

Abstract
The heart is capable of balancing the rate of mitochondrial ATP production with utilization continuously over a wide range of activity. This results in a constant phosphorylation potential despite a large change in metabolite turnover. The molecular mechanisms responsible for generating this energy homeostasis are poorly understood. The best candidate for a cytosolic signaling molecule reflecting ATP hydrolysis is Ca2+. Since Ca2+ initiates and powers muscle contraction as well as serves as the primary substrate for SERCA, Ca2+ is an ideal feed-forward signal for priming ATP production. With the sarcoplasmic reticulum to cytosolic Ca2+ gradient near equilibrium with the free energy of ATP, cytosolic Ca2+ release is exquisitely sensitive to the cellular energy state providing a feedback signal. Thus, Ca2+ can serve as a feed-forward and feedback regulator of ATP production. Consistent with this notion is the correlation of cytosolic and mitochondrial Ca2+ with work in numerous preparations as well as the localization of mitochondria near Ca2+ release sites. How cytosolic Ca2+ signaling might regulate oxidative phosphorylation is a focus of this review. The relevant Ca2+ sensitive sites include several dehydrogenases and substrate transporters together with a post-translational modification of F1-FO-ATPase and cytochrome oxidase. Thus, Ca2+ apparently activates both the generation of the mitochondrial membrane potential as well as utilization to produce ATP. This balanced activation extends the energy homeostasis observed in the cytosol into the mitochondria matrix in the never resting heart.
Keywords: Dehydrogenase, F1-FO-ATPase, Membrane Potential, Oxidative Phosphorylation, NADH, Oxygen Consumption, Sarcoplasmic Reticulum, Starling Effect, Energy Homeostasis, Aralar, Citrin
Introduction
The requirement to continuously pump blood to the body makes the heart one of the most active tissues in the body when integrated over any significant amount of time. This continuous work requirement results in an energy conversion system that is capable of supporting this activity with little or no mismatch between energy conversion and utilization, or the system will fail. To get some perspective in this process, the entire ATP pool in the canine heart is turned over in ∼1 minute at “rest” (∼5 μmol/gram ATP [127], respiratory rate ∼5μmol/gram/min[64]) and less than 10 sec at maximum workloads[64,76]. Thus, even a small mis-match between ATP production and utilization would rapidly lead to energy failure in the heart. This requirement likely generates a cytosolic energy conversion control network that is not only accurate, but has several parallel/backup systems to prevent a sustained mismatch between energy conversion and utilization for work.
The major source of cardiac energy conversion in the steady state is ATP generated by oxidative phosphorylation. This was appreciated many years ago when the rate of respiration was found to be linear with the level of work[76,118,139]. Glycolysis may play a role during burst activity[36,50], providing additional ATP during transition phases helping to maintain the energy homeostasis as well as provide pyruvate as a substrate for oxidative phosphorylation. However, in the steady state oxidative phosphorylation is clearly the major source of ATP that must be balanced with the rate of hydrolysis by the myosin ATPase for contraction and active ion transport in the sarcoplasmic reticulum used to control the contraction process.
In addition to maintaining the balance of ATP production with utilization the heart is also capable of supporting large changes in ATP turnover without altering the effective free energy of ATP hydrolysis (for recent review see[13]. That is, the hydrolysis products of ATP, ADP and Pi, are essentially held constant over most physiological workloads resulting in a near constant free energy of hydrolysis independent of the workload. Thus, the heart is able to balance the rate of ATP production with the rate of utilization while maintaining the energy generating capacity of ATP hydrolysis by not having [ATP] decrease or [ADP] and [Pi] increase during increases in workload [17,24,67,73,84,101,111,120,141]}. An example of this phenomenon is presented in Figure 1 in a canine heart in vivo[61]. In this example an NMR coil was placed directly on the heart and the 31P NMR spectra of ATP, creatine phosphate (CrP), and inorganic phosphate (Pi) were collected under controlled and paced up conditions. The pace up roughly increased the rate pressure product by a factor of 2. As seen in the different spectra between these conditions, no significant change in the high energy phosphates occurred despite the fact that the turnover of these molecules doubled under these conditions.
Figure 1.
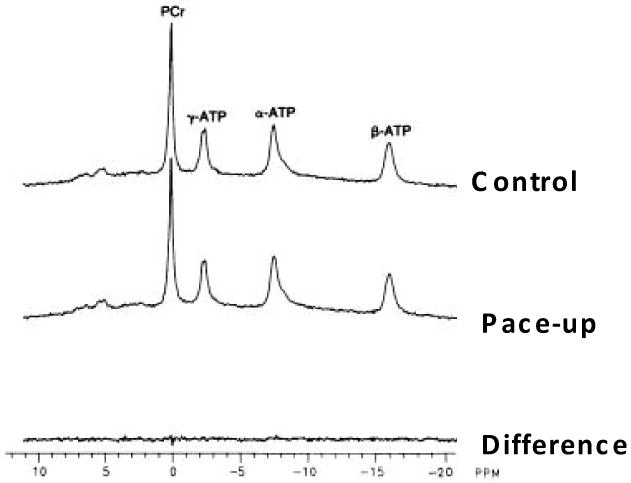
Effect of pacing workload on the 31P NMR detected high energy phosphates in the canine heart in vivo. The post spectrum was collected after a pacing that increased the rate pressure product by ∼2 fold. Data adapted from Heineman and Balaban [61]
What is the cytosolic control network that orchestrates the complex ATP hydrolysis at the filaments and sarcoplasmic reticulum (SR) with ATP production at the mitochondrion? The earliest model of the regulation of oxidative phosphorylation with work was a simple feedback of the ATP hydrolysis products ADP and Pi back to the mitochondrion[33,66]. That is, as ATP hydrolysis increases the concentration of ADP and Pi in the cytosol increases driving oxidative phosphorylation faster via simple enzyme kinetic driving forces. This has often been coupled to a facilitated diffusion or shuttle mechanism associated with the compartmentation of different isoforms of creatine kinase[20,21,117,137]. The feedback of ADP and Pi or creatine back to the mitochondrion clearly does play a role in the regulation of oxidative phosphorylation under a variety of different specialized conditions[13,14]. However, as discussed above (Figure 1), over the last two decades evidence has been building that the concentrations of ATP, ADP, Pi and CrP are maintained in the myocardium in the face of physiological challenges, in vivo. Thus, other cytoplasmic regulatory mechanisms have been looked at to supplement or modify the simple metabolite feedback mechanism of respiratory control. The two major, non-exclusive, models currently being investigated include a metabolite compartmentation model that relies on small pools of ADP at the interface of the mitochondria and cytosolic elements (for example see[1]). This is usually supplemented by the distribution of creatine kinase isoforms in the different compartments of the cell, relying on the diffusion of creatine in the free cytosol [21,137]. Compartmentation or “channeling” of ADP at the sarcoplasmic reticulum/mitochondria interface has also been proposed[5,71], much like demonstrated for Ca2+ signaling[92,114]. The second, again non-exclusive, model relies on a feed forward or parallel activation scheme using Ca2+ in many of the same compartments proposed for metabolite compartmentation. It is important to note that these models are not exclusive. That is it is likely that elements of both models, compartmentalized metabolic substrate feedback and Ca2+ feed-forward and feedback, are likely important under different conditions in the heart. The role of Ca2+ in the orchestration of oxidative phosphorylation with cytosolic ATP hydrolysis will be evaluated in this review. A special focus will be applied to the putative control sites for Ca2+ regulating oxidative phosphorylation in the mitochondrial matrix.
