

Abstract
Transgenic liver-specific inactivation of the carcinoembryonic antigen-related cell adhesion molecule (CEACAM1) impairs hepatic insulin clearance and causes hyperinsulinemia, insulin resistance, elevation in hepatic and serum triglyceride levels, and visceral obesity. It also predisposes to nonalchoholic steatohepatitis (NASH) in response to a high-fat diet. To discern whether this phenotype reflects a physiological function of CEACAM1 rather than the effect of the dominant-negative transgene, we investigated whether Ceacam1 (gene encoding CEACAM1 protein) null mice with impaired insulin clearance also develop a NASH-like phenotype on a prolonged high-fat diet. Three-month-old male null and wild-type mice were fed a high-fat diet for 3 months and their NASH phenotype was examined. While high-fat feeding elevated hepatic triglyceride content in both strains of mice, it exacerbated macrosteatosis and caused NASH-characteristic fibrogenic changes and inflammatory responses more intensely in the null mouse. This demonstrates that CEACAM1-dependent insulin clearance pathways are linked with NASH pathogenesis.
Keywords: nonalcoholic steatohepatitis, CEACAM1, high-fat diet, inflammation, apoptosis, fibrosis
Introduction
Nonalcoholic fatty liver disease (NAFLD) constitutes a spectrum of diseases ranging from benign hepatic steatosis to chronic nonalcoholic steatohepatitis (NASH) and cirrhosis. The incidence of nonalchoholic fatty liver disease (NAFLD) is on the rise worldwide, in part due to the increase in obesity.1 The prevalence of NAFLD in the United States is currently estimated at ~20%–25%, and in the morbidly obese population, at ~75%–90%. NASH, its progressive form, is estimated to be present in ~3%–5% of the general population.2,3 Of these, ~3%–5% have progressed to cirrhosis, including those with end-stage liver disease and/or primary liver cancer.4,5
Despite intense research efforts, mechanisms of the etiology and progression of NAFLD remain poorly understood. This could be due in part, to the limitation of the experimental animal models, which have been used to study the pathogenesis of the disease.6,7 Clearly, NAFLD is multifaceted and includes both genetic and environmental factors.1 It is generally agreed that visceral obesity and dysregulation in lipid metabolism and hepatic immune response are involved in progression to NASH.8 Although it is likely that NAFLD shares common pathophysiology with type 2 diabetes and metabolic syndrome, the role of insulin resistance in its pathogenesis remains controversial.9,10
Upon phosphorylation by the insulin receptor, carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) promotes receptor-mediated insulin endocytosis and degradation in the hepatocyte, the main mechanism of insulin clearance in the liver.11 Mice with liver-specific dominant-negative transgenic inactivation of CEACAM1 (L-SACC1) and mice with global null mutation of the Ceacam1 gene (Cc1−/−) develop insulin resistance due to impaired hepatic insulin extraction and hyperinsulinemia.12 Chronic hyperinsulinemia results in increased hepatic lipid production and output followed by substrate redistribution to white adipose tissue and visceral obesity.
We have recently shown that L-SACC1 mice develop spontaneous hepatic fibrosis, which becomes more intense when the mice are fed a high-fat (HF) diet for 3 months.13 Moreover, other key features of NASH (inflammation, oxidative stress, and apoptosis) are induced by sustained HF intake.13 This could be due to complete loss of function of CEACAM1 or to the inability of the transgene-encoded protein to become phosphorylated. To gain further insight into the role of CEACAM1 in the etiology and progression of NAFLD, we have assessed the effect of whole body Ceacam1 gene deletion on the pathogenesis of NASH in response to HF feeding. We herein report that Ceacam1 null mice (Cc1−/−) exhibit key features of obesity-related NASH when fed a HF diet for three months.
Materials and methods
Cell culture
Cc1−/− mice were backcrossed twelve times onto the C57BL/6 (BL6) genetic background,14 kept in a 12-hour dark/light cycle and fed a standard chow, ad libitum. All procedures were approved by the Institutional Animal Care and Utilization Committee. Three-month-old male mice were fed ad libitum either a standard chow (2016 Teklad, Harlan Laboratories), low fat (12%) regular diet (RD) or a 45% HF diet (Research Diets, Catalog #D12451, New Brunswick, NJ) for 3 months prior to sacrifice and phenotypic characterization.
Metabolic analysis
Following an overnight fast, mice were anesthetized with sodium pentobarbital at 1100 h. Whole venous blood was drawn from the retro-orbital sinuses to measure serum insulin by radioimmunoassays (Linco Research, St Charles, MO), serum-free fatty acids (FFA) by NEFA C kit (Wako Diagnostics, Richmond, VA) and serum triglycerides by Infinity® Triglycerides (Sigma, St Louis, MO). Hepatic total cholesterol and free cholesterol were measured using Infinity™ cholesterol reagent (Thermo Electron, Waltham, MA) and free cholesterol reagent (Wako Diagnostics), respectively,15 and triglyceride content, as described previously.16 Visceral adipose tissue was excised and weighed and visceral adiposity expressed as percentage of total body weight.
Liver histology
Formalin-fixed, paraffin-embedded liver sections were stained with hematoxylin-eosin (H&E) and assessed for the degree of steatosis and lobular inflammation according to the NASH scoring system recently proposed by the NIDDKNASH Clinical Research Network.17 Fibrosis was assessed on deparaffinized and rehydrated slides in 0.1% solution of Sirius Red (Sigma, Direct Red 80, St Louis, MO). TUNEL assay was performed using the ApopTag Plus Peroxidase Apoptosis Detection Kit (Chemicon International, Temecula, CA). Tissue sections on the other half of the same slide served as controls without terminal deoxynucleotidyl transferase (TDT) enzyme following digestion with proteinase K to account for nonspecific binding of enzyme conjugate.
Glutathione assay
Liver concentrations of reduced glutathione (GSH) were assayed using the Bioxytech® GSH-400 kit (OXISResearch, Portland, OR), as previously done.13
Western analysis
Tissue lysates or serum (for apolipoprotein B [ApoB]) were analyzed by 4%–12% gradient SDS-PAGE (Invitrogen, Carlsbad, CA) prior to Western analysis with polyclonal antibodies against caspase 3 (Cell Signaling Technology-Cat#9662, Beverly, MA), ApoB48/100 (Chemicon International, Temecula, CA), fatty acid synthase (FAS),18 Niemann Pick type C1 (NPC1) (Abcam, Cambridge, MA), p65 NF-kB phosphoserine (Ser 536) and p65 NF-kB (Cell Signaling Technology, Beverly, MA), as previously done.13 For normalization, membranes were reprobed with monoclonal antibodies against RPS3 (ProteinTech Group, Inc., Chicago, IL) and α-actin (Sigma). All blots were incubated with horseradish peroxidase-conjugated anti-IgG antibody prior to protein detection by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Piscataway, NJ) and quantification by densitometry and Image J software (v. 1.40, NIH).
Semiquantitative real time polymerase chain reaction-based gene expression
Total RNA in the liver was isolated with PerfectPure RNA Tissue Kit (5 Prime, Gaithersburg, MD) and total RNA in the adipose tissue was isolated with RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. cDNA was synthesized using ImProm-II™ Reverse Transcriptase (Promega, Madison, WI) using 1 μg of total RNA and primers for lipid catabolism, fibrosis and inflammatory markers, and as reference control, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Table 1). cDNA was evaluated with real time quantitative polymerase chain reaction (StepOnePlus™; Applied Biosystems, Foster City CA). The relative amounts of mRNA were calculated by comparison to the corresponding standards and normalized relative to GAPDH. Results are expressed in fold change as mean ± SEM.
Table 1.
Primer sequences used for quantitative real-time polymerase chain reaction (qRT-PCR)
| Primer | Forward sequence | Reverse sequence |
|---|---|---|
| GAPDH | CCAGGTTGTCTCCTGCGACT | ATACCAGGAAATGAGCTTGACAAAGT |
| PPARα | TGCTGGTATCGGCTCAATAA | TCCTGCCACTTGCTCACTAC |
| CYP2E1 | CCATCGGCACCATGGCGGTT | GCCCGAAGCGCTTTGCCAAC |
| F4/80 | CTTTGGCTATGGGCTTCCAGTC | CAAGGAGGACAGAGTTTATCGTG |
| CD3 | AGAGCAGCTGGCAAAGGTGGTGTC | CAGCCATGGTGCCCGAGTCTAGC |
| CD4 | TCACCTGGAAGTTCTCTGACC | GGAATCAAAACGATCAAACTGCG |
| CD8 | CTCTGGCTGGTCTTCAGTATGA | TCTTTGCCGTATGGTTGGTTT |
| TNFα | ACGGCATGGATCTCAAAGAC | CGGACTCCGCAAAGTCTAAG |
| IL-6 | GGCCTTCCCTACTTCACAAG | ATTTCCACGATTTCCCAGAG |
| IL-4 | AGGTCACAGGAGAAGGGACGCC | TGCGAAGCACCTTGGAAGCCC |
| IL-13 | TGTTTCGCCACGGCCCCTTC | TGCTCAAGCTGCTGCCTGCC |
| IFNγ | ATGAACGCTACACACTGCATC | CCATCCTTTTGCCAGTTCCTC |
| TGFβ | CAACAATTCCTGGCGTTACCTTGG | GAAAGCCCTGTATTCCGTCTCCTT |
Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PPARα, peroxisome proliferator-activated receptor α; CYP2E1, cytochrome p450 enzyme; TNFα, tumor necrosis factor-α; TGFβ, transforming growth factor-β; IL, interleukins; IFNγ, interferon gamma; TGFβ, transforming growth factor-β.
Statistical analysis
Data were analyzed with SPSS software using one-factor analysis of variance (ANOVA) and graphed with GraphPad Prism 4 software. P < 0.05 was statistically significant.
Results
High-fat diet alters hepatic insulin and lipid metabolism in Cc1−/− null mice
As previously reported19,20 6-month-old male Cc1−/− mice exhibited visceral obesity and hyperinsulinemia (Table 2). Consistent with increased visceral obesity, they also developed elevated serum levels of leptin and FFA (Table 2). HF feeding for 3 months induced serum insulin levels by ~2-fold in both strains of mice (Table 2).
Table 2.
Effect of HF intake on serum and tissue biochemistry of 6-month-old male Cc1+/+ and Cc1−/− mice
|
Cc1+/+
|
Cc1−/−
|
|||
|---|---|---|---|---|
| RD | HF | RD | HF | |
| Body weight, g | 26.2 ± 0.35 | 42.4 ± 0.49a | 29.3 ± 0.78b | 43.6 ± 1.32a |
| Visceral fat, % Bwt | 1.33 ± 0.15 | 3.87 ± 0.39a | 2.57 ± 0.44b | 4.27 ± 0.37a |
| Serum insulin, pM | 50.0 ± 2.00 | 123.0 ± 37.0a | 160.0 ± 48.0b | 387.0 ± 70.0a,b |
| Serum FFA, mEq/L | 0.50 ± 0.10 | 1.09 ± 0.11a | 1.08 ± 0.10b | 0.82 ± 0.13 |
| Serum TG, mg/dL | 46.9 ± 7.00 | 46.4 ± 4.84 | 38.1 ± 1.66 | 30.9 ± 1.43a,b |
| Serum leptin, ng/mL | 2.46 ± 0.24 | 67.2 ± 1.39a | 8.01 ± 2.65b | 72.0 ± 2.22a |
| Hepatic TG, mg/g protein | 97.9 ± 13.2 | 224. ± 37.9a | 169.0 ± 22.9b | 296.0 ± 48.8a |
| Hepatic total cholesterol (mg/g protein) | 14.6 ± 0.88 | 59.6 ± 11.1a | 14.7 ± 0.99 | 57.6 ± 9.39a |
| Hepatic free cholesterol (mg/g protein) | 10.7 ± 0.68 | 12.1 ± 1.01 | 9.75 ± 0.86 | 10.9 ± 0.29 |
| Hepatic cholesterol esters (mg/g protein) | 3.86 ± 0.68 | 47.5 ± 10.1a | 4.96 ± 1.35 | 46.7 ± 9.23a |
Notes: Male mice (n > 7; 3 months of age) were fed RD or HF for 3 months. Mice were fasted overnight and the serum and tissues removed and analyzed. Values are expressed as mean ± SEM.
P < 0.05 HF versus RD;
P < 0.05 Cc1−/− versus Cc1+/+ in the same feeding group.
Abbreviations: Cc1−/−, global ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet; TG, triglyceride; FFA, free fatty acids.
Western analysis revealed hepatic FAS protein was ~2-fold higher in null than wild-type mice, regardless of diet (Figure 1a). This could contribute to the significantly higher hepatic triglyceride content in RD-fed null mice (Table 2). Sustained HF intake induced hepatic triglyceride level in both mouse strains, but decreased serum triglyceride in Cc1−/− null mice (Table 2). The lower gain in adiposity in Cc1−/− null mice in response to HF diet (1.5- versus 3-fold increase in Cc1+/+ wild-type mice from the same genetic background [Table 2]) suggests that the lower serum triglyceride in Cc1−/− null mice was due to a reduction in hepatic triglyceride output. This notion is, in part, supported by the lower serum ApoB100/ApoB48 protein levels in HF-fed null compared to wild-type mice (Figure 1b).
Figure 1.
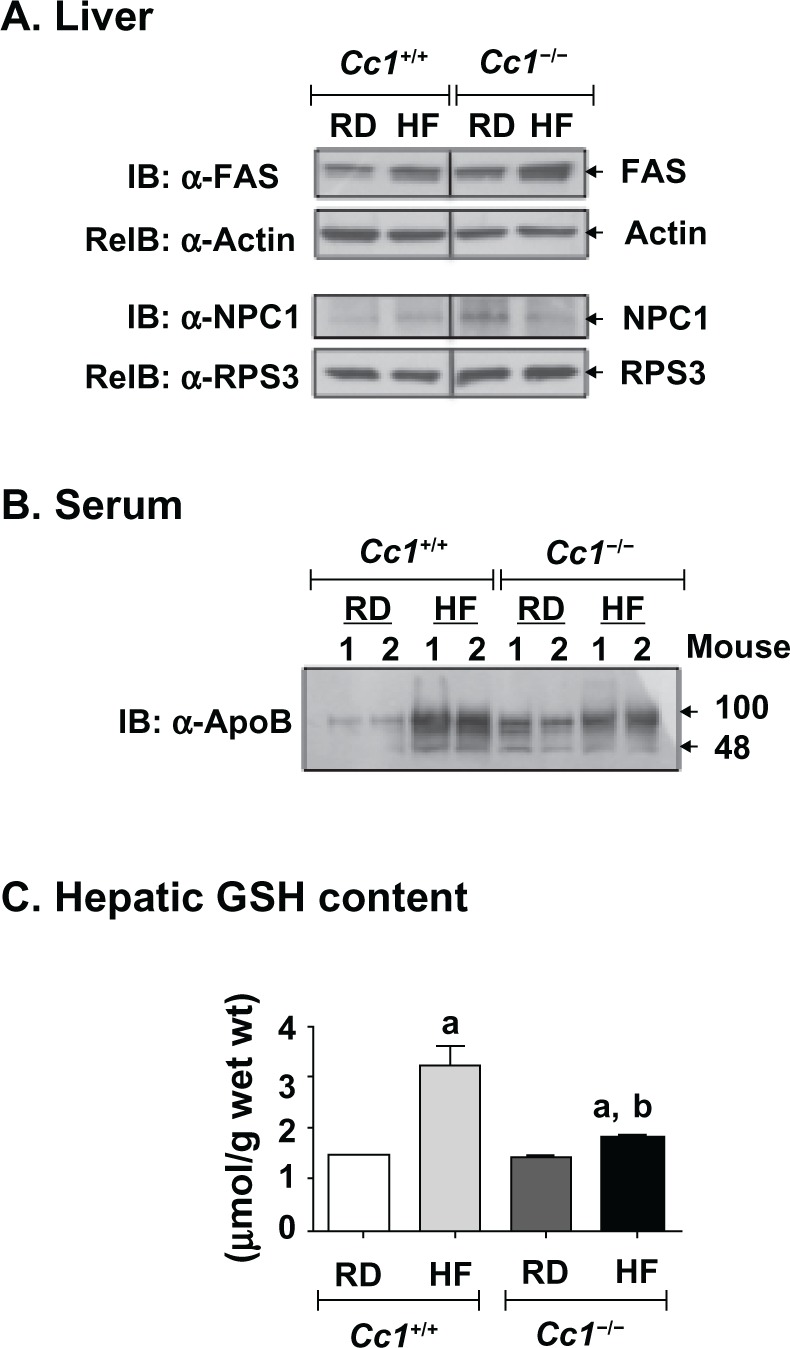
Effect of HF diet on proteins related to hepatic lipid metabolism and on hepatic glutathione (GSH) levels in male Cc1+/+ and Cc1−/− mice. Male wild-type Cc1+/+ and Cc1−/− mice were fed a regular chow (RD) or a HF diet for 3 months, starting at 3 months of age. A) Livers were then removed and analyzed by immunoblotting (IB) with α-FAS and α-NPC1, followed by reimmunoblotting (reIB) with α-actin and α-RPS3 antibodies, respectively, to normalize for the amount of proteins loaded, as described in Materials and Methods. The gel represents at least 3 mice per feeding group. B) Serum (10 μg) was analyzed, as above, by immunoblotting using α-ApoB antibody to detect ApoB100 and ApoB48 proteins. Although 2 mice are included in the figure, these experiments were performed on at least 5 mice per feeding group. C) Effect on hepatic GSH levels. N > 7 mice per feeding group. Values are mean ± SEM. aP < 0.005 HF versus RD. bP < 0.05 Cc1−/− versus Cc1+/+ in the same feeding group.
Abbreviations: Cc1−/−, global Ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet; FAS, fatty acid synthase; NPC1, niemann Pick type C1; ApoB, apolipoprotein B; GSH, glutathione; IB, immunoblotting; reIB, reimmunoblotting.
Liver total and free cholesterol were comparable in both mouse strains on the low fat regular diet. Total cholesterol was increased ~4-fold in both on the HF diet, with the increase due to increased cholesterol esters (Table 2). The protein level of hepatic NPC1, a late endosomal cholesterol traffic protein, was decreased ~50% by HF diet in null, but not wild-type mice (Figure 1a). Consistently, hepatic GSH content in Cc1−/− mice was almost half that of Cc1+/+ mice on the HF diet (Figure 1c).
Hepatic steatosis was increased in both strains on the HF diet (Figure 2). Histological evaluation from different fields revealed that fat infiltration was more diffuse in livers from Cc1−/− as compared to Cc1+/+ wild-type mice (panel 4 versus 2), which showed fat-free parenchyma alternating with affected areas. In HF-fed Cc1−/− null mice, the steatosis appeared to be predominantly macrovesicular in addition to microvesicular hepatocyte fat accumulation. However, the HF-fed wild-type Cc1+/+ livers exhibited predominantly microvesicular steatosis with a mix of macrosteatosis. No ballooning injury of the hepatocyte was noted.
Figure 2.
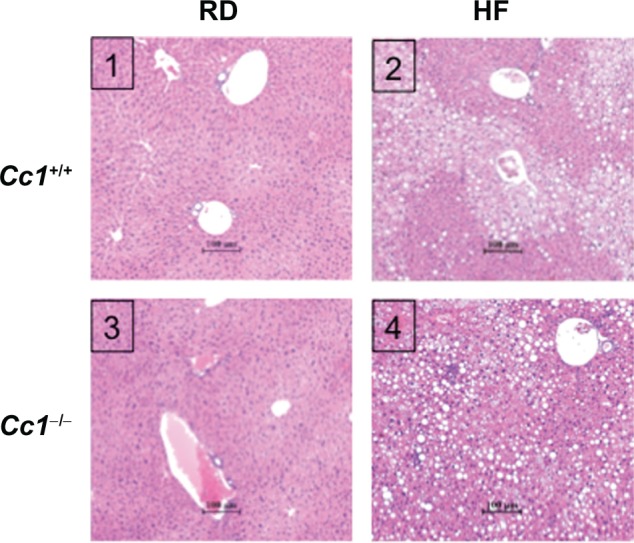
Effect of HF diet on liver histology in male wild-type Cc1+/+ and Cc1−/− mice. Liver histology was assessed in H&E stained sections (n > 4 mice per feeding group), as described in Materials and Methods. Panel 1: Cc1+/+, regular diet; Panel 2: Cc1+/+, HF diet; Panel 3: Cc1−/−, regular diet; Panel 4: Cc1−/−, diet. Representative sections are shown. in HF-fed Cc1+/+ (panel 2), the lipid infiltration alternates with normal liver parenchyma and is predominantly microvesicular. In contrast, in HF-fed Cc1−/− (panel 4), the lipid infiltration appears more diffuse and predominantly macrovesicular.
Abbreviations: Cc1−/−, global ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet; H&E, hematoxylin-eosin.
High-fat diet induces an inflammatory response in liver and adipose tissue of Cc1−/− null mice
H&E staining of liver sections revealed few inflammatory islands with no significant change in hepatocellular architecture in both strains of mice on the RD (Figure 2, panels 1 and 3). Upon HF feeding, Cc1+/+ wild-type mice showed occasional foci of inflammatory cells, mostly periportal (Figure 2, panel 2 and Figure 3a, left panel), as opposed to Cc1−/− null mice, which exhibited multiple foci of inflammatory cell infiltrates in the hepatic lobules, as well as in the periportal areas (Figure 2, panel 4 and Figure 3a, right panel). Consistently, HF diet activated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to a higher extent in Cc1−/− than Cc1+/+ livers, as indicated by a ~10-fold versus ~1.5-fold increase in NF-κB phosphorylation (Figure 3b).
Figure 3.
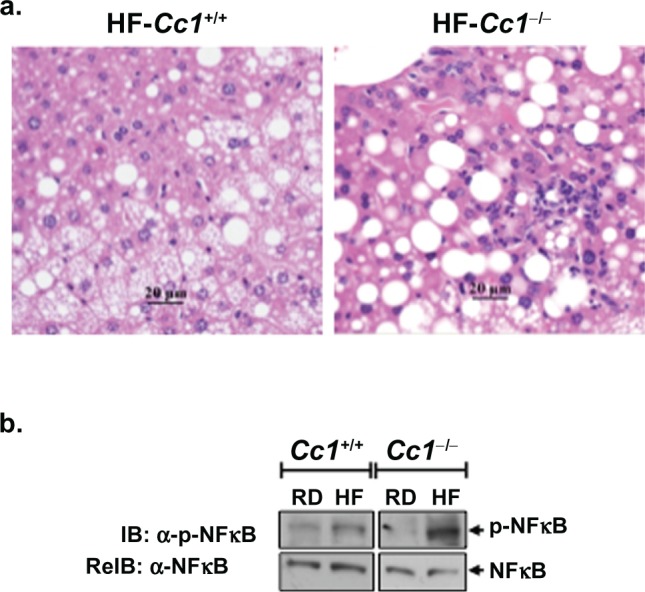
Effect of HF diet and genotype on the presence of liver inflammatory infiltrate. a) Representative liver sections from n > 4 mice stained with H&E. HF-fed Cc1−/− mouse livers (right panel) reveal increased perivascular and lobular inflammatory cell infiltrate relative to livers from HF-fed Cc1+/+ mice (left panel) that had fewer and mostly portal inflammatory cell foci. b) Liver lysates were analyzed by sequential immunoblotting (IB) with α-phospho-NF-κB (upper gel), followed by α-NF-κB (lower gel) antibodies as an index of inflammation. The gel represents at least 3 mice from each feeding group.
Abbreviations: Cc1−/−, global ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet; H&E staining, hematoxylin-eosin staining; IB, immunoblotting; reIB, reimmunoblotting.
Consistent with macrophage recruitment to white adipose tissue (WAT) in visceral obesity,21,22 RD-fed Cc1−/− null mice exhibited higher F4/80 and tumor necrosis factor-α (TNFα) mRNA levels in their WAT than their wild-type counterparts, as assessed by semiquantitative reverse transcription polymerase chain reaction (RT-PCR) analysis (Table 3). Similarly, hepatic TNFα mRNA content is elevated in the Cc1−/− null mouse. This could derive from resident hepatic macrophages, the population of which appeared to be elevated based on the higher F4/80 mRNA content in Cc1−/− mouse liver (Table 3), in addition to hepatic WAT infiltrates. Sustained HF diet induced a further increase in the macrophage pool (F4/80) and TNFα mRNA content in WAT and liver of both groups of mice (Table 3).
Table 3.
Effect of HF intake on the expression of selected genes related to inflammation in Cc1+/+ and Cc1−/− mouse livers and white adipose tissue (WAT)
|
Cc1+/+
|
Cc1−/−
|
||||
|---|---|---|---|---|---|
| RD | HF | RD | HF | ||
| WAT | F4/80 | 42.2 ± 5.55 | 262. ± 57.8a | 67.6 ± 7.90b | 610. ± 76.7a,b |
| TNFα | 0.03 ± 0.00 | 0.06 ± 0.00a | 0.05 ± 0.01b | 0.08 ± 0.01a | |
| Liver | F4/80 | 0.22 ± 0.08 | 0.42 ± 0.06a | 0.37 ± 0.05b | 0.74 ± 0.18a,b |
| TNFα | 0.04 ± 0.01 | 0.14 ± 0.02a | 0.09 ± 0.01b | 0.19 ± 0.02a | |
| IL-6 (x 10–1) | 0.06 ± 0.02 | 0.07 ± 0.02 | 0.17 ± 0.03b | 0.13 ± 0.00b | |
| CD3 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.01 | |
| CD8 | 0.26 ± 0.09 | 0.42 ± 0.06 | 0.41 ± 0.04 | 0.48 ± 0.05 | |
| CD4 | 0.34 ± 0.05 | 0.47 ± 0.08 | 0.76 ± 0.07b | 0.83 ± 0.08b | |
| IL-4 | 0.12 ± 0.01 | 0.12 ± 0.01 | 0.13 ± 0.02 | 0.14 ± 0.01 | |
| IL-13 | 0.18 ± 0.01 | 0.15 ± 0.02 | 0.18 ± 0.01 | 0.15 ± 0.02 | |
| IFNγ (x 10–2) | 0.07 ± 0.02 | 0.20 ± 0.04a | 0.20 ± 0.02b | 0.20 ± 0.04 | |
| TGFβ | 0.29 ± 0.07 | 0.56 ± 0.05a | 0.54 ± 0.06b | 0.56 ± 0.02 | |
Notes: Male mice (n > 7; 3 months of age) were fed RD or HF for 3 months. Mice were fasted overnight, the tissues removed and gene expression assessed by semiquantitative real-time PCR analysis. Values are normalized to GAPDH and expressed as mean units ± SEM.
P < 0.05 HF vs RD;
P < 0.05 Cc1−/− vs Cc1+/+ in the same feeding group.
Abbreviations: Cc1−/−, global Ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF high-fat diet; WAT, white adipose tissue; TNFα, tumor necrosis factor-α; TGFβ, transforming growth factor-β; IL, interleukins; IFNγ, interferon gamma; IL, interleukins; TGFβ, transforming growth factor-β.
mRNA content of CD4 was elevated in RD-fed Cc1−/− null mice and remained elevated without further increase in response to HF diet (Table 3). This suggests basal elevation in CD4+ T cell pool in null mice. Hepatic mRNA content of transforming growth factor β (TGFβ) and interferon-gamma (IFNγ) followed a similar change. Elevation in basal IFNγ, but not interleukin (IL)-4/IL-13 mRNA levels, suggests a CD4+ Th1, but not CD4+ Th2 response, in RD-fed Cc1−/− mouse livers.
In agreement with the small degree of periportal inflammation in HF-fed Cc1+/+ wild-type mice (Figure 3a, left panel), HF diet induced hepatic mRNA levels of proinflammatory TNFα and IFNγ cytokines (Table 3).
High-fat diet causes apoptosis in Cc1−/− null mice
Excessive fatty acid β-oxidation and lipid ω-peroxidation promote oxidative stress in obesity.23,24 Consistently, semiquantitative RT-PCR analysis revealed higher hepatic mRNA levels of peroxisome proliferator-activated receptor α (PPARα) in RD-fed Cc1−/− null than Cc1+/+ wild-type mice, and a further increase by HF intake in both strains of mice (Table 4). This suggests that fatty acid β-oxidation was elevated in Cc1−/− mouse liver. The mRNA levels of hepatic CYP2E1, a member of the microsomal cytochrome p450 family involved in metabolism of long chain fatty acids (lipooxygenation) and microsomal lipid ω-peroxidation, were comparable in both RD-fed mouse strains, but underwent a ~2-fold increase by HF diet only in Cc1−/− null mice (Table 4). Thus, it is likely that HF induced oxidative changes in the null mouse,25 which in the presence of high TNFα levels, predispose to cell death.26 Consistently, Western analysis (Figure 4a) revealed higher caspase 3 cleavage in HF-fed Cc1−/− liver than other groups of mice. In addition to TUNEL stain (Figure 4b, panel 2 versus 1), this reveals enhanced apoptosis in HF-fed null, but not wild-type mice.
Table 4.
Effect of HF intake on hepatic expression of selected genes related to lipid catabolism in Cc1+/+ and Cc1−/− mice
|
Cc1