathway approaching Vmax.13, 15 The extent of the potential energy homeostasis within the energy conversion metabolic pathway in vivo, and its limits, provided key information on the possible cellular control mechanisms for cardiac cellular respiration as well as coronary blood flow control.
The Cellular Potential Energy Homeostasis
As discussed above, 31P NMRS and chemical extraction studies demonstrated that in many systems, including man, that the ATP, ADP and Pi concentrations are very stable with variations in ATP utilization rates. This also concerned Hill shortly after the discovery of ATP challenging biochemist to demonstrate a breakdown of ATP during muscle contraction in a famous paper in 1950 29. Indeed, this challenge was essentially left unmet for a decade until multiple inhibitors of metabolism, iodoacetic acid, glycolytic inhibition, and fluoro-dinitrobenzene, creatine kinase inhibition, were employed by Cain and Davies30 in 1962. This historical result together with the modern non-invasive techniques, demonstrate that the energy metabolism control network is poised to maintain ATP as constant as possible in the face of changes in ATP utilization.
What is the advantage of a potential energy homeostasis? The importance of phosphorylation potential in the function of the heart has been the subject of numerous articles in this journal31-34. There are numerous advantages of maintaining the free energy of ATP constant during changes in ATP turnover. First, and most obvious, is that the amount of energy available for work is determined by the free energy of ATP hydrolysis. If during increases in work the hydrolysis products of ATP are allowed to increase, then the free energy available for contractile work decreases when you need it most. This is demonstrated in Figure 2a where the effect of a 2% mismatch of ATP production with ATP utilization is plotted for the apparent ΔGATP and [ATP] in the canine heart, in vivo. As seen in Figure 2a, even small changes in ATP levels (∼5% drop), and associated increases in hydrolysis products, will decrease the free energy available for contraction and ion transport by 5 to 6 kJ/mol. While a 20% change in ATP results in a drop of ∼10 kJ/mole. In addition to decreasing the energy for contraction, the most sensitive energy dependent parameter may be the maintenance of the sarcolemma calcium gradient by the calcium pump (SERCA) as well as the Ca-Na exchange at the plasma membrane35. This calcium gradient is critical for the initiation and power of cardiac muscle contractions. It has been appreciated for many years that one of the highest potential energy “stores” in the heart cell is in the calcium gradient 35, and any decrease in the free energy of ATP would impede SERCA from generating a Ca signaling gradient36. This is also reflected in Figure 2b where the maximum sarcoplasmic reticulum calcium gradient is plotted along with the changes in ΔGATP. Thus, a small change in ATP content and ΔGATP results in a large decrease in the ability of the cell to accumulate Ca in the SR for signaling.
Figure 2.

Simulations of the changes in the approximated ΔGATP with ATP hydrolysis and potential for SR calcium retention. During the arbitrary time points ATP breakdown was made to exceed production by 2%. The approximated ΔGATP (see 34) was determined using the following equation: ΔGATP= ΔGATP°+RTln([ADP][Pi]/[ATP]). ΔGATP° was assumed to be 33kJ/mol. T was 310K. Resting metabolite levels were taken from literature for the canine heart 13, 135. A) Presents the ATP values in absolute values (M) solid line. Simulation is only shown for a 20% decrease in ATP. ΔGATP (kJ/M) is represented in a dashed line. B) A replot of ΔGATP (dashed line) with the calculated maximum ratio of [Ca] cytosol/[Ca]SR(solid line) calculated from the following equation: [Ca] cytosol/[Ca]SR = e(ΔGATP/2RT).
Secondary effects of having ATP and its hydrolysis products labile to changes in workloads would be reflected in the allosteric effects of these molecules on other reactions within the cell including numerous signaling processes associated with protein phosphorylation, redox state and synthetic reactions. If the ATP levels were not maintained all of these reactions would be modified by routine changes in cardiac workload, likely not a desirable condition.
Most of the measurements discussed to this point have been the gross NMR measures or grind and find determinations of ATP and its metabolites. What about the potential energy within the mitochondrion during work transitions? In most animals, the mitochondria volume is on the order of 25% of the cellular volume thus if large changes in ATP content in the mitochondria were occurring, one would suspect that this would be reflected in the gross ATP measurements and as discussed above no significant changes are observed. However, the ATP concentrations within the matrix could certainly be more labile than the total ATP content and be missed by the aforementioned volume and concentration differences as well as a broaden line width in 31P NMR measurements of metabolites in the viscous metal rich mitochondrial matrix.
More direct measures of the mitochondrial potential energy status can be obtained from optical measures of the NADH or FAD fluorescence in vitro or cytochrome c absorbance, in vivo. The most extensively studied system has been the mitochondrial NADH or FAD fluorescence signal. Recently we demonstrated that Complex 1 binding of NADH is one of the major sources of the NADH fluorescence lifetime enhancement in the heart mitochondrion 37 supporting the notion that using this signal is effective in determining the potential energy available for ATP production. Chance et al 38-40 showed that the NADH redox state is highly dependent on the ADP and Pi levels, generally oxidizing with increases in ATP production rate driven by increases in ADP and Pi. Thus, the NADH redox state can be used as a monitor of the ADP and PI levels at the mitochondrion in the intact cell. In several studies the mitochondrial NADH fluorescence signal or chemically determined NADH/NAD ratio in perfused hearts in vitro has been found to be essentially constant over normal work load transitions23, 41-43 suggesting that the potential energy homeostasis with workload extends into the mitochondrial matrix. Similar results have also be obtained by Brandes et al in isolated cardiac muscle 44 and by O'Rourke's lab in isolated myocytes 45. In contrast, Ashruf et al showed that a massive work jump from a vented non-working heart to a fully working model transition that did result in a net oxidation of NADH 46. However, it is unclear whether even the cytosol phosphorylation potential was maintained under these conditions since others have shown that cardiac arrest dramatically increases the phosphorylation potential47-49 and increases mitochondrial NADH 50 by removing most of the work in a system that is normally never at rest.
No measure of the mitochondria NADH fluorescence has been determined in vivo in large animals during work transitions. Our own efforts failed in this regard due to the highly fluorescent elastin of the visceral pericardium interfering with the measurement51. However, optical measures of the cytochrome redox state have been made during work transitions in the canine heart. Arai et al 52 using optical reflectance spectroscopy determined that the redox state of cytochrome c remained constant during a tripling of the metabolic rate. These data taken together suggest that the potential energy homeostasis extends to the mitochondria matrix where most of the energy conversion processes of ATP production occur.
This extension of the potential energy homeostasis to the mitochondria matrix is important since it limits the types of models that may explain the regulation of mitochondrial ATP production. Extensive mathematical modeling has revealed that small micro-compartments of metabolites at diffusional barriers could exist that would result in a diffusion limited delivery of ADP and Pi without large changes in the bulk cytosol as detected with NMR or extraction methods (for example see 53). However the fact that the potential energy homeostasis extends to the mitochondrial matrix implies that ADP and Pi delivery alone is not being increased under this conditions. That is, if only local increases in ADP and Pi were increasing oxidative phosphorylation during work increases, then the classical oxidation of NADH and cytochrome c should be observed, no matter how small the metabolite pools are. Having said that, some experimental data consistent with metabolite compartmentation is compelling 54 and, like Ca2+ in the cytosol, the geometry of the heart cell may be generating metabolite gradients that play a role in this process. However, also like Ca2+, the direct imaging of these metabolite gradients would greatly move this field forward as did the cytosolic [Ca2+] indicators55, 56. New imaging modalities or metabolite sensitive probes may prove useful in this regard 57, 58. The simplest probe to develop may simply involve a fluorescence molecule sensitive to free phosphate for analysis of work transitions in the intact cell. Brune et al 59 has described a fluorescent protein capable sensitive to μM concentrations of Pi however the affinity is much too high for resting cytosolic levels. Potentially modifying this base protein may provide a novel tool in the investigation of compartmentation in the regulation of cellular energetics.
Another important component in this process is the creatine kinase reaction. It has been appreciated since the early experiments by Cain and Davies 30 discussed above that the creatine kinase reaction is essentially a limited buffer for ATP and ADP. Its limitation is linked to the fact that this reaction does not rely on a constant supply of energy but only redistributes energy from the dead end metabolites, creatine and creatine phosphate. Neither creatine or creatine phosphate has been demonstrated to have any allosteric or regulatory activity outside of being substrates for creatine kinase. Thus, the creatine kinase reaction is an effective temporal buffer of ATP contributing the potential energy homeostasis60. Adding the creatine kinase reaction to the 2% imbalance between ATP production and synthesis presented in Figure 2, clearly reduces the short term impact of this imbalance in the calculated aΔGATP as well as ADP and ATP concentrations (Figure 3a). However, the actual changes in Pi are enhanced (See Figure 3b), suggesting that any short term imbalance between ATP production and breakdown will be reflected primarily in Pi and creatine concentrations over the short term and not ADP or ATP in the presence of a rapid creatine kinase reaction. Consequences of this relationship will be discussed further below. Another near equilibrium enzyme distributed through the cytosol is adenylate kinase61 which removes 2 ADP molecules to reform ATP and AMP. In combination with creatine kinase and ATPase activity, this reaction generates another free Pi molecule by hydrolyzing ATP to AMP instead of just ADP, and limits the formation of ADP in the cytosol.
Figure 3.
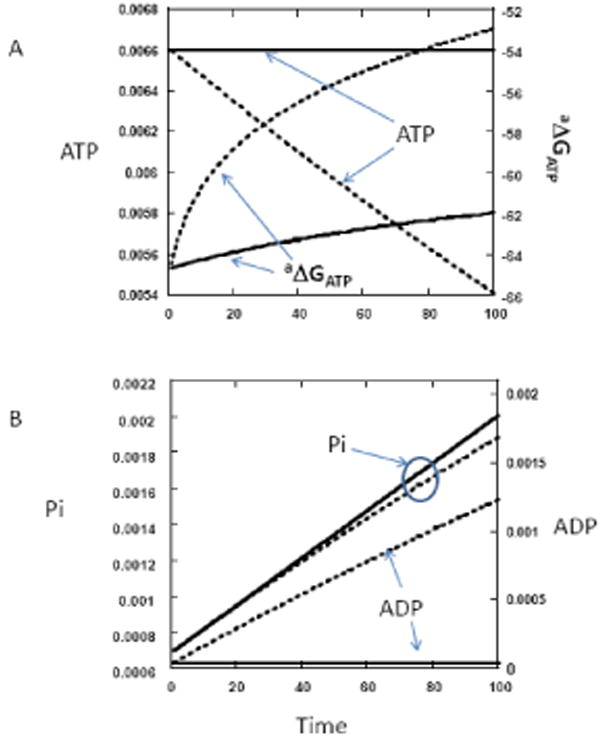
Simulations as conducted in Figure 2 but with and without the addition of a creatine kinase equilibrium reaction. After each step in ATP hydrolysis (2% hydrolysis steps), the creatine kinase reaction was permitted to reach equilibrium through a numerical approximation. The creatine kinase equilibrium constant was assumed to be 1.66×109. pH was 7.0. Other parameters were identical to those in Figure 2. A) Plot of ATP and the ΔGATP with, solid line, and without the creatine kinase reaction, dotted line. B) Plot of ADP and Pi with, solid line, and without the creatine kinase reaction, dotted line.
Creatine kinase (CPK) is located both at the myofibrils and mitochondria as different isoforms. Since the CPK equilibrium converts uM changes in ADP to mM changes in creatine the creatine could serve as a “shuttle”, or facilitated diffusion, of ADP contributing to the dissipation of any metabolite gradients. This property of CPK has been well appreciated in the cytosol or even within the mitochondria membranes coupled to other transport mechanisms 62-67. Indeed, a compromise of the CPK activity and associated facilitated diffusion has been implicated as a contributing factor in heart failure 68. However, it is quite surprising that the near total knockout of the CPK enzyme isoforms in mice results in modest if no measurable cardiac phenotype 69-71. The cardiac mitochondrial enzyme contents and total cellular ΔGATP was essentially unchanged in the knockouts as well as the ability to perform work transitions in vitro and in vivo where essentially unaffected. This is particularly surprising in the mouse where the same pressure volume work is being done as a large animal at 10 times the rate, placing the heart in the “resting state” near its theoretic Vmax of ATP production 28. Thus, the strain on the ATP delivery system in the mouse at rest is equivalent to the ATP turnover of a sprinting dog5 and would predictably be highly susceptible to significant alterations in the ATP delivery system. The fact that even in skeletal muscle only burst performance is apparently affected in these CPK knockout mice along with aspects of Ca2+ handling and modest remodeling of the muscle structure might imply that the creatine kinase system is most critical in short burst activity, supporting the notion of a temporal buffer of the cytosolic potential energy. The impact on Ca2+ processing in these knockouts might reflect the high sensitivity to ΔGATP and rapid temporal responses required for Ca2+ sequestration discussed above.
There is another analogous finding related to a facilitated diffusion system for oxygen involving myoglobin 72. Again in the highly active mouse where oxygen delivery is close to Vmax conditions, the knockout of myoglobin has little or no effect on the normal function of the heart and is only associated with minor remodeling of the tissue73, 74. Thus, these two facilitated diffusion systems, one for ADP and the other for oxygen seem not to be required for “normal” function or survival but may be more involved in transient peak performance issues that do not impact survival of a mouse living in a laboratory cage. This peak performance may have significant evolutionary importance since large amounts of both CPK and myoglobin protein is present in the heart and it is unlikely that these proteins are a biochemical appendix. The modest remodeling of the hearts in these situations do imply some impact on the normal functions that will need further investigation in these very interesting animals.
How is the Potential Energy Homeostasis Maintained in the Steady State?
As discussed earlier the major source of ATP in the muscle is oxidative phosphorylation and if the heart is able to vary the production of ATP at near constant levels of ADP and Pi then the simplest solution is simply to have the Vmax of mitochondrial oxidative phosphorylation vary as a function of ATP hydrolysis. This would permit the generation of ATP at an appropriate rate without requirements for large changes in the reaction substrates ADP or Pi. Though this concept has been suggested for quite some time 12 the mechanisms associated with this balance must be quite complex since we now know that this homeostasis extends into the mitochondria matrix at the level of the NADH redox state, or primary driving force for ATP production, and the redox state of the cytochromes. This fact alone suggests that the control has to occur at multiple levels as suggested by numerous investigators over the years using control theory or more mechanistic models 24, 75-81. To maintain the NADH/NAD ratio, cytochrome c redox state and the phosphorylation potential during large changes in electron flow in the cytochrome chain and generation of ATP at the F1-FoATPase would require a coordinated activation of multiple steps of oxidative phosphorylation within the matrix. In addition, this process has to occur rapidly (<1s) since even transient changes NADH 41 or high energy phosphates, including creatine phosphate, are not detected during work jumps 16. Despite the recognition that there are multiple sites of control within the oxidative phosphorylation reaction sequence, very little insight has been provide on the actual mechanisms associated with this distributed control. Currently, there is only one viable candidate for orchestrating this complex process, cytosolic free Ca2+82.
Role of cytoplasmic Ca2+ in regulating the potential energy homeostasis
It has been appreciated for years that the release of Ca2+ is critical for the initiation and control of cardiac contraction. By Ca2+ controlling contractile activity ATPase activity and being the primary substrate for the transport ATPase's of the SR and plasma membranes (via Na/Ca exchange and Na-K-ATPase) it is clear that cytoplasmic Ca2+ is an excellent marker of work related ATPase activity 83-85. Indeed the analysis of Duncker and Bache6 suggest that over 80% of the work associated with exercise (i.e. heart rate and contractility) is under cytosolic Ca2+ control. In addition, as shown in Figure 2b, any mismatch between ATP production and utilization would decrease ΔGATP. This would result in less Ca2+ uptake also increasing diastolic Ca2+ and decreasing the peak systolic Ca2+ level. The decrease in peak systolic Ca2+ provides a negative feedback signal for muscle contraction slowing ATP hydrolysis, while the increased average cytosolic [Ca2+] would provide a signal of extensive ATP hydrolytic activity to stimulate mitochondrial ATP production. Thus, the cytosol [Ca2+] is an appropriate cytosolic signal to reflect the extent of ATP hydrolysis in both a feedforward and feedback model. That is, whether cytosolic [Ca2+] is high from inducing contractile activity (feedforward) or in response to a decrease in ΔGATP due to excessive ATP hydrolysis (feedback) the mitochondria is receiving a positive signal to increase ATP production.
Randle provided a classical enzyme regulation model by demonstrating that pyruvate dehydrogenase(PDH) 86 is activated by a Ca2+ dependent dephosphorylation in the matrix. These studies were followed by the extensive studies by Denton and McCormack 87 and others 88 that clearly established that Ca2+ could activate several dehydrogenase activities in the intact heart through a variety of mechanisms. These dehydrogenases included PDH, αketoglutaratedehydrogenase (KDH) and isocitrate dehydrogenase. This leaves only malate dehydrogenase and succinate dehydrogenase as the two sources of reducing equivalents not activated by Ca2+. Panov et al 89 suggested that the dominate effect of Ca2+ on NADH generation in heart mitochondria is the allosteric activation of KDH which is consistent with data from our laboratory.
The Ca2+ dependent dehydrogenase activation by work has been demonstrated in several intact cell systems. Kobayashi and Neely demonstrated an increase in PDH activity with work in the intact perfused heart90. This activation of PDH demonstrates that the alterations in matrix Ca2+ were sufficient to activate PDH in this simple work transition experiment. Similar results have been obtained in human skeletal muscle with exercise91. These results suggest that the alterations in cytoplasmic Ca2+ and the mitochondrial Ca2+ transport mechanisms are sufficient for this enzyme activation. This is likely the result of the compartmentalization of the Ca2+ release sites of the SR with mitochondria as proposed and demonstrated by Rizzuto and Pozzan et al55, 56, 92. The activation of the dehydrogenases via Ca2+ has been demonstrated in the intact heart or heart cells to modulate the NADH redox state using a variety of methods 41, 44, 45, 83, 84, 93. Most notable is the disruption of the potential energy homeostasis in the presence of mitochondria Ca2+ transport inhibitors such as ruthenium red94, 95. Thus, it seems plausible given these extensive results that the supply of NADH can be matched to the demand for reducing equivalents for ATP production by using the same molecule, Ca2, that activates ATP hydrolysis to enhance mitochondrial NADH generation, in parallel.
How does the F1-F0 ATPase increase ATP production during work transitions when the substrates, ADP and Pi, as well as the driving force, reflected in the NADH redox state and cytochrome redox state, is constant? The simplest manner to accomplish this task would be to essentially increase the Vmax, or decrease the effective “resistance”, of the F1-F0 ATPase permitting an increase in ATP production at a constant driving force.
Does Ca2+ modulate the F1-F0-ATPase? Das and Harris 96 found that Ca2+ activated the overall ATP hydrolytic activity in extracted F1-F0-ATPase from heart cells. This was reproduced in serial biopsy samples from dobutamine treated canine hearts, in vivo97. Thus, changes in cytosolic Ca2+ apparently generated a post-translational modification of the F1-F0-ATPase that persisted as an alteration in ATP hydrolytic activity, in vitro. It is important to note that the addition of Ca2+ directly to isolated F1-F0-ATPase does not affect activity, thus the effect is more than a direct association of Ca2+ with the enzyme98. Territo et al in a series of studies demonstrated that Ca2+ added to previously Ca2+ depleted cardiac mitochondria rapidly increased the velocity of ATP production by the F1-F0-ATPase at a given driving force(i.e. membrane potential)99-101. Thus, Ca is capable of increasing the capacity of ATP production by the F1-F0-ATPase at a constant driving force simultaneously with the increase delivery of reducing equivalents to the cytochrome chain via several Ca2+ sensitive dehydrogenases.
The dose dependence of the different actions of the Ca2+ on mitochondria and the SERCA pump was illustrated in experiments were the Ca2+ was added to mixtures of mitochondria and purified SR vesicles from the same heart 82. Ca2+was added to activate the dehydrogenases, F1-F0-ATPase and SERCA ATP hydrolytic activity to establish whether a potential energy homeostasis could be simulated in under these reconstituted conditions. Adding Ca2+ increased ATP turnover in this system, monitored as oxygen consumption, by activating SERCA. However, the increase in ATP generation was not associated with the classical decrease in NADH as electrons are withdrawn to support ATP production, the mitochondrial NADH was shown to be held constant or slightly increase (see Figure 4) with the addition of Ca2+. The addition of Ca2+ in this system was contrasted with the graded addition of an ATPase (Apyrase) directly to the suspension generation ADP and Pi alone. Not only did NADH become more oxidized with increasing ATP turnover, but the overall maximum rate of ATP production was reduced due to the lack of activation of the dehydrogenases and F1-F0ATPase by Ca2+. This rather simple reconstitution illustrates that extramitochondria Ca2+ driving the SERCA and activating oxidative phosphorylation can result in the required balanced activation to create the potential energy homeostasis during large increases in ATP production. In this reconstituted system a potential energy homeostasis was maintained over a 5 fold increase in ATP turnover.
Figure 4.

Effect of Ca2+ (closed symbols) or Apyrase (open symbols) additions on the oxygen consumption and NADH/NAD ratio of an isolated porcine heart mitochondria and sarcoplasmic reticulum (SR) vesicle suspension. The Ca2+ was varied from a nominal value of 0 to 492nm free Ca2+ (calculated). The SR to mitochondria protein content was 0.5 in the suspension. Data adapted from 136.
Taken together these data imply that Ca2+ may be significantly contributing to the balancing of ATP production capacity during increases in ATP utilization, triggered by Ca2+ in the heart cell.
What is still yet to be resolved is the molecular mechanism associated with the activation of the F1-F0-ATPase by Ca2+. Since this effect apparently persists in the purified protein extracted from mitochondria or cells activated with Ca2+ it is likely that some type of post-translational modification is responsible for this action.
Post-translational Modifications in Mitochondrial Energy Conversion Enzymes
One of the first examples of protein phosphorylation modifying enzymatic activity was the inhibitory phosphorylation of PDH, discussed earlier, that is modified by Ca2+ levels. Recently several screening studies have demonstrated that the dynamic phosphorylation of mitochondrial proteins may be much more extensive than previously appreciated 102-106.
Hopper et al 102 demonstrated that protein phosphorylation is extensive in the mitochondria matrix using a combination of protein phosphorylation dyes and 32P labeling. Protein phosphorylation sites within all of the complexes of oxidative phosphorylation have been identified that may provide a mechanism of controlling multiple sites of oxidative phosphorylation using protein phosphorylation alone. Several of the subunits of the F1-F0-ATPase were found to be phosphorylated in the matrix. Extramitochondrial Ca2+ was found to dephosphorylate the gamma subunit along with PDH and MnSOD. Possibly this dephosphorylation of the gamma subunit may be partially responsible for the modulation of F1-F0-ATPase activity by Ca2+. Others have also identified protein phosphorylation sites in the F1-F0-ATPase complex that could also contribute to activity modulation106, 107. The identification and functional significance of these protein phosphorylation sites in the F1-F0-ATPase are being actively studied in several laboratories around the world.
Another important issue with the growing evidence of an extensive protein phosphorylation network in the mitochondria matrix is the regulatory kinase/phosphatase system that is really quite poorly defined. Outside of a handful of kinases and phosphatases very few of these enzymes have been unequivocally located within the mitochondria matrix. The protein screens of the mitochondria reveal a very low number of kinases and phosphatases with uncertainty with regard to its location in the mitochondria or as contaminates from cytosolic structures102, 108-111. Thus, together with the analysis of the functional significance of these matrix protein phosphorylations, the associated kinases and phosphatases will likely be a significant subject of investigation over the next several years.
Another Ca2+ sensitive post-translational modification of the F1-F0-ATPase could be the association of regulatory peptides112. The F1 inhibitory protein is one of these regulatory proteins113 but its association with F1-F0-ATPase has not been linked to Ca2+ levels. Yamada et al 114, 115 described a protein that inhibits F1-ATPase activity in vitro that is reversed by μM Ca2+. This would provide a mechanism for Ca2+ to activate F1-ATPase activity by modulating this inhibitory protein activity. However, to my knowledge, no follow-up studies on this calcium sensitive inhibitory protein have been published. Boerries et al described a protein, S100A1, that has a Ca2+ dependent association with the F1-ATPase improving ATP production capacity 116. Interestingly, S100A1 over expression also facilitates the recovery from heart failure in model systems117 possibly linked to this activity in the mitochondrion. The role of this protein in normal work transitions in the heart has not been investigated.
Over the last several years numerous other post-translational modifications, in addition to protein phosphorylation, have been identified in the energy conversion network of the mitochondrion using a variety of screening methods. These include acylation118, 119, palmitoylation 120, nitrosylation121-123, oxidation122, 124, ADP ribosylation125, 126, and glycosylation 127. Many of these post-translational modifications occur in the complexes of oxidative phosphorylation most notable in Complex 1 and Complex V, however, the functional significance of these interactions is still under investigation. Many of these post-translational modifications have been evaluated under extreme conditions such as heart failure and diabetes, while very little work has been conducted on the normal work transitions of the heart, where the role of most of these modifications is unknown. Clearly, there are many more post-translational modifications within the matrix energy conversion machinery than was imagined even a few years ago, with more likely to be discovered in the near future, increasing the potential complexity of the energy regulatory network. However, given the complex outcome of the maintenance of the potential energy throughout the cell, an extensive control network is likely necessary.
Feedback Role of ADP and Pi
Despite the fact that the concentrations of ADP and Pi remain essentially constant over normal workloads in the heart, at very high workloads13, 15 or different clinical conditions128, the rate of ATP hydrolysis begins to exceed the ability of the control network to generate sufficient ATP without increasing the substrates ADP and Pi as well as reducing the ΔGATP to facilitate the rate of production. Another condition is when the potential energy generation capacity of the mitochondrion is externally influenced by the addition of substrates or hormones that induce a decrease in cytosolic ADP and Pi increasing the ΔGATP to “throttle” back the ATP production rate to match hydrolysis. This phenomenon has been observed with the addition of ketones in vivo129 and with numerous substrates in the perfused heart 14, 130-132. These high work and substrate studies clearly show that ADP and Pi exert control on the ATP production rate in both directions as the classical feedback model suggested. What are the sites of activation of ATP production by ADP and Pi? Pi is transported into the mitochondria primarily in exchange for hydroxyl while ADP enters via the electrogenic adenylate translocase. Thus, both dissipate to some extent the overall proton-motive force with entry. Though ADP has been demonstrated to regulate numerous enzymes associated with NADH generation in vitro, Bose et al 133 found no evidence of increased NADH generation with the addition of ADP in the absence of Pi (Figure 5). Indeed a decrease in NADH is observed with ADP consistent with the reduction in residual matrix Pi by ADP phosphorylation (see below). Thus, the influence of ADP seems to be limited as a substrate for the F1-F0-ATPase. This is a somewhat surprisingly limited effect of ADP for what many believe is a key metabolite in the regulation of mitochondria function.
Figure 5.
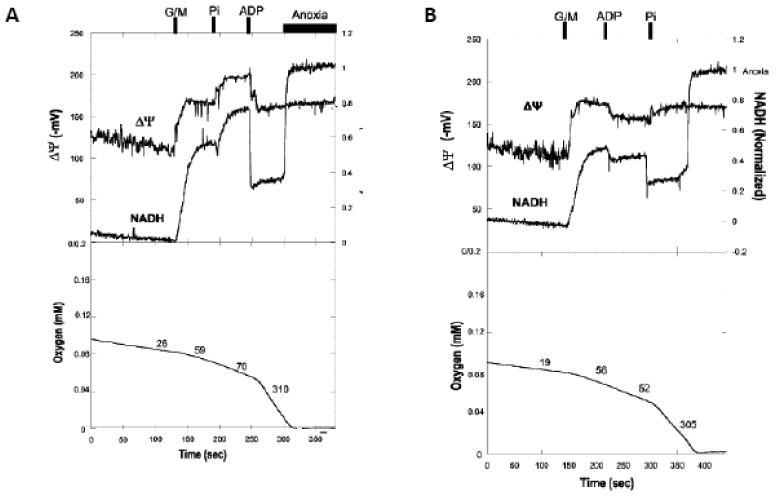
Effect of additions of Pi and ADP alone to State 4 porcine heart mitochondria. A) ADP added to State 4 mitochondria at the time indicated. The addition of Pi then initiated oxidative phosphorylation and associated oxygen consumption. B) Addition of Pi to State 4 mitochondria at the time indicated. Addition of ADP then initiated oxidative phosphorylation and associated oxygen consumption. Data is adapted from 133.
In contrast, the addition of Pi in the absence of ADP reveals a large increase in NADH generation and membrane potential (Figure 5A) as well as modifications to the redox equilibrium in the cytochrome chain limitations. The uncoupled respiratory rate is also dependent on Pi 133. The activation of electron transport by Pi is illustrated by the hyperpolarization of the mitochondrial membrane potential when oxidative phosphorylation is initiated by the addition of Pi after ADP (Figure 5B). Siess et al 134 noted a dependence of Kreb cycle function over physiological levels of phosphate in liver mitochondria with a cross-over point near succinate-CoA synthetase. Several other dehydrogenases have also been reported to be phosphate sensitive 133. Thus, it appears that Pi not only serves as a substrate for ATP synthesis but also stimulates the production of NADH in parallel resulting in an enhanced activation of ATP production capacity when compared to ADP alone.
The extensive nature of Pi's activation of oxidative phosphorylation is interesting when one considers which metabolites are likely to be changing during early mis-matches of ATP hydrolysis and production in the heart. As shown in Figure 3, in the presence of creatine kinase the changes in Pi are much earlier and larger than ADP in response to a mismatch of ATP production and utilization. Thus, the largest cytosolic error signal is Pi and not ADP. The regulatory network may be taking advantage of this fact by utilizing alterations in Pi to generate a balanced stimulation of oxidative phosphorylation, while the stimulatory sites available to ADP are limited. One issue with the notion of Pi playing a significant role in regulating oxidative phosphorylation is the apparent Km for these processes in isolated mitochondria (∼0.5 mM) is generally lower than the resting Pi levels in most tissues133. Thus, either the overall gain of the Pi process needs to be very high so that less than half of the activation curve is significant, or the kinetics of phosphate activation are much different in the intact cell. No information on the apparent affinity of oxidative phosphorylation for Pi is currently available in the intact cell.
A summary of the activation of ATP hydrolysis and oxidative phosphorylation by localized sarcoplasmic reticulum Ca2+ release is shown in Figure 6a. The only demonstrated Ca2+ activation sites are within the dehydrogenases and the F1-F0-ATPase. It is likely that other activation sites will be revealed in the future along with better molecular mechanisms associated with the system level activations demonstrated in the literature. I have not discussed the mitochondrial transport mechanisms since these were discussed in a previous review 27 in this journal. With a developing mis-match between ATP hydrolysis and synthesis, the earliest signal is predictably cytosolic Pi to supplement the effects of Ca2+ and to some extent ADP and creatine resulting from the creatine kinase reaction. The decrease in cytosolic ΔGATP will increase diastolic Ca2+ by inhibiting SERCA. The metabolic effects of Pi have been demonstrated to be even more distributed than Ca2+, activating NADH generation capacity, electron flux in the cytochrome chain, and a substrate for the F1-F0-ATPase as illustrated in Figure 6b. The specific interactions of Pi on these sites of oxidative phosphorylation are still under active investigation. These models suggest that both Pi and Ca2+ have the capacity to lead to an increase in energy conversion providing the potential energy required for ATP production under different physiological conditions. It is likely that this is still an oversimplification of this complex process that will be unraveled by future experimentation.
Figure 6.
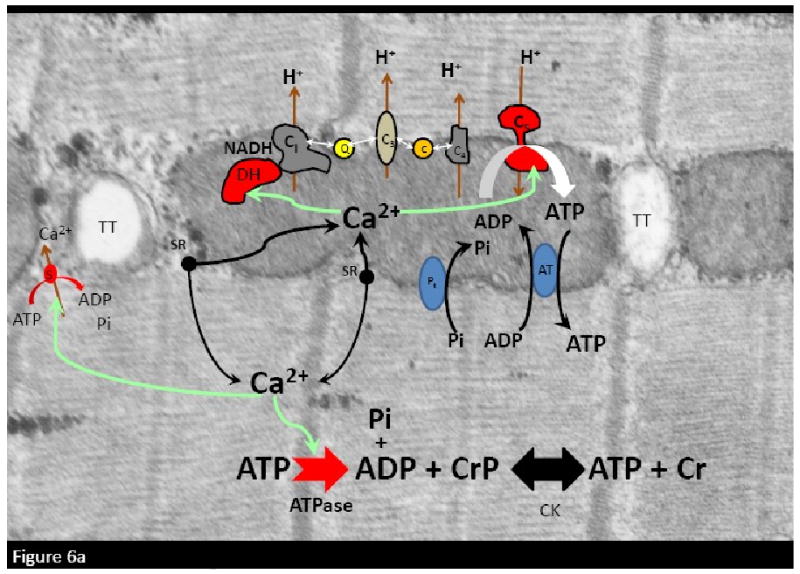

Schematic diagrams of the cellular energetic effects of Ca2+ and Pi in the heart cell. A) effect of sarcoplasmic reticulum (sr) release of Ca2+ on the ATP hydrolysis and oxidative phosphorylation. Activated sites are marked in red. Green lines indicate activation pathways. B) With a mismatch of ATP production with utilization Pi and creatine will increase. The metabolic effects of Pi are highlighted in green with green lined indicating the path. Schemes are depicted on a electron microgram of a porcine heart cell fixed in vivo. Legend: TT: T-Tubules. DH: Dehydrogenases. C1: Complex 1.Q: Co-enzyme Q. C: Cytochrome c. C3: Complex 3. C4; Complex 4; C5: Complex 5 (F1-F0-ATPase). CPK: Creatine Kinase. ATPase: primarily myosin ATPase. s: SERCA Calcium pump. Pt:phosphate transporter. AT: Adenylate kinase. CrP: creatine phosphate. Cr: Creatine. Pi: inorganic free phosphate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Proulx KL, Woodard SI, Dailey HA. In situ conversion of coproporphyrinogen to heme by murine mitochondria: terminal steps of the heme biosynthetic pathway. Protein Sci. 1993;2(7):1092–8. doi: 10.1002/pro.5560020703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39(3):257–88. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 3.Gutterman DD. Mitochondria and reactive oxygen species: an evolution in function. Circ Res. 2005;97(4):302–4. doi: 10.1161/01.RES.0000179773.18195.12. [DOI] [PubMed] [Google Scholar]
- 4.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 5.Khouri EM, Gregg DE, Rayford CR. Effect of excercise on cardiac output, left coronary flow and myocardial metabolism in the unanesthetized dog. Circ Res. 1965;17:427–37. doi: 10.1161/01.res.17.5.427. [DOI] [PubMed] [Google Scholar]
- 6.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88(3):1009–86. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 7.Devin A, Rigoulet M. Mechanisms of mitochondrial response to variations in energy demand in eukaryotic cells. Am J Physiol Cell Physiol. 2007;292(1):C52–C58. doi: 10.1152/ajpcell.00208.2006. [DOI] [PubMed] [Google Scholar]
- 8.Lardy HA, Wellman H. Oxidative phosphorylations: Role of inorganic phosphate as an acceptor systems in control of metabolic rate. J Biol Chem. 1952;185:215–24. [PubMed] [Google Scholar]
- 9.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 10.Whittam R. Active cation transport as a pace-maker of respiration. Nature. 1961;191:603–4. doi: 10.1038/191603a0. [DOI] [PubMed] [Google Scholar]
- 11.Berne RM. The role of adenosine in the regulation of coronary blood flow. Circ Res. 1980;47:807–13. doi: 10.1161/01.res.47.6.807. [DOI] [PubMed] [Google Scholar]
- 12.Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science. 1986;232:1121–3. doi: 10.1126/science.3704638. [DOI] [PubMed] [Google Scholar]
- 13.Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol. 1989;256:H265–H274. doi: 10.1152/ajpheart.1989.256.1.H265. [DOI] [PubMed] [Google Scholar]
- 14.From AHL, Petein MA, Michurski SP, Zimmer SD, Ugurbil K. 31P-NMR studies of respiratory regulation in the intact myocardium. FEBS Lett. 1986;206:257–61. doi: 10.1016/0014-5793(86)80992-9. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Duncker DJ, Xu Y, Zhang Y, Path G, Merkle H, et al. Transmural bioenergetic responses of normal myocardium to high workstates. Am J Physiol. 1995;268:H1891–H1911. doi: 10.1152/ajpheart.1995.268.5.H1891. [DOI] [PubMed] [Google Scholar]
- 16.Heineman FW, Balaban RS. Phosphorus-31 nuclear magnetic resonance analysis of transient changes of canine myocardial metabolism in vivo. J Clin Invest. 1990;85:843–52. doi: 10.1172/JCI114511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robitaille PM, Merkle H, Lew B, Path G, Hendrich K, Lindstrom P, et al. Transmural high energy phosphate distribution and response to alterations in workload in the normal canine myocardium as studied with spatially localized 31P NMR spectroscopy. Mag Res Med. 1990;16:91–116. doi: 10.1002/mrm.1910160110. [DOI] [PubMed] [Google Scholar]
- 18.Detre JA, Koretsky AP, Williams DS, Ho C. Absence of pH changes during altered work in the IN VIVO sheep heart: A 31P NMR investigation. J Mol Cell Cardiol. 1990;22:543–53. doi: 10.1016/0022-2828(90)90956-3. [DOI] [PubMed] [Google Scholar]
- 19.Koretsky AP, Wang S, Murphy-Boesch J, Klein J, James TL, Weiner MW. 31P NMR spectroscopy of rat organs in situ, using chronically implanted radiofrequency coils. Proc Natl Acad Sci USA. 1983;80:7491–5. doi: 10.1073/pnas.80.24.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaefer S, Schwartz GG, Steinman SK, Meyerhoff DJ, Massie BM, Weiner MW. Metabolic response of the human heart to inotropic stimulation: in vivo phosphorus-31 studies of normal and cardiomyopathic myocardium. Mag Res Med. 1992;25:260–72. doi: 10.1002/mrm.1910250205. [DOI] [PubMed] [Google Scholar]
- 21.Wollenberger A. Relation between work and labile phosphate content in the isolated dog heart. Circ Res. 1957;V:175–8. doi: 10.1161/01.res.5.2.175. [DOI] [PubMed] [Google Scholar]
- 22.Boerth RC, Covell JW, Seagren SC, Pool PE. High -energy phosphate concentrations in dog myocardium during stress. Am J Physiol. 1969;216(5):1103–6. doi: 10.1152/ajplegacy.1969.216.5.1103. [DOI] [PubMed] [Google Scholar]
- 23.Neely JR, Denton RM, England PJ, Randle PJ. The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem J. 1972;128:147–59. doi: 10.1042/bj1280147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou L, Huang H, Yuan CL, Keung W, Lopaschuk GD, Stanley WC. Metabolic response to an acute jump in cardiac workload: effects on malonyl-CoA, mechanical efficiency, and fatty acid oxidation. Am J Physiol Heart Circ Physiol. 2008;294(2):H954–H960. doi: 10.1152/ajpheart.00557.2007. [DOI] [PubMed] [Google Scholar]
- 25.Hochachka PW, McClelland GB. Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J Exp Biol. 1997;200(Pt 2):381–6. doi: 10.1242/jeb.200.2.381. [DOI] [PubMed] [Google Scholar]
- 26.Balaban RS. Regulation of oxidative phosphorylation in the mammalian cell. Am J Physiol. 1989;258:C377–C389. doi: 10.1152/ajpcell.1990.258.3.C377. [DOI] [PubMed] [Google Scholar]
- 27.Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol. 2002;34(10):1259. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 28.Balaban RS. Maintenance of the metabolic homeostasis of the heart: developing a systems analysis approach. Ann N Y Acad Sci. 2006;1080:140–53. doi: 10.1196/annals.1380.013. [DOI] [PubMed] [Google Scholar]
- 29.Hill AV. A challange to biochemists. Biochim Biophys Acta. 1950;4:4–11. doi: 10.1016/0006-3002(50)90003-5. [DOI] [PubMed] [Google Scholar]
- 30.Cain DF, Davies RE. Breakdown of adenosine triphosphate during a single contraction of working muscle. Biochem Biophys Res Commun. 1962;8:361–6. doi: 10.1016/0006-291x(62)90008-6. [DOI] [PubMed] [Google Scholar]
- 31.Giesen J, Kammermeier H. Relationship of Phosphorylation potential and oxygen consumption in isolated perfused rat hearts. Journ Molecular and Cellular Cardiology. 1980;12:891–907. doi: 10.1016/0022-2828(80)90058-9. [DOI] [PubMed] [Google Scholar]
- 32.Kentish JC, Allen DG. Is force production in the myocardium directly dependent upon the free energy change of ATP hydrolysis? J Mol Cell Cardiol. 1986;18(9):879–84. doi: 10.1016/s0022-2828(86)80001-3. [DOI] [PubMed] [Google Scholar]
- 33.Gibbs C. The cytoplasmic phosphorylation potential. Its possible role in the control of myocardial respiration and cardiac contractility. J Mol Cell Cardiol. 1985;17(8):727–31. doi: 10.1016/s0022-2828(85)80034-1. [DOI] [PubMed] [Google Scholar]
- 34.Fiolet JW. The interpretation of experimentally obtained values of the cytoplasmic phosphorylation potential. J Mol Cell Cardiol. 1988;20(12):1203–9. doi: 10.1016/0022-2828(88)90599-8. [DOI] [PubMed] [Google Scholar]
- 35.Fiolet JW, Baartscheer A. Cellular calcium homeostasis during ischemia; a thermodynamic approach. Cardiovasc Res. 2000;45(1):100–6. doi: 10.1016/s0008-6363(99)00294-1. [DOI] [PubMed] [Google Scholar]
- 36.Chen W, London R, Murphy E, Steenbergen C. Regulation of the Ca2+ gradient across the sarcoplasmic reticulum in perfused rabbit heart. A 19F nuclear magnetic resonance study. Circ Res. 1998;83(9):898–907. doi: 10.1161/01.res.83.9.898. [DOI] [PubMed] [Google Scholar]
- 37.Blinova K, Levine RL, Boja ES, Griffiths GL, Shi ZD, Ruddy B, et al. Mitochondrial NADH Fluorescence is Enhanced by Complex I Binding. Biochem. 2008;47(36):9636–9645. doi: 10.1021/bi800307y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chance B, Thorell B. Fluorescence measurements of mitochondrial pyridine nucleotide in aerobiosis and anaerobiosis. Nature. 1959;184:931–4. doi: 10.1038/184931a0. [DOI] [PubMed] [Google Scholar]
- 39.Chance B, Williamson JR, Famieson D, Schoener B. Properties and kinetics of reduced pyridine nucleotide fluorescence of the isolated and in vivo rat heart. Biochemische Zeitschrift. 1965;341:357–77. [Google Scholar]
- 40.Chance B, Williams CM. The respiratory chain and oxidative phosphorylation. Adv Enzymol. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 41.Heineman FW, Balaban RS. Effects of afterload and heart rate on NAD(P)H redox state in the isolated rabbit heart. Am J Physiol. 1993;264:H433–H440. doi: 10.1152/ajpheart.1993.264.2.H433. [DOI] [PubMed] [Google Scholar]
- 42.Wan B, Doumen C, Duszynski J, Salama G, Vary TC, LaNoue KF. Effect of cardiac work on electrical potential gradient across mitochondrial membrane in perfused rat hearts. Amer J Physiol. 1993;265:453–60. doi: 10.1152/ajpheart.1993.265.2.H453. [DOI] [PubMed] [Google Scholar]
- 43.Vuorinen K, Alarami A, Yan Y, Ingman P, Hassinen IE. Respiratory control in heart muscle during fatty acid oxidation: Energy-state or substrate-level regulation by Ca2+ J Mol Cell Cardiol. 1995;27(8):1581–91. doi: 10.1016/s0022-2828(95)90458-1. [DOI] [PubMed] [Google Scholar]
- 44.Brandes R, Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res. 1997;80(1):82–7. doi: 10.1161/01.res.80.1.82. [DOI] [PubMed] [Google Scholar]
- 45.Liu T, O'Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103(3):279–88. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashruf JF, Coremans JMCC, Bruining HA, Ince C. Increase of cardiac work is associated with decrease of mitochondrial NADH. Am J Physiol. 1995;269:H856–H862. doi: 10.1152/ajpheart.1995.269.3.H856. [DOI] [PubMed] [Google Scholar]
- 47.Jilkina O, Kuzio B, Grover GJ, Folmes CD, Kong HJ, Kupriyanov VV. Sarcolemmal and mitochondrial effects of a KATP opener, P-1075, in “polarized” and “depolarized” Langendorff-perfused rat hearts. Biochim Biophys Acta. 2003;1618(1):39–50. doi: 10.1016/j.bbamem.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 48.Knott EM, Ryou MG, Sun J, Heymann A, Sharma AB, Lei Y, et al. Pyruvate-fortified cardioplegia suppresses oxidative stress and enhances phosphorylation potential of arrested myocardium. Am J Physiol Heart Circ Physiol. 2005;289(3):H1123–H1130. doi: 10.1152/ajpheart.00322.2005. [DOI] [PubMed] [Google Scholar]
- 49.Knott EM, Sun J, Lei Y, Ryou MG, Olivencia-Yurvati AH, Mallet RT. Pyruvate mitigates oxidative stress during reperfusion of cardioplegia-arrested myocardium. Ann Thorac Surg. 2006;81(3):928–34. doi: 10.1016/j.athoracsur.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 50.Brachmanski M, Gebhard MM, Nobiling R. Separation of fluorescence signals from Ca2+ and NADH during cardioplegic arrest and cardiac ischemia. Cell Calcium. 2004;35(4):381–91. doi: 10.1016/j.ceca.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 51.Jobsis PD, Ashikaga H, Wen H, Rothstein EC, Horvath KA, McVeigh ER, et al. The visceral pericardium: macromolecular structure and contribution to passive mechanical properties of the left ventricle. Am J Physiol Heart Circ Physiol. 2007;293(6):H3379–H3387. doi: 10.1152/ajpheart.00967.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arai AE, Kasserra CE, Territo PR, Gandjbakhche AH, Balaban RS. Myocardial oxygenation in vivo: optical spectroscopy of cytoplasmic myoglobin and mitochondrial cytochromes. Am J Physiol. 1999;277(2 Pt 2):H683–H697. doi: 10.1152/ajpheart.1999.277.2.H683. [DOI] [PubMed] [Google Scholar]
- 53.Aliev MK, van Dorsten FA, Nederhoff MG, Van Echteld CJ, Veksler V, Nicolay K, et al. Mathematical model of compartmentalized energy transfer: its use for analysis and interpretation of 31P-NMR studies of isolated heart of creatine kinase deficient mice. Mol Cell Biochem. 1998;184(1-2):209–29. [PubMed] [Google Scholar]
- 54.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res. 2001;89(2):153–159. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- 55.Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994;126(5):1183–94. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hajnoczky G, Csordas G, Madesh M, Pacher P. The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J Physiol. 2000;529(Pt 1):69–81. doi: 10.1111/j.1469-7793.2000.00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothstein EC, Nauman M, Chesnick S, Balaban RS. Multi-photon excitation microscopy in intact animals. J Microsc. 2006;222(Pt 1):58–64. doi: 10.1111/j.1365-2818.2006.01570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Combs CA, Balaban RS. Direct imaging of dehydrogenase activity within living cells using enzyme-dependent fluorescence recovery after photobleaching (ED-FRAP) Biophys J. 2001;80(4):2018–28. doi: 10.1016/S0006-3495(01)76172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brune M, Hunter JL, Corrie JE, Webb MR. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994;33(27):8262–71. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 60.Funk C, Clark A, Connett RJ. How phosphocreatine buffers cyclic changes in ATP demand in working muscle. Adv Exp Med Biol. 1989;248:687–92. doi: 10.1007/978-1-4684-5643-1_76. [DOI] [PubMed] [Google Scholar]
- 61.Dzeja PP, Zeleznikar RJ, Goldberg ND. Adenylate kinase: kinetic behavior in intact cells indicates it is integral to multiple cellular processes. Mol Cell Biochem. 1998;184(1-2):169–82. [PubMed] [Google Scholar]
- 62.Bessman SP, Geiger PJ. Transport of energy in muscle: The phosphorylcreatine shuttle. Science. 1981;211:448–52. doi: 10.1126/science.6450446. [DOI] [PubMed] [Google Scholar]
- 63.Meyer RA, Sweeney HL, Kushmerick MJ. A simple analysis of the “Phosphocreatine Shuttle.”. Am J Physiol. 1984;242:1–11. doi: 10.1152/ajpcell.1984.246.5.C365. [DOI] [PubMed] [Google Scholar]
- 64.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(Pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallimann T, Dolder M, Schlattner U, Eder M, Hornemann T, Kraft T, et al. Creatine kinase: an enzyme with a central role in cellular energy metabolism. MAGMA. 1998;6(2-3):116–9. doi: 10.1007/BF02660927. [DOI] [PubMed] [Google Scholar]
- 66.Saks VA, Lipina NV, Smirnov VN, Chazov EI. Studies of energy transport in heart cells. The functional coupling between mitochondrial cretine phosphokinase and ATP-ADP translocase: Kinetic evidence. Ach Biochem Biophys. 1976;173:34–41. doi: 10.1016/0003-9861(76)90231-9. [DOI] [PubMed] [Google Scholar]
- 67.Saks VA, Vasil'eva E, Belikova YO, Kuznetsov AV, Lyapina S, Petrova L, et al. Retarded diffusion of ADP in cardiomyocytes: possible role of mitochondrial outer membrane and creatine kinase in cellular regulation of oxidative phosphorylation. Biochimica et Biophysica Acta. 1993;1144:134–48. doi: 10.1016/0005-2728(93)90166-d. [DOI] [PubMed] [Google Scholar]
- 68.Ingwall JS, Atkinson DE, Clarke K, Fetters JK. Energetic correlates of cardiac failure: changes in the creatine kinase system in the failing myocardium. Eur Heart J. 1990;11(Suppl B):108–15. doi: 10.1093/eurheartj/11.suppl_b.108. [DOI] [PubMed] [Google Scholar]
- 69.Ingwall JS. Creatine kinase knockout mice--what is the phenotype: heart. MAGMA. 1998;6(2-3):120–1. [PubMed] [Google Scholar]
- 70.Lygate CA, Hunyor I, Medway D, de Bono JP, Dawson D, Wallis J, et al. Cardiac phenotype of mitochondrial creatine kinase knockout mice is modified on a pure C57BL/6 genetic background. J Mol Cell Cardiol. 2009;46(1):93–9. doi: 10.1016/j.yjmcc.2008.09.710. [DOI] [PubMed] [Google Scholar]
- 71.Steeghs K, Benders A, Oerlemans F, de HA, Heerschap A, Ruitenbeek W, et al. Altered Ca2+ responses in muscles with combined mitochondrial and cytosolic creatine kinase deficiencies. Cell. 1997;89(1):93–103. doi: 10.1016/s0092-8674(00)80186-5. [DOI] [PubMed] [Google Scholar]
- 72.Wittenberg JB. Myoglobin-faclitiated oxygen diffusion:Role of myoglobin in oxygen entry into muscle. Physiol Rev. 1970;50(4):559–632. doi: 10.1152/physrev.1970.50.4.559. [DOI] [PubMed] [Google Scholar]
- 73.Garry DJ, Ordway GA, Lorenz JN, Radford NB, Chin ER, Grange RW, et al. Mice without myoglobin. Nature. 1998;395(6705):905–8. doi: 10.1038/27681. [DOI] [PubMed] [Google Scholar]
- 74.Liimatta EV, Godecke A, Schrader J, Hassinen IE. Regulation of cellular respiration in myoglobin-deficient mouse heart. Mol Cell Biochem. 2004;256-257(1-2):201–8. doi: 10.1023/b:mcbi.0000009887.35254.61. [DOI] [PubMed] [Google Scholar]
- 75.Groen AK, Wanders RJA, Westerhoff HV, van der Meer R, Tager JM. Quantification of the contribution of various steps to the control of mitochondrial respiration. J Biol Chem. 1982;257(6):2754–7. [PubMed] [Google Scholar]
- 76.Tager JM, Wanders RJ, Groen AK, Kunz W, Bohnensack R, Kuster U, et al. Control of mitochondrial respiration. FEBS Lett. 1983;151:1–9. doi: 10.1016/0014-5793(83)80330-5. [DOI] [PubMed] [Google Scholar]
- 77.Bohnensack R. Mathematical modeling of mitochondrial energy metabolism. Biochim Biophys Acta. 1985;44:113–25. [PubMed] [Google Scholar]
- 78.Brand MD, Vallis BP, Kesseler A. The sum of flux control coefficents in the electron-transport chain of mitochondria. Eur J Biochem. 1994;226:819–29. doi: 10.1111/j.1432-1033.1994.00819.x. [DOI] [PubMed] [Google Scholar]
- 79.Korzeniewski B, Brown GC. Quantification of the relative contribution of parallel pathways to signal transfer: application to cellular energy transduction. Biophys Chem. 1998;75(1):73–80. doi: 10.1016/s0301-4622(98)00193-8. [DOI] [PubMed] [Google Scholar]
- 80.Korzeniewski B. Theoretical studies on how ATP supply meets ATP demand. Biochem Soc Trans. 1999;27(2):271–6. doi: 10.1042/bst0270271. [DOI] [PubMed] [Google Scholar]
- 81.Zhou L, Cabrera ME, Huang H, Yuan CL, Monika DK, Sharma N, et al. Parallel activation of mitochondrial oxidative metabolism with increased cardiac energy expenditure is not dependent on fatty acid oxidation in pigs. J Physiol. 2007;579(Pt 3):811–21. doi: 10.1113/jphysiol.2006.123828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Balaban RS, Bose S, French SA, Territo PR. Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. American Journal of Physiology-Cell Physiology. 2003;284(2):C285–C293. doi: 10.1152/ajpcell.00129.2002. [DOI] [PubMed] [Google Scholar]
- 83.Fralix TA, Heineman FW, Balaban RS. Effect of work on intracellular calcium of the intact heart. Am J Physiol. 1991;261:54–9. doi: 10.1152/ajpheart.1991.261.4.54. [DOI] [PubMed] [Google Scholar]
- 84.Brandes R, Maier LS, Bers DM. Regulation of mitochondrial [NADH] by cytosolic [Ca2+] and work in trabeculae from hypertrophic and normal rat hearts. Circ Res. 1998;82(11):1189–98. doi: 10.1161/01.res.82.11.1189. [DOI] [PubMed] [Google Scholar]
- 85.Backx PH, Gao W, Asam-Backx MD, Marban E. The relationship between contractile force and intracellular [Ca2+] in intact cardiac trabeculae. J Gen Physiol. 1995;105:1–19. doi: 10.1085/jgp.105.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Randle PJ, Denton RM, Pask HT, Severson DL. Calcium ions and the regulation of pyruvate dehydrogenase. Biochem Soc Symp. 1974;39:75–87. [PubMed] [Google Scholar]
- 87.Denton RM, McCormack JG. The calcium sensitive dehydrogenases of vertebrate mitochondria. Cell Calcium. 1986;7:377–86. doi: 10.1016/0143-4160(86)90040-0. [DOI] [PubMed] [Google Scholar]
- 88.Hansford RG. Role of calcium in respiratory control. Med Sci Sports Exerc. 1994;26:44–51. [PubMed] [Google Scholar]
- 89.Panov AV, Scaduto RC., Jr Substrate specific effects of calcium on metabolism of rat heart mitochondria. Am J Physiol. 1996;270(4 Pt 2):H1398–H1406. doi: 10.1152/ajpheart.1996.270.4.H1398. [DOI] [PubMed] [Google Scholar]
- 90.Kobayashi K, Neely JR. Mechanism of pyruvate dehydrogenase activation by increased cardiac work. J Mol Cell Cardiol. 1983;15:369–82. doi: 10.1016/0022-2828(83)90321-8. [DOI] [PubMed] [Google Scholar]
- 91.Parolin ML, Chesley A, Matsos MP, Spriet LL, Jones NL, Heigenhauser GJ. Regulation of skeletal muscle glycogen phosphorylase and PDH during maximal intermittent exercise. Am J Physiol. 1999;277(5 Pt 1):E890–E900. doi: 10.1152/ajpendo.1999.277.5.E890. [DOI] [PubMed] [Google Scholar]
- 92.Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, et al. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci U S A. 1996;93(11):5489–94. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Di Lisa F, Gambassi G, Spurgeon H, Hansford RG. Intramitochondrial free calcium in cardiac myocytes in relation to dehydrogenase activation. Cardiovasc Res. 1993;27(10):1840–4. doi: 10.1093/cvr/27.10.1840. [DOI] [PubMed] [Google Scholar]
- 94.Unitt JF, McCormack JG, Reid D, Maclachlan LK. Direct evidence for a role of intramitochondrial Ca+2 in the regulation of oxidative phosphorylation in the stimulated rat heart. Biochem J. 1989;262:293–301. doi: 10.1042/bj2620293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Katz LA, Koretsky AP, Balaban RS. Activation of dehydrogenase activity and cardiac respiration: a 31P-NMR study. Amer J Physiol. 1988;255:185–8. doi: 10.1152/ajpheart.1988.255.1.H185. [DOI] [PubMed] [Google Scholar]
- 96.Harris DA. Regulation of the mitochondrial ATP synthase in rat heart. Biochem Soc Trans. 1993;21(Pt 3):778–81. doi: 10.1042/bst0210778. [DOI] [PubMed] [Google Scholar]
- 97.Scholz TD, Balaban RS. Mitochondrial F1-ATPase activity of canine myocardium: effects of hypoxia and stimulation. Am J Physiol. 1994;266:H2396–H2403. doi: 10.1152/ajpheart.1994.266.6.H2396. [DOI] [PubMed] [Google Scholar]
- 98.Hubbard MJ, McHugh NJ. Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Lett. 1996;391(3):323–9. doi: 10.1016/0014-5793(96)00767-3. [DOI] [PubMed] [Google Scholar]
- 99.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation:role of F0/F1ATPase. Am J Physiol. 2000;278(2):c423–c435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 100.Territo PR, French SA, Balaban RS. Simulation of cardiac work transitions, in vitro: effects of simultaneous Ca(2+)and ATPase additions on isolated porcine heart mitochondria. Cell Calcium. 2001;30(1):19–27. doi: 10.1054/ceca.2001.0211. [DOI] [PubMed] [Google Scholar]
- 101.Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, and light scattering. J Biol Chem. 2001;276(4):2586–99. doi: 10.1074/jbc.M002923200. [DOI] [PubMed] [Google Scholar]
- 102.Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, et al. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45(8):2524–36. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schulenberg B, Aggeler R, Beechem JM, Capaldi RA, Patton WF. Analysis of steady-state protein phosphorylation in mitochondria using a novel fluorescent phosphosensor dye. J Biol Chem. 2003;278(29):27251–5. doi: 10.1074/jbc.C300189200. [DOI] [PubMed] [Google Scholar]
- 104.Struglics A, Fredlund KM, Konstantinov YM, Allen JF, Moller IM. Protein phosphorylation/dephosphorylation in the inner membrane of potato tuber mitochondria. FEBS Letters. 2000;475(3):213–7. doi: 10.1016/s0014-5793(00)01680-x. [DOI] [PubMed] [Google Scholar]
- 105.Bykova NV, Egsgaard H, Moller IM. Identification of 14 new phosphoproteins involved in important plant mitochondrial processes. FEBS Lett. 2003;540(1-3):141–6. doi: 10.1016/s0014-5793(03)00250-3. [DOI] [PubMed] [Google Scholar]
- 106.Augereau O, Claverol S, Boudes N, Basurko MJ, Bonneu M, Rossignol R, et al. Identification of tyrosine-phosphorylated proteins of the mitochondrial oxidative phosphorylation machinery. Cell Mol Life Sci. 2005;62(13):1478–88. doi: 10.1007/s00018-005-5005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McDonald T, Sheng S, Stanley B, Chen D, Ko Y, Cole RN, et al. Expanding the subproteome of the inner mitochondria using protein separation technologies: one- and two-dimensional liquid chromatography and two-dimensional gel electrophoresis. Mol Cell Proteomics. 2006;5(12):2392–411. doi: 10.1074/mcp.T500036-MCP200. [DOI] [PubMed] [Google Scholar]
- 108.Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, et al. Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol. 2007;292(2):C689–C697. doi: 10.1152/ajpcell.00108.2006. [DOI] [PubMed] [Google Scholar]
- 109.Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, et al. Characterization of the human heart mitochondrial proteome. Nature Biotechnology. 2003;21(3):281–6. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 110.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115(5):629–40. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 111.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134(1):112–23. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harris DA, Das AM. Control of mitochondrial ATP synthesis in the heart. Biochem J. 1991;280(Pt 3):561–73. doi: 10.1042/bj2800561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cabezon E, Montgomery MG, Leslie AG, Walker JE. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat Struct Biol. 2003;10(9):744–50. doi: 10.1038/nsb966. [DOI] [PubMed] [Google Scholar]
- 114.Yamada EW, Huzel NJ. Calcium-binding ATPase inhibitor protein of bovine heart mitochondria. Role in ATP synthesis and effect of Ca2+ Biochemistry. 1989;28:9714–8. doi: 10.1021/bi00451a026. [DOI] [PubMed] [Google Scholar]
- 115.Yamada EW, Huzel NJ. The calcium binding ATPase inhibitor protein from bovine heart mitochondria. Purification and properties. J Biol Chem. 1988;263:11498–503. [PubMed] [Google Scholar]
- 116.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, et al. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27(12):4365–73. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004;114(11):1550–63. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stucki JW, Lehmann LH, Siegel E. Acylation of proteins by myristic acid in isolated mitochondria. J Biol Chem. 1989;264(11):6376–80. [PubMed] [Google Scholar]
- 119.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105(38):14447–52. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kostiuk MA, Corvi MM, Keller BO, Plummer G, Prescher JA, Hangauer MJ, et al. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. 2008;22(3):721–32. doi: 10.1096/fj.07-9199com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem. 2004;279(24):25891–7. doi: 10.1074/jbc.M313853200. [DOI] [PubMed] [Google Scholar]
- 122.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, et al. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44(5):1218–30. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 123.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101(11):1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 124.Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, rley-Usmar V, et al. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem. 2002;277(19):17048–56. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- 125.Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, et al. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104(6):1700–11. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- 126.Scovassi AI. Mitochondrial poly(ADP-ribosylation): from old data to new perspectives. FASEB J. 2004;18(13):1487–8. doi: 10.1096/fj.04-1841rev. [DOI] [PubMed] [Google Scholar]
- 127.Anello M, Spampinato D, Piro S, Purrello F, Rabuazzo AM. Glucosamine-induced alterations of mitochondrial function in pancreatic beta-cells: possible role of protein glycosylation. Am J Physiol Endocrinol Metab. 2004;287(4):E602–E608. doi: 10.1152/ajpendo.00320.2003. [DOI] [PubMed] [Google Scholar]
- 128.ten HM, Neubauer S. The application of NMR spectroscopy for the study of heart failure. Curr Pharm Des. 2008;14(18):1787–97. doi: 10.2174/138161208784746743. [DOI] [PubMed] [Google Scholar]
- 129.Kim DK, Heineman FW, Balaban RS. Effects of B-hydroxybutyrate on oxidative metabolism and phosphorylation potential in canine heart in vivo. Am J Physiol. 1991;260:H1767–H1773. doi: 10.1152/ajpheart.1991.260.6.H1767. [DOI] [PubMed] [Google Scholar]
- 130.Scholz TD, Laughlin MR, Balaban RS, Kupriyanov VV, Heineman FW. Effect of substrate on mitochondrial NADH, cytosolic redox state, and phosphorylated compounds in isolated hearts. Am J Physiol. 1995;268:H82–H91. doi: 10.1152/ajpheart.1995.268.1.H82. [DOI] [PubMed] [Google Scholar]
- 131.Matthews PM, Williams SR, Seymour AM, Schwartz A, Dube G, Gadian DG, et al. A 31P-NMR study of some metabolic and functional effects of the inotropic agents epinephrine and ouabain, and the ionophore R02- 2985 (X537A) in the isolated, perfused rat heart. Biochimica et Biophysica Acta. 1982;720:163–71. doi: 10.1016/0167-4889(82)90008-8. [DOI] [PubMed] [Google Scholar]
- 132.Zweier JL, Jacobus WE. Substrate-induced alterations of high energy phosphate metabolism and contractile function in the perfused heart. J Biol Chem. 1987;262:8015–21. [PubMed] [Google Scholar]
- 133.Bose S, French S, Evans FJ, Joubert F, Balaban RS. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J Biol Chem. 2003;278(40):39155–65. doi: 10.1074/jbc.M306409200. [DOI] [PubMed] [Google Scholar]
- 134.Siess EA, Kientsch-Engel RI, Fahimi FM, Wieland OH. Possible role of Pi supply in mitochondrial actions of glucagon. Eur J Biochem. 1984;141(3):543–8. doi: 10.1111/j.1432-1033.1984.tb08227.x. [DOI] [PubMed] [Google Scholar]
- 135.Katz LA, Swain JA, Portman MA, Balaban RS. Intracellular pH and inorganic phosphate content of the heart in vivo: A 31P NMR study. Am J Physiol. 1988;255:H189–H196. doi: 10.1152/ajpheart.1988.255.1.H189. [DOI] [PubMed] [Google Scholar]
- 136.Balaban RS, Bose S, French S, Territo PR. Work-related cytosolic signaling between cardiac sarcoplasmic reticulum and mitochondra: Role of Ca2+ Mol Biol Cell. 2001;12:2101. [Google Scholar]