Ca2+ as a Feed-Forward and Feed Back Signaling Molecule for the rate of ATP hydrolysis
Ca2+ is very unique as a signaling molecule in that it can serve as a feed-forward or a feed-back signaling molecule reflecting the rate of ATP hydrolysis. A feed-forward signaling molecule for ATP hydrolysis provides information about this process either in parallel or before the change in rate occurs. Since Ca2+ is the primary signaling molecule driving ATP hydrolysis in muscle contraction and ion transport, it is ideally suited as a feed-forward signaling molecule for ATP hydrolysis. Duncker and Bache [45] recently reviewed the major sources of cardiac work with exercise as with regard to rate, contractility and ventricular work (Starling). These effects are summarized in a normalized fashion in Figure 2. The black bars represent the rate and contractility terms that are controlled by Ca2+ or reflected by the integrated level of cytoplasmic Ca2+. The contractility improvement associated with heart rate, treppe, is incorporated into the contractility term. Heart rate is the major driver of the overall metabolic load during exercise in the heart, supporting the earlier analysis of using pacing as a work transition in the 31P NMR analysis as presented in Figure 1[17,61]. From this analysis, 80 percent of the exercise work transition is clearly dependent on integrated increases in cytosolic Ca2+, supporting the notion that Ca2+ is an would be an excellent feed forward signal.
Figure 2.

Estimated relative contributions of heart rate, contractility and Starling relationship to the overall increase in myocardial oxygen consumption associated with exercise. Data adapted from [45].
The relatively small contribution of the Starling effect, or change in end-diastolic volume, to the cardiac work associated with exercise has been appreciated for years. Though the Starling phenomenon itself has been well characterized in vitro as an increase in Ca2+ sensitivity of the contractile apparatus with stretch [83] and is thus not dependent on an increase in cytosolic Ca2+, the large majority of studies in dogs [136] and man [35,62,102,112,125] find little or no change in end diastolic volume with exercise even using the latest MRI techniques. Thus, as suggested by Duncker and Bache, the Starling phenomenon apparently plays a small role in the overall stimulation of cardiac output with exercise.
The “grey” area represents the Starling component of the increase in work that is generally believed to be initially independent of cytosolic Ca2+ as described by Allen and Kurihara in the isolated rat and cat heart papillary muscles and trabeculae [3]. However, in a subsequent study in the ferret Allen et al found a strong correlation of fiber length with resting Ca2+ concluding: “We suggest that muscle length influences resting [Ca2+]i and this in turn affects the Ca2+ transients and developed tension.” [4]. A minority of subsequent studies have found an increase in resting Ca2+ with stretch[86] [82] using sensitive optical methods for detecting diastolic Ca2+ levels, while most only detect increases in the “slow response” to stretch[74]. The relationship between stretch and Ca2+ was reviewed by Calaghan and White [32]. As mentioned above, it is generally accepted that increases in muscle stretch enhances the sensitivity of the muscle fibers to Ca2+ [83]. Without a coordinated increase in Ca2+ sensitivity of ATP production with stretch an unraveling of the metabolic homeostasis would occur if it only depended on changes in cytosolic calcium. It is interesting to note that some increases in the end-diastolic volume occurs in canines near maximum exercise [136] where the metabolic homeostasis has been shown to breakdown[73] independent of oxygen tension [146]. Possibly, the Starling effect resulting from an increase in Ca2+ sensitivity of the myofilaments may be contributing to imbalance of ATP production with utilization at high workloads resulting in a requirement for the free energy of ATP to decrease. However, it is still unclear whether the slow component of Ca2+ increase seen with muscle stretch in most preparations could also influence the overall metabolic network. Thus, intracellular Ca2+ may also play a direct role in the smaller Starling component of exercise induced increases in cardiac work through the slow component of Ca2+ increase, a few beats after the change in stretch.
Feedback implies that the Ca2+ levels can increase after an increase in ATPase activity. The Ca2+ gradient generated by the sarcoplasmic reticulum Ca2+ pump (SERCA) is one of the largest potential energies in the cell[34] from 70% to 90% of the theoretical thermodynamic limits as defined by ΔGATP+nRTln([Ca2+]out/[Ca2+]in)[80,133] Being so close to its thermodynamic limits, it is not surprising the alterations in the ΔGATP due to a mismatch between ATP production and utilization either locally or across the entire cell would result in an increase in diastolic Ca2+ [48,133]. This sensing of the ΔGATP can also occur just in the region of the sarcoplasmic reticulum providing a compartmentalized signal of ATP depletion [71] if such gradients exist. Thus, under conditions where ΔGATP decreases with large increases in workload[73,145] or compromised substrate oxidation, cytoplasmic Ca2+ levels would predictably increase providing a signal that oxidative phosphorylation is not keeping up with ATP hydrolysis, a classical feedback indicator. This mechanism could be applicable to the whole cytosol or regional variations in ΔGATP at the SR.
Direct experimental evidence that cytosolic Ca2+ follows the work level of the heart has been provided by many investigators in the intact heart[27,28,49,81,87,128,142] and cells or trabeculae[27,28,51,94] [2,12,44]. An example of the correlation of intracellular Ca2+ and oxygen consumption in the perfused rat heart treated with different catecholamines is presented in Figure 3 adapted from Wu et al [142]. As predicted by the mechanisms of action of Ca2+, there is a tight correlation between oxygen consumption and free intracellular Ca2+ determined by fluorescent probes in the cytosol. Thus, there is significant evidence, from molecular mechanisms to whole heart and cellular measurements, that cytosolic [Ca2+] correlates with myocardial workload and would make an excellent element in a metabolic control network regulating energy conversion with utilization.
Figure 3.

Correlation of peak systolic Ca2+ levels and oxygen consumption in the perfused rat heart. Heart work was increased using a combination of inotropic agents. Data adapted from [142].
Are changes in cytosolic Ca2+ reflected in the mitochondrial matrix? This has been a controversial topic over the years since the kinetics for Ca2+ uptake and more importantly efflux were believed to be much to slow at physiological concentrations to account for reasonable tracking of matrix Ca2+ with cytosolic transients. Even if the matrix did not track transients in the cytosol, it is clear that the matrix could integrate both systolic and diastolic Ca2+ levels even without rapid transport systems[90]. However, recently evidence has been gaining that more rapid mitochondrial transport mechanisms are present. Gunter and Sheu[53] recently reviewed the potential fast mechanisms for Ca2+ import including the Ca2+ uniporter, RaM and a ryanodine sensitive transporter. However the export mechanism is still limited to rather slow Na/Ca2+ exchange and some poorly defined Na independent mechanisms. Again, a rapid export mechanism against the substantial electrochemical potential for Ca2+ is still one of the unsolved problems in this field along with the lack of definitive identification of the Ca2+ uniporter.
One of the major advances in this field was the realization that the systolic local Ca2+ concentration around the intrafibrillar mitochondria may be much higher than the average cytosolic concentration, resulting in a sufficient high Ca2+ concentration that the conventional uniporter kinetics could result in rapid Ca2+ mitochondrial uptake. In a clever set of experiments by Rizzuto and Pozzan [108,109] these local gradients in Ca2+ were visualized and correlated with mitochondrial Ca2+ levels as well as activation of PDH[114]. The local release of Ca2+ from the SR associated with the mitochondria in the intrafibrillar mitochondria is a reasonable mechanism for the observed close coupling of cytosolic and mitochondrial Ca2+ observed by numerous investigators using both genetically inserted as well as extrinsic optical probes[104,110,114,134]. However the compartmentation of Ca2+ at the mitochondria still does not resolve the mechanism for rapid Ca2+ efflux since it is unclear how a compartment could be used to enhance this process, significantly. Thus, the Ca2+ efflux pathway remains a nagging unresolved question.
Supporting the notion that mitochondrial [Ca2+] tracks the pacemaker cytosolic [Ca2+] are numerous less direct experiments demonstrating that the altering the mitochondrial transport of Ca2+ with ruthenium red and similar molecules severely alters the metabolic homeostasis associated with increases in cardiac work in the intact heart[72,96,135] and isolated preparations [91]. Furthermore, a surrogate marker of matrix Ca2+ is the activation of PDH (see below). Numerous studies have confirmed that an activation of PDH mirrors cardiac work [30,79,96] supporting the notion that the matrix is tracking cytosolic Ca2+ levels. Both of these classes of experiments support the hypothesis that matrix Ca+2 tracks cytosolic Ca2+ and is partially responsible for the metabolic homeostasis observed.
These data, direct observation of mitochondrial Ca2+ tracking cytosolic Ca2+ and the correlation with other surrogate markers of matrix Ca2, support the notion that the matrix Ca2+ tracks the pacemaker cytosolic Ca2+. However, the molecular mechanisms associated with this process are not fully described. The remainder of this discussion will focus on the effects of Ca2+ on oxidative phosphorylation.
Ca2+ modulation of oxidative phosphorylation
To regulate oxidative phosphorylation in a feed forward or feedback mode Ca2+ must have the appropriate sites within the mitochondria to properly alter the rate of ATP synthesis. The regulation of oxidative phosphorylation by Ca2+ is extremely complex and has been an area of study for many years. The different areas of interaction sites can be grouped as dehydrogenase activity, substrate transport, F1-FO-ATPase and cytochrome oxidase. Other mechanism associated with Ca2+ activation of liver mitochondria include modifications of volume and pyrophosphate levels (for review see[97]). However, neither of these mechanisms seem to play a role in the heart based on the work of Griffiths and Halestrap[52] and will not be discussed in this review.
Ca2+ Modulated Mitochondria Substrate Transport
Some aspects of substrate exchange and import into the mitochondria is regulated by Ca2+ regulated mitochondrial carriers (CaMC)[39,119]. These pathways are particularly interesting since cytosolic Ca2+ could alter mitochondrial metabolism without entering the matrix via the uniporter, which has been a controversial topic in past. Both citrin and aralar are present in the heart [38,68] with more aralar present in the atria than the ventricles. Aralar and citrin are aspartate/glutamate carriers involved in the malate-aspartate NADH shuttle across the inner membrane possibly capable of equilibrating the NAD/NADH ratio from the cytosol to the matrix across the inner membrane. The activation of these transporters occurs at physiological levels of Ca2+ on the order of 300 nM [38]. Based on these substrate transport mechanisms, and subsequent metabolism, the cytsolic Ca2+ activation citrin and aralar could result in the transfer of cytosolic reducing equivalents into the mitochondria matrix to increase the capacity for ATP production. However, the capacity of this pathway is limited, Williams et al estimated the maximum flux on the order of 3μmol NADH/min/gm wt[140], while the maximum exercise NADH consumption of the heart approaches 50 μmol NADH/min/gm wt [100]. In addition, no evidence of the cytosolic NADH/NAD ratio on the mitochondrial NADH/NAD ratio has been demonstrated in the heart[122]. Thus, the direct support of oxidative phosphorylation by this shuttle is likely minimal. However, the shuttle removing cytosolic NADH may improve the oxidation of lactate and glucose in the cytosol improving the delivery of pyruvate for oxidation[116] which could be very important in burst activity [37,50]. Thus, it is more likely that the activation of the NADH shuttle system by Ca2+ is likely related to keeping pyruvate entering the TCA cycle rather than being “wasted” to lactate via lactate dehydrogenase regeneration of NAD.
Based on the original work by Aprille et al [8] the ATP-Mg/Pi (APC) carrier is a method of modifying the net adenosine pool in the matrix. The ATP-Mg/Pi carrier is electroneutral exchanging divalent phosphate with divalent ATP and in some situations divalent ADP. This transport mechanism is sensitive to extra-mitochondrial Ca in the μM range increasing the affinity for ATP[60,103]. In the APC2 and AP3 isoforms of the carrier are present in the heart[47], but at a rather low activity and likely only plays a significant role in long term regulation of matrix adenosine pools[123].
Mitochondrial glycerol phosphate dehydrogenase (GPDH) is an inner membrane protein that responds to physiological levels of cytosolic Ca2+ by lowering its Km for reactants which increased the delivery of reducing equivalents to Coenzyme Q via FADH[29,46,93]. However the content [68,106] and activity[116] of this enzyme is very low in the heart. Thus, it is unlikely that this pathway significantly contributes as a source of reducing equivalents to oxidative phosphorylation in the heart.
Ca2+ Modulated Dehydrogenases
Since the early experiments in Randle's lab [42,107] on pyruvate dehydrogenase (PDH), it has been appreciated that the citric acid cycle dehydrogenases are extremely sensitive to Ca2+. Subsequently Ca2+ activation of isocitrate dehydrogenase (ICDH) and a- ketoglutarate dehydrogenase (KDH) was demonstrated[43,95,97] and reviewed by McCormack and Denton[97]. The mechanism of action of Ca2+ is surprisingly different for each of the dehydrogenase systems. PDH is deactivated by phosphorylation by PDH kinases and activated by the dephosphorylation via Ca2+ sensitive phosphatase [41,42,59,75]. The activation of PDH seems to be primarily an alteration in Vmax of the enzyme. In contrast to PDH, Ca2+ apparently binds directly to ICDH and KDH[115] resulting in alterations in the kinetics of both substrates and inhibitory metabolites[85]. In the heart it has been proposed that the KDH reaction is the most significant Ca2+ activated dehydrogenase under physiological conditions[138]. This is consistent with the observations by Hansford and Castro[55] as well as our experience since we find that the oxidation of α-ketoglutarate is nearly obligatorily linked to the presence of Ca2+ in porcine heart mitochondria. It is interesting that all three of these dehydrogenases are activated through different mechanisms suggesting that the origins of the activation are very different or the specific mechanisms required are very dissimilar.
Some evidence that Ca2+ may be involved in other oxidative pathways. Malstrom and Carafoli [77] found the oxidation of β-hydroxybutyrate, even under uncoupled conditions, was critically dependent on Ca2+, but no mechanism has been subsequently generated. This is particularly interesting since the ketones are the preferred substrate of the heart, in vivo [77]. Otto and Ontko [105] found an activation of fatty acid oxidation to β-hydroxybutyrate, not oxidation to CO2 by Kreb cycle, was doubled by the addition of Ca2+ to liver mitochondria. This was accompanied with an increase in mitochondrial NADH, again consistent with the increased oxidation of fatty acids. The specific mechanisms involved in these rather fascinating results are also still unknown.
No evidence is currently available on the role of Ca2+ at complex 1 where the reducing equivalents enter the cytochrome chain and is a potent source of reactive oxygen species[18]. Thus, it is apparent that the control of the delivery of NADH, via the dehydrogenase regulation, is the major mechanism of Ca2+ regulation. This is notion is consistent with the direct measurement of NADH formation on Complex 1 using the photo-oxidation of NADH to monitor the rate of re-reduction using ED-FRAP[70] relying on the assumption that most of the fluorescently enhanced matrix NADH is within Complex 1[22]. These studies reveal that the rate of reduction or binding of NADH to Complex 1 essentially matches the rate of electron flow through the cytochrome chain, implying this reaction is far from equilibrium and dependent on the generation of NADH by the dehydrogenases. The advantage of this approach is that electrons are not permitted into the ROS generating Complex 1, or the cytochrome chain, during periods of low flux, reducing the potential for damaging oxygen free radial generation[18]. These observation underscores the importance of the Ca2+ regulation of dehydrogenases in the regulation of oxidative phosphorylation as reviewed by Denton, McCormack, Hansford and Halestrap in several reviews[40,54,56,97].
It is also important to note that there is no evidence that Ca2+ alters the resistance or ability to convert the Complex 1NADH/NAD redox potential into membrane potential via the cytochrome chain as demonstrated for inorganic phosphate[26]. Even though a strong binding site for Ca2+ has been indentified in Complex 4, no functional correlation has been made[78]. Also Ca2+ has been shown not to alter the proton leak across the mitochondrial membrane at physiological levels[98]. These observations suggest the effects of Ca2+ are limited to the dehydrogenases and F1-FO-ATPase, but given the complexity of the interaction and difficulty of the experimental methods available, this author would not be surprised to see an even more distributed interaction of Ca2+ on oxidative phosphorylation with continuing discovery.
Since the mitochondria is essentially dependent on cytosolic sources of carbon substrates to oxidize in the form of pyruvate, fats, amino acids or ketones. The regulation the supply of these substrates from the cytosol by Ca2+ should not be ignored. One of the classical regulatory sites of Ca2+ in the cytosol is the activation of glycogen phosphorylase kinase that subsequent phosphorylates and activates glycogen phosphorylase increasing the delivery of glucose to glycolysis. In addition, Ca2+ has also been linked to GLUT-4 mobilization via a calmodulin dependent protein kinase and AMPK (for review see[113]). Like GLUT-4 the fatty acid transporter FAT/CD36 is also mobilized to the plasma membrane [25] and modulated by AMPK (for review see [69]) however, no direct link between FATCD/36 translocation and cytosolic Ca2+ has yet been made. Thus, Ca2+ activity in the cytosol is also linked to the transport and metabolism of metabolic substrates to support oxidative phosphorylation.
F1-FO-ATPase
The first suggestion that the F1-FO-ATPase may be directly affected by matrix Ca2+ came from the studies of Das and Harris [57] that demonstrated Ca2+ activated the ATP hydrolytic activity in extracted F1-FO-ATPase from pretreated heart cells. This was reproduced in serial biopsy samples from dobutamine treated canine hearts, in vivo[121]. Thus, changes in cytosolic Ca2+ apparently generated a post-translational modification of the F1-FO-ATPase that persisted as an alteration in ATP hydrolytic activity, in vitro. It is important to note that the addition of Ca2+ directly to isolated F1-FO-ATPase does not affect activity, thus the effect is more than a direct association of Ca2+ with the enzyme[65]. We have also confirmed that Ca2+ does not directly activate F1-FO-ATPase complex activity in blue native gels or from immune captured complexes from porcine heart mitochondria (Phillips and Balaban, unpublished data). Thus it is likely that Ca2+ regulates a post-translational process that occurs in the mitochondrial matrix in combination with others proteins or enzymes. What is the evidence that this post-translational modification that alters ATPase activity in vitro affects normal ATP production by this enzyme in the intact mitochondria? Territo et al in a series of studies[15,130-132] demonstrated that Ca2+ can rapidly increase the velocity of ATP production by the F1-FO-ATPase at a given driving force(i.e. membrane potential). Similar conclusions were reached by Mildazien using control theory analysis during Ca2+ additions [98]. These studies show that the addition of Ca2+ essentially reduced the resistance of the F1-FOATPase to form ATP using the membrane potential. Thus, Ca2+ is capable of increasing the capacity of ATP production by the F1-FO-ATPase at a given driving force simultaneously with the increase delivery of reducing equivalents to the cytochrome chain via the several Ca2+ sensitive dehydrogenases discussed above.
An example of this “balanced” activation by Ca2+ in intact mitochondria is given by experiments where Ca2+ was added to mixtures of mitochondria and purified SR vesicles from the same heart [16]. Ca2+ was added to activate the dehydrogenases, F1-FO-ATPase and SERCA ATP hydrolytic activity simultaneously to establish whether a potential energy homeostasis could be simulated in under these reconstituted conditions. Ca2+ additions increased ATP turnover by activating SERCA while the mitochondrial NADH and membrane potential was shown to be held constant or slightly increase with the addition of Ca2+. The addition of Ca2+ in this system was contrasted with the graded addition of a non-Ca2+ sensitive ATPase directly generation ADP and Pi alone. Not only did NADH become more oxidized and the membrane potential depolarize with increasing ATP turnover, but the overall maximum rate of ATP production was reduced due to the lack of activation of the dehydrogenases and F1-FO-ATPase by Ca2+. This rather simple reconstitution illustrates that extramitochondria Ca2+ driving the SERCA and activating dehydrogenases and the F1-FO-ATPase can result in the required balanced activation to create the potential energy homeostasis during large, 5 fold, increases in ATP production.
What is still yet to be resolved is the molecular mechanism associated with the activation of the F1-FO-ATPase by Ca2+. Since this effect apparently persists in the purified protein extracted from mitochondria or cells activated with Ca2+ it is likely that some type of post-translational modification is responsible for this action. There are several published post-translational modifications of the F1-FO-ATPase that might be responsible for the Ca2+ induced change. These include s-nitrosylation[126], phosphorylation[10,11,124], oxidation[99,129] and glycosylation[6]. Aponte et al[7]have recently demonstrated dynamic 32P association with several of the F1-FO-ATPase sub-units in the intact mitochondria including, α β, γ, OSCP and d. Hopper et al [63] showed that the γ subunit phosphorylation was sensitive to mitochondrial Ca2+ levels, however, a definitive link between γ subunit phosphorylation and the function of the enzyme has yet to be established.
Another post-translational modification of the F1-FO-ATPase is the association of regulatory peptides [58]. The F1 inhibitory protein is the classic example of a regulatory protein on the F1-FO-ATPase [31] but its association has not been linked to Ca2+ levels. Yamada et al [143,144] described a protein that associates with the F1-FO-ATPase that inhibits activity and that reversed by Ca2+. While Boerries et al described a protein, S100A1, that has a Ca2+ dependent association with the F1-FO-ATPase improving ATP production capacity [23]. The role of both of these proteins in normal work transitions in the heart has not been investigated, but both proteins in vitro seem to have the proper dynamic range and Ca2+ sensitivity to regulate the F1-FO-ATPase.
Cytochrome Oxidase
Bender and Kadenbach [19,88] demonstrated a Ca2+ dependent dephosphorylation of cytochrome oxidase (COX) in intact bovine heart mitochondria that they suggested removed the inhibition of ATP increasing net ATP production. This COX phosphorylation site was also shown to be cAMP dependent. Lee et al[89] demonstrated a cAMP dependent tyrosine phosphorylation site in liver COX subunit I that decreased the Vmax as well as the affinity for cytochrome c. In the original studies, Ca2+ only dephosphorylated COX in intact mitochondria and did not dephosphorylate purified samples. However, the Ca2+ doses used in these studies were excessive at 1 mM [19] and 100 μM[88] where the ATP content of the matrix could have been severely depleted via uncoupling and damage. Thus, some of the effects observed may be due to depleted ATP rather than specific effects of Ca2+. To my knowledge, a dose dependent Ca2+ study on COX dephosphorylation has not been conducted, thus it is difficult to establish its physiological role. Though a phosphorylation of COX has been detected in mitochondria phosphorylation screens [7,63]. A Ca2+ dependent dephosphorylation of COX was not detected [63], however this screening study did not focus on subunit I let alone COX. A Ca2+ activation of COX would provide another downstream activation of oxidative phosphorylation by cytoplasmic Ca2+ aiding to a balanced activation of ATP production. It would potentially help explain the redox homeostasis of the cytochrome chain through cytochrome c with alterations of work as observed by Arai et al [9]. Based on the important role of COX in oxidative phosphorylation further study on this interaction with Ca2+ is likely warranted.
Summary
Ca2+ is an excellent candidate as a feed-forward and feedback cytosolic signaling molecule for the rate of ATP hydrolysis. Indeed, the positioning of the mitochondria within the cytosol intertwined with the Ca2+ activation machinery may enhance the coupling of the cytosolic signals with the mitochondria. Within the mitochondria Ca2+ exerts a complex activation of mitochondrial oxidative phosphorylation by simultaneously activating several dehydrogenases and the F1-FO-ATPase. The six major Ca2+ sensitive sites in the mitochondria matrix are outlined in Figure 4. Note that most of the sites generating NADH are either directly or indirectly regulated by Ca2+, with the exception of MDH. The glutamate dehydrogenase (GDH) is indirectly regulated by the delivery of glutamate via citrin/Aralar. The mechanisms of activating the dehydrogenases are rather well defined while the precise molecular mechanisms involved in the activation of the F1-FO-ATPase are yet to be resolved. This balanced activation of oxidative phosphorylation by Ca2+, along with other regulatory mechanisms, likely plays a significant role in the balancing the rate of ATP production with utilization in the constantly working heart.
Figure 4.

Summary of the six Ca2+ regulatory sites in the heart mitochondria. The seven sites, outlined in white, are 1) F1-FO-ATPase, 2) APC, 3) Aralar/Citrin, 4) PDH, 5)ICDH, 6) αKDH and 7) COX. Of these sites COX is the least studied. Cit: Citrate. ICit: Isocitrate. OAA: Oxaloacetate. MDH Malate dehydrogenase. Succ: Succinate. ASP: Aspartate SCoA: Succinyl-CoA Fum: Fumarate. MAL: Malate. Other abbreviations are in the Text.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Aliev MK, van Dorsten FA, Nederhoff MG, Van Echteld CJ, Veksler V, Nicolay K, Saks VA. Mathematical model of compartmentalized energy transfer: its use for analysis and interpretation of 31P-NMR studies of isolated heart of creatine kinase deficient mice. Mol Cell Biochem. 1998;184:209. [PubMed] [Google Scholar]
- 2.Allen DG, Kurihara S. Calcium transients in mammalian ventricular muscle. Eur Heart J. 1980 A:5. doi: 10.1093/eurheartj/1.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- 3.Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982;327:79. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen DG, Nichols CG, Smith GL. The Effects of Changes in Muscle Length During Diastole on the Calcium Transient in Ferret Ventricular Muscle. J Physiol. 1988;406:359. doi: 10.1113/jphysiol.1988.sp017385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrienko T, Kuznetsov AV, Kaambre T, Usson Y, Orosco A, Appaix F, Tiivel T, Sikk P, Vendelin M, Margreiter R, Saks VA. Metabolic consequences of functional complexes of mitochondria, myofibrils and sarcoplasmic reticulum in muscle cells. J Exp Biol. 2003;206:2059. doi: 10.1242/jeb.00242. [DOI] [PubMed] [Google Scholar]
- 6.Anello M, Spampinato D, Piro S, Purrello F, Rabuazzo AM. Glucosamine-induced alterations of mitochondrial function in pancreatic beta-cells: possible role of protein glycosylation. Am J Physiol Endocrinol Metab. 2004;287:E602–E608. doi: 10.1152/ajpendo.00320.2003. [DOI] [PubMed] [Google Scholar]
- 7.Aponte AM, Phillips D, Hopper RK, Johnson DT, Harris RA, Blinova K, Boja ES, French S, Balaban RS. Use of 32P to Study Dynamics of the Mitochondrial Phosphoproteome. Journal of Proteomic Research. 2009 doi: 10.1021/pr800913j. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aprille JR. Mechanism and regulation of the mitochondrial ATP-Mg/P(i) carrier. J Bioenerg Biomembr. 1993;25:473. doi: 10.1007/BF01108404. [DOI] [PubMed] [Google Scholar]
- 9.Arai AE, Kasserra CE, Territo PR, Gandjbakhche AH, Balaban RS. Myocardial oxygenation in vivo: optical spectroscopy of cytoplasmic myoglobin and mitochondrial cytochromes. Am J Physiol. 1999;277:H683–H697. doi: 10.1152/ajpheart.1999.277.2.H683. [DOI] [PubMed] [Google Scholar]
- 10.Azarashvili TS, Tyynela J, Odinokova IV, Grigorjev PA, Baumann M, Evtodienko YV, Saris NE. Phosphorylation of a peptide related to subunit c of the F0F1-ATPase/ATP synthase and relationship to permeability transition pore opening in mitochondria. J Bioenerg Biomembr. 2002;34:279. doi: 10.1023/a:1020204518513. [DOI] [PubMed] [Google Scholar]
- 11.Azarashvily TS, Tyynela J, Baumann M, Evtodienko YV, Saris NE. Ca(2+)-modulated phosphorylation of a low-molecular-mass polypeptide in rat liver mitochondria: evidence that it is identical with subunit c of F(0)F(1)-ATPase. Biochem Biophys Res Commun. 2000;270:741. doi: 10.1006/bbrc.2000.2488. [DOI] [PubMed] [Google Scholar]
- 12.Backx PH, Gao W, Asam-Backx MD, Marban E. The relationship between contractile force and intracellular [Ca2+] in intact cardiac trabeculae. J Gen Physiol. 1995;105:1. doi: 10.1085/jgp.105.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaban RS. Domestication of the Cardiac Mitochondrion for Energy Conversion. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.02.018. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balaban RS. Maintenance of the metabolic homeostasis of the heart: developing a systems analysis approach. Ann N Y Acad Sci. 2006;1080:140. doi: 10.1196/annals.1380.013. [DOI] [PubMed] [Google Scholar]
- 15.Balaban RS, Bose S, French S, Territo PR. Work-related cytosolic signaling between cardiac sarcoplasmic reticulum and mitochondra: Role of Ca2+ Mol Biol Cell. 2001;12:2101. [Google Scholar]
- 16.Balaban RS, Bose S, French SA, Territo PR. Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. American Journal of Physiology-Cell Physiology. 2003;284:C285–C293. doi: 10.1152/ajpcell.00129.2002. [DOI] [PubMed] [Google Scholar]
- 17.Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science. 1986;232:1121. doi: 10.1126/science.3704638. [DOI] [PubMed] [Google Scholar]
- 18.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466:130. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- 20.Bessman SP, Fonyo A. The possible role of the mitochondrial bound creatine kinase in regulation of mitochondrial respiration. Biochem Biophys Res Commun. 1966;22:597. doi: 10.1016/0006-291x(66)90317-2. [DOI] [PubMed] [Google Scholar]
- 21.Bessman SP, Geiger PJ. Transport of energy in muscle: The phosphorylcreatine shuttle. Science. 1981;211:448. doi: 10.1126/science.6450446. [DOI] [PubMed] [Google Scholar]
- 22.Blinova K, Levine RL, Boja ES, Griffiths GL, Shi ZD, Ruddy B, Balaban RS. Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry. 2008;47:9636. doi: 10.1021/bi800307y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27:4365. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boerth RC, Covell JW, Seagren SC, Pool PE. High -energy phosphate concentrations in dog myocardium during stress. Am J Physiol. 1969;216:1103. doi: 10.1152/ajplegacy.1969.216.5.1103. [DOI] [PubMed] [Google Scholar]
- 25.Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- 26.Bose S, French S, Evans FJ, Joubert F, Balaban RS. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J Biol Chem. 2003;278:39155. doi: 10.1074/jbc.M306409200. [DOI] [PubMed] [Google Scholar]
- 27.Brandes R, Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res. 1997;80:82. doi: 10.1161/01.res.80.1.82. [DOI] [PubMed] [Google Scholar]
- 28.Brandes R, Maier LS, Bers DM. Regulation of mitochondrial [NADH] by cytosolic [Ca2+] and work in trabeculae from hypertrophic and normal rat hearts. Circ Res. 1998;82:1189. doi: 10.1161/01.res.82.11.1189. [DOI] [PubMed] [Google Scholar]
- 29.Brown LJ, MacDonald MJ, Lehn DA, Moran SM. Sequence of rat mitochondrial glycerol-3-phosphate dehydrogenase cDNA. Evidence for EF-hand calcium-binding domains. J Biol Chem. 1994;269:14363. [PubMed] [Google Scholar]
- 30.Bunger R, Permanetter B, Yaffe S. Energy-utilization and pyruvate as determinants of pyruvate-dehydrogenase in norepinephrine-stimulated heart. Pfluegers Arch. 1983;397:214. doi: 10.1007/BF00584360. [DOI] [PubMed] [Google Scholar]
- 31.Cabezon E, Montgomery MG, Leslie AG, Walker JE. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat Struct Biol. 2003;10:744. doi: 10.1038/nsb966. [DOI] [PubMed] [Google Scholar]
- 32.Calaghan SC, White E. The role of calcium in the response of cardiac muscle to stretch. Prog Biophys Mol Biol. 1999;71:59. doi: 10.1016/s0079-6107(98)00037-6. [DOI] [PubMed] [Google Scholar]
- 33.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383. [PubMed] [Google Scholar]
- 34.Chen W, London R, Murphy E, Steenbergen C. Regulation of the Ca2+ gradient across the sarcoplasmic reticulum in perfused rabbit heart. A 19F nuclear magnetic resonance study. Circ Res. 1998;83:898. doi: 10.1161/01.res.83.9.898. [DOI] [PubMed] [Google Scholar]
- 35.Christie J, Sheldahl LM, Tristani FE, Sagar KB, Ptacin MJ, Wann S. Determination of stroke volume and cardiac output during exercise: comparison of two-dimensional and Doppler echocardiography, Fick oximetry, and thermodilution. Circulation. 1987;76:539. doi: 10.1161/01.cir.76.3.539. [DOI] [PubMed] [Google Scholar]
- 36.Connett RJ. Why is there a delay in the increased oxygen consumption during the rest-work transition in skeletal muscle? Adv Exp Med Biol. 1986;194:271. doi: 10.1007/978-1-4684-5107-8_20. [DOI] [PubMed] [Google Scholar]
- 37.Connett RJ. Glycolytic regulation during an aerobic rest-to-work transition in dog gracilis muscle. J Appl Physiol. 1987;63:2366. doi: 10.1152/jappl.1987.63.6.2366. [DOI] [PubMed] [Google Scholar]
- 38.Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrustegui J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282:7098. doi: 10.1074/jbc.M610491200. [DOI] [PubMed] [Google Scholar]
- 39.del Arco A, Satrustegui J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem. 1998;273:23327. doi: 10.1074/jbc.273.36.23327. [DOI] [PubMed] [Google Scholar]
- 40.Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian imtochondria. FEBS Lett. 1980;119:1. doi: 10.1016/0014-5793(80)80986-0. [DOI] [PubMed] [Google Scholar]
- 41.Denton RM, McCormack JG, Rutter GA, Burnett P, Edgell NJ, Moule SK, Diggle TA. The hormonal regulation of pyruvate dehydrogenase complex. Adv Enzyme Regul. 1996;36:183–98. 183. doi: 10.1016/0065-2571(95)00020-8. [DOI] [PubMed] [Google Scholar]
- 42.Denton RM, Randle P, Martin BR. Stimulation by Ca2+ of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128:163. doi: 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176:899. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doeller JE, Wittenberg BA. Inracellular calcium and high-energy phosphates in isolated cardiac myocytes. Am J Physiol. 1990;259:H1851–H1859. doi: 10.1152/ajpheart.1990.259.6.H1851. [DOI] [PubMed] [Google Scholar]
- 45.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 46.Estabrook RW, Sacktor B. alpha-Glycerophosphate oxidase of flight muscle mitochondria. J Biol Chem. 1958;233:1014. [PubMed] [Google Scholar]
- 47.Fiermonte G, De LF, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279:30722. doi: 10.1074/jbc.M400445200. [DOI] [PubMed] [Google Scholar]
- 48.Fiolet JW, Baartscheer A. Cellular calcium homeostasis during ischemia; a thermodynamic approach. Cardiovasc Res. 2000;45:100. doi: 10.1016/s0008-6363(99)00294-1. [DOI] [PubMed] [Google Scholar]
- 49.Fralix TA, Heineman FW, Balaban RS. Effect of work on intracellular calcium of the intact heart. Am J Physiol. 1991;261:54. doi: 10.1152/ajpheart.1991.261.4.54. [DOI] [PubMed] [Google Scholar]
- 50.Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. Journal of Biological Chemistry. 1998;273:29530. doi: 10.1074/jbc.273.45.29530. [DOI] [PubMed] [Google Scholar]
- 51.Griffiths EJ. Species dependence of mitochondrial calcium transients during excitation-contraction coupling in isolated cardiomyocytes. Biochem Biophys Res Commun. 1999;263:554. doi: 10.1006/bbrc.1999.1311. [DOI] [PubMed] [Google Scholar]
- 52.Griffiths EJ, Halestrap AP. Pyrophosphate metabolism in the perfused heart and isolated heart mitochondria and its role in regulation of mitochondrial function by calcium. Biochem J. 1993;290:489. doi: 10.1042/bj2900489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbabio.2008.12.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansford RG. Role of calcium in respiratory control. Med Sci Sports Exerc. 1994;26:44. [PubMed] [Google Scholar]
- 55.Hansford RG, Castro F. Effects of micromolar concentrations of free calcium ions on the reduction of heart mitochondrial NAD(P) by 2-oxoglutarate. Biochem J. 1981;198:525. doi: 10.1042/bj1980525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem. 1998;184:359. [PubMed] [Google Scholar]
- 57.Harris DA. Regulation of the mitochondrial ATP synthase in rat heart. Biochem Soc Trans. 1993;21:778. doi: 10.1042/bst0210778. [DOI] [PubMed] [Google Scholar]
- 58.Harris DA, Das AM. Control of mitochondrial ATP synthesis in the heart. Biochem J. 1991;280(Pt 3):561. doi: 10.1042/bj2800561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris RA, Popov KM, Zhao Y, Kedishvili NY, Shimomura Y, Crabb DW. A new family of protein kinases--the mitochondrial protein kinases. Adv Enzyme Regul. 1995;35:147. doi: 10.1016/0065-2571(94)00020-4. [DOI] [PubMed] [Google Scholar]
- 60.Haynes RC, Jr, Picking RA, Zaks WJ. Control of mitochondrial content of adenine nucleotides by submicromolar calcium concentrations and its relationship to hormonal effects. J Biol Chem. 1986;261:16121. [PubMed] [Google Scholar]
- 61.Heineman FW, Balaban RS. Phosphorus-31 nuclear magnetic resonance analysis of transient changes of canine myocardial metabolism in vivo. J Clin Invest. 1990;85:843. doi: 10.1172/JCI114511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higginbotham MB, Morris KG, Williams RS, McHale pA, Coleman RE, Cobb FR. Regulation of stroke volume during submaximal and maximal upright exercise in normal man. Circ Res. 1986;58:281. doi: 10.1161/01.res.58.2.281. [DOI] [PubMed] [Google Scholar]
- 63.Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45:2524. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang AH, Feigl EO. Adrenergic coronary vascoconstriction helps maintain uniform transmural blood flow distribution during exercise. Circ Res. 1988;62:286. doi: 10.1161/01.res.62.2.286. [DOI] [PubMed] [Google Scholar]
- 65.Hubbard MJ, McHugh NJ. Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Lett. 1996;391:323. doi: 10.1016/0014-5793(96)00767-3. [DOI] [PubMed] [Google Scholar]
- 66.Jacobus WE, Moreadity RW, Vandegaer KM. Mitochondrial respiratory control: Evidence against the regulation of respiration by extramitochondrial phosphorylation potentials or by ATP/ADP ratios. J Biol Chem. 1982;257:2397. [PubMed] [Google Scholar]
- 67.Jeffrey FMH, Malloy CR. Respiratory control substrate effects in the working rat heart. Biochem J. 1992;287:117. doi: 10.1042/bj2870117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, Wang M, Balaban RS. Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol. 2007;292:C689–C697. doi: 10.1152/ajpcell.00108.2006. [DOI] [PubMed] [Google Scholar]
- 69.Jorgensen SB, Richter EA, Wojtaszewski JF. Role of AMPK in skeletal muscle metabolic regulation and adaptation in relation to exercise. J Physiol. 2006;574:17. doi: 10.1113/jphysiol.2006.109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joubert F, Fales HM, Wen H, Combs CA, Balaban RS. NADH enzyme-dependent fluorescence recovery after photobleaching (ED-FRAP): applications to enzyme and mitochondrial reaction kinetics, in vitro. Biophys J. 2004;86:629. doi: 10.1016/S0006-3495(04)74141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res. 2001;89:153. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- 72.Katz LA, Koretsky AP, Balaban RS. Activation of dehydrogenase activity and cardiac respiration: a 31P-NMR study. Amer J Physiol. 1988;255:185. doi: 10.1152/ajpheart.1988.255.1.H185. [DOI] [PubMed] [Google Scholar]
- 73.Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol. 1989;256:H265–H274. doi: 10.1152/ajpheart.1989.256.1.H265. [DOI] [PubMed] [Google Scholar]
- 74.Kentish JC, Wrzosek A. Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J Physiol. 1998;506(Pt 2):431. doi: 10.1111/j.1469-7793.1998.431bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kerbey AL, Randle PJ, Cooper RH, Whitehouse S, Pask HT, Denton RM. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem J. 1976;154:327. doi: 10.1042/bj1540327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khouri EM, Gregg DE, Rayford CR. Effect of excercise on cardiac output, left coronary flow and myocardial metabolism in the unanesthetized dog. Circ Res. 1965;17:427. doi: 10.1161/01.res.17.5.427. [DOI] [PubMed] [Google Scholar]
- 77.Kim DK, Heineman FW, Balaban RS. Effects of B-hydroxybutyrate on oxidative metabolism and phosphorylation potential in canine heart in vivo. Am J Physiol. 1991;260:H1767–H1773. doi: 10.1152/ajpheart.1991.260.6.H1767. [DOI] [PubMed] [Google Scholar]
- 78.Kirichenko A, pfitzner U, Ludwig B, Soares CM, Vygodina TV, Konstantinov A. Cytochrome c Oxidase as a Calcium Binding Protein. Studies on the Role of a Conserved Aspartate in Helices XI-XII Cytoplasmic Loop in Cation Binding. Biochemistry. 2005;44:12391. doi: 10.1021/bi050376v. [DOI] [PubMed] [Google Scholar]
- 79.Kobayashi K, Neely JR. Mechanism of pyruvate dehydrogenase activation by increased cardiac work. J Mol Cell Cardiol. 1983;15:369. doi: 10.1016/0022-2828(83)90321-8. [DOI] [PubMed] [Google Scholar]
- 80.Kodama T. Thermodynamic analysis of muscle ATPase mechanisms. Physiol Rev. 1985;65:467. doi: 10.1152/physrev.1985.65.2.467. [DOI] [PubMed] [Google Scholar]
- 81.Kojima S, Wu ST, Parmley WW, Wikman-Coffelt J. Relationship between intracellular calcium and oxygen consumption: effects of perfusion pressure, extracellular calcium, dobutamine, and nifedipine. Am Heart J. 1994;127:386. doi: 10.1016/0002-8703(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 82.Kojima S, Wu ST, Watters TA, Parmley WW, Wikman-Coffelt J. Effects of perfusion pressure on intracellular calcium, energetics, and function in perfused rat hearts. Am J Physiol. 1993;264:H183–H189. doi: 10.1152/ajpheart.1993.264.1.H183. [DOI] [PubMed] [Google Scholar]
- 83.Konhilas JP, Irving TC, de Tombe PP. Frank-Starling law of the heart and the cellular mechanisms of length-dependent activation. Pflugers Arch. 2002;445:305. doi: 10.1007/s00424-002-0902-1. [DOI] [PubMed] [Google Scholar]
- 84.Koretsky AP, Wang S, Murphy-Boesch J, Klein J, James TL, Weiner MW. 31P NMR spectroscopy of rat organs in situ, using chronically implanted radiofrequency coils. Proc Natl Acad Sci USA. 1983;80:7491. doi: 10.1073/pnas.80.24.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lawlis VB, Roche TE. Effect of micromolar Ca2+ on NADH inhibition of bovine kidney alpha ketoglurarate dehydrogenase complex and possible role of Ca2+ in signal amplification. Mol Cell Biochem. 1980;32:147. doi: 10.1007/BF00227441. [DOI] [PubMed] [Google Scholar]
- 86.Le Guennec JY, White E, Gannier F, Argibay JA, Garnier D. Stretch-induced increase of resting intracellular calcium concentration in single guinea-pig ventricular myocytes. Exp Physiol. 1991;76:975. doi: 10.1113/expphysiol.1991.sp003560. [DOI] [PubMed] [Google Scholar]
- 87.Lee HC, Smith N, Mohabir R, Clusin WT. Cytosolic calcium transients from the beating mammalian heart. Proc Natl Acad Sci. 1987;84:7793. doi: 10.1073/pnas.84.21.7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee I, Bender E, Kadenbach B. Control of mitochondrial membrane potential and ROS formation by reversible phosphorylation of cytochrome c oxidase. Mol Cell Biochem. 2002;234-235:63. [PubMed] [Google Scholar]
- 89.Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Huttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- 90.Leisey JR, Grotyohann LW, Scott DA, Scaduto RC., Jr Regulation of cardiac mitochondrial calcium by average extramitochondrial calcium. Am J Physiol. 1993;265:H1203–H1208. doi: 10.1152/ajpheart.1993.265.4.H1203. [DOI] [PubMed] [Google Scholar]
- 91.Liu T, O'Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maack C, O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.MacDonald MJ, Brown LJ. Calcium activation of mitochondrial glycerol phosphate dehydrogenase restudied. Arch Biochem Biophys. 1996;326:79. doi: 10.1006/abbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- 94.Martin BJ, Valdivia HH, Bunger R, Lasley RD, Mentzer RM., Jr Pyruvate augments calcium transients and cell shortening in rat ventricular myocytes. Am J Physiol. 1998;274:H8. doi: 10.1152/ajpheart.1998.274.1.H8. [DOI] [PubMed] [Google Scholar]
- 95.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McCormack JG, England PJ. Ruthenium Red inhibits the action of pyruvate dehydrogenase caused by positive inotropic agents in the perfused rat heart. Biochem J. 1983;214:581. doi: 10.1042/bj2140581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 98.Mildazien V, Baniene R, Nauciene Z, Marcinkeviciute A, Morkuniene R, Borutaite V, Kholodenko B, Brown GC. Ca2+ stimulates both the respiratory and phosphorylation subsystems in rat heart mitochondria. Biochem J. 1996;320:329. doi: 10.1042/bj3200329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 100.Mootha VK, Arai AE, Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol. 1997;272:H769. doi: 10.1152/ajpheart.1997.272.2.H769. [DOI] [PubMed] [Google Scholar]
- 101.Neely JR, Denton RM, England PJ, Randle PJ. The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem J. 1972;128:147. doi: 10.1042/bj1280147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nobrega AC, Williamson JW, Mitchell JH. Left ventricular volumes and hemodynamic responses at onset of dynamic exercise with reduced venous return. J Appl Physiol. 1995;79:1405. doi: 10.1152/jappl.1995.79.5.1405. [DOI] [PubMed] [Google Scholar]
- 103.Nosek MT, Dransfield DT, Aprille JR. Calcium stimulates ATP-Mg/Pi carrier activity in rat liver mitochondria. J Biol Chem. 1990;265:8444. [PubMed] [Google Scholar]
- 104.Ohata H, Chacon E, Tesfai SA, Harper IS, Herman B, Lemasters JJ. Mitochondrial Ca2+ transients in cardiac myocytes during the excitation-contraction cycle: effects of pacing and hormonal stimulation. J Bioenerg Biomembr. 1998;30:207. doi: 10.1023/a:1020588618496. [DOI] [PubMed] [Google Scholar]
- 105.Otto DA, Ontko JA. Activation of mitochondrial fatty acid oxidation by calcium. Conversion to the energized state. J Biol Chem. 1978;253:789. [PubMed] [Google Scholar]
- 106.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Randle PJ, Denton RM, Pask HT, Severson DL. Calcium ions and the regulation of pyruvate dehydrogenase. Biochem Soc Symp. 1974;39:75. [PubMed] [Google Scholar]
- 108.Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994;126:1183. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004 doi: 10.1126/stke.2152004re1. [DOI] [PubMed] [Google Scholar]
- 110.Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20:4998. doi: 10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Robitaille PM, Merkle H, Lew B, Path G, Hendrich K, Lindstrom P, From AHL, Garwood M, Bache RJ, Ugurbil K. Transmural high energy phosphate distribution and response to alterations in workload in the normal canine myocardium as studied with spatially localized 31P NMR spectroscopy. Mag Res Med. 1990;16:91. doi: 10.1002/mrm.1910160110. [DOI] [PubMed] [Google Scholar]
- 112.Roest AA, Lamb HJ, van der Wall EE, Vliegen HW, van den Aardweg JG, Kunz P, de RA, Helbing WA. Cardiovascular response to physical exercise in adult patients after atrial correction for transposition of the great arteries assessed with magnetic resonance imaging. Heart. 2004;90:678. doi: 10.1136/hrt.2003.023499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology (Bethesda) 2005;20:260. doi: 10.1152/physiol.00012.2005. [DOI] [PubMed] [Google Scholar]
- 114.Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavare JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci U S A. 1996;93:5489. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rutter GA, Denton RM. The binding of Ca2+ ions to pig heart NAD+-isocitrate dehydrogenase and the 2-oxoglutarate dehydrogenase complex. Biochem J. 1989;263:453. doi: 10.1042/bj2630453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Safer B, Smith CM, Williamson JR. Control of the transport of reducing equivalents across the mitochondrial membrane in perfused rat heart. J Mol Cell Cardiol. 1971;2:111. doi: 10.1016/0022-2828(71)90065-4. [DOI] [PubMed] [Google Scholar]
- 117.Saks VA, Kongas O, Vendelin M, Kay L. Role of the creatine/phosphocreatine system in the regulation of mitochondrial respiration. Acta Physiol Scand. 2000;168:635. doi: 10.1046/j.1365-201x.2000.00715.x. [DOI] [PubMed] [Google Scholar]
- 118.Sarnoff SJ, Braunwald E, Welch GH, Jr, Case RB, Stainsby WN, Macruz R. Hemodynamic determinants of oxygen consumption of the heart with special reference to the tension-time index. Am J Physiol. 1958;192:148. doi: 10.1152/ajplegacy.1957.192.1.148. [DOI] [PubMed] [Google Scholar]
- 119.Satrustegui J, Pardo B, del AA. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev. 2007;87:29. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- 120.Schaefer S, Schwartz GG, Steinman SK, Meyerhoff DJ, Massie BM, Weiner MW. Metabolic response of the human heart to inotropic stimulation: in vivo phosphorus-31 studies of normal and cardiomyopathic myocardium. Mag Res Med. 1992;25:260. doi: 10.1002/mrm.1910250205. [DOI] [PubMed] [Google Scholar]
- 121.Scholz TD, Balaban RS. Mitochondrial F1-ATPase activity of canine myocardium: effects of hypoxia and stimulation. Am J Physiol. 1994;266:H2396–H2403. doi: 10.1152/ajpheart.1994.266.6.H2396. [DOI] [PubMed] [Google Scholar]
- 122.Scholz TD, Laughlin MR, Balaban RS, Kupriyanov VV, Heineman FW. Effect of substrate on mitochondrial NADH, cytosolic redox state, and phosphorylated compounds in isolated hearts. Am J Physiol. 1995;268:H82–H91. doi: 10.1152/ajpheart.1995.268.1.H82. [DOI] [PubMed] [Google Scholar]
- 123.Schonfeld P, Schild L, Bohnensack R. Expression of the ADP/ATP carrier and expansion of the mitochondrial (ATP ADP) pool contribute to postnatal maturation of the rat heart. Eur J Biochem. 1996;241:895. doi: 10.1111/j.1432-1033.1996.00895.x. [DOI] [PubMed] [Google Scholar]
- 124.Schulenberg B, Aggeler R, Beechem JM, Capaldi RA, Patton WF. Analysis of steady-state protein phosphorylation in mitochondria using a novel fluorescent phosphosensor dye. J Biol Chem. 2003;278:27251. doi: 10.1074/jbc.C300189200. [DOI] [PubMed] [Google Scholar]
- 125.Stoylen A, Wisloff U, Slordahl S. Left ventricular mechanics during exercise: a Doppler and tissue Doppler study. Eur J Echocardiogr. 2003;4:286. doi: 10.1016/s1525-2167(03)00008-8. [DOI] [PubMed] [Google Scholar]
- 126.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 127.Swain JL, Sabina RL, McHale pA, Greenfield JC, Holmes EW. Prolonged myocardial nucleotide depletion after brief ischemia in the open chest dog. Am J Physiol. 1982;242:H818–H826. doi: 10.1152/ajpheart.1982.242.5.H818. [DOI] [PubMed] [Google Scholar]
- 128.Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R, Lee J, Plan P, Wilson J, Xin HB, Sanbe A, Gulick J, Mathai J, Robbins J, Salama G, Nakai J, Kotlikoff MI. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci U S A. 2006;103:4753. doi: 10.1073/pnas.0509378103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J Biol Chem. 2003;278:19587. doi: 10.1074/jbc.C300135200. [DOI] [PubMed] [Google Scholar]
- 130.Territo PR, French SA, Balaban RS. Simulation of cardiac work transitions, in vitro: effects of simultaneous Ca(2+) and ATPase additions on isolated porcine heart mitochondria. Cell Calcium. 2001;30:19. doi: 10.1054/ceca.2001.0211. [DOI] [PubMed] [Google Scholar]
- 131.Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, and light scattering. J Biol Chem. 2001;276:2586. doi: 10.1074/jbc.M002923200. [DOI] [PubMed] [Google Scholar]
- 132.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of F0/F1ATPase. Am J Physiol. 2000;278:c423–c435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 133.Tian R. Thermodynamic limitation for the sarcoplasmic reticulum Ca(2+)-ATPase contributes to impaired contractile reserve in hearts. Ann N Y Acad Sci. 1998;853:322. doi: 10.1111/j.1749-6632.1998.tb08290.x. [DOI] [PubMed] [Google Scholar]
- 134.Trollinger DR, Cascio WE, Lemasters JJ. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca(2+)-indicating fluorophores. Biophys J. 2000;79:39. doi: 10.1016/S0006-3495(00)76272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Unitt JF, McCormack JG, Reid D, Maclachlan LK. Direct evidence for a role of intramitochondrial Ca+2 in the regulation of oxidative phosphorylation in the stimulated rat heart. Biochem J. 1989;262:293. doi: 10.1042/bj2620293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vatner SF, Franklin D, Higgins CB, Patrick T, Braunwald E. Left ventricular response to serve exertion in untethered dogs. J Clin Invest. 1972;51:3052. doi: 10.1172/JCI107132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wallimann T, Dolder M, Schlattner U, Eder M, Hornemann T, Kraft T, Stolz M. Creatine kinase: an enzyme with a central role in cellular energy metabolism. MAGMA. 1998;6:116. doi: 10.1007/BF02660927. [DOI] [PubMed] [Google Scholar]
- 138.Wan B, LaNoue KF, Cheung JY, Scaduto RC., Jr Regulation of citric acid cycle by calcium. J Biol Chem. 1989;264:13430. [PubMed] [Google Scholar]
- 139.Welsh GH, Jr, Sarnoff SJ, Braunwald E, Stainsby WN, Case RB, Macruz R. The influence of cardiac output, aortic pressure and heart rate on myocardial oxygen utilization. Surg Forum. 1957;8:294. [PubMed] [Google Scholar]
- 140.Williamson JR, Safer B, LaNoue KF, Smith CM, Walajtys E. Mitochondrial-cytosolic interactions in cardiac tissue: role of the malate-aspartate cycle in the removal of glycolytic NADH from the cytosol. Symp Soc Exp Biol. 1973;27:241. [PubMed] [Google Scholar]
- 141.Wollenberger A. Relation between work and labile phosphate content in the isolated dog heart. Circ Res. 1957;5:175. doi: 10.1161/01.res.5.2.175. [DOI] [PubMed] [Google Scholar]
- 142.Wu ST, Kojima S, Parmley WW, Wikman-Coffelt J. Relationship between cytosolic calcium and oxygen consumption in isolated rat hearts. Cell Calcium. 1992;13:235. doi: 10.1016/0143-4160(92)90012-h. [DOI] [PubMed] [Google Scholar]
- 143.Yamada EW, Huzel NJ. The calcium binding ATPase inhibitor protein from bovine heart mitochondria. Purification and properties. J Biol Chem. 1988;263:11498. [PubMed] [Google Scholar]
- 144.Yamada EW, Huzel NJ. Calcium-binding ATPase inhibitor protein of bovine heart mitochondria. Role in ATP synthesis and effect of Ca2+ Biochemistry. 1989;28:9714. doi: 10.1021/bi00451a026. [DOI] [PubMed] [Google Scholar]
- 145.Zhang J, Duncker DJ, Xu Y, Zhang Y, Path G, Merkle H, Hendrich K, From AHL, Bache RJ, Ugurbil K. Transmural bioenergetic responses of normal myocardium to high workstates. Am J Physiol. 1995;268:H1891–H1911. doi: 10.1152/ajpheart.1995.268.5.H1891. [DOI] [PubMed] [Google Scholar]
- 146.Zhang J, Murakami Y, Zhang Y, Cho YK, Ye Y, Gong G, Bache RJ, Ugurbil K, From AH. Oxygen delivery does not limit cardiac performance during high work states. Am J Physiol. 1999;277:H50–H57. doi: 10.1152/ajpheart.1999.277.1.H50. [DOI] [PubMed] [Google Scholar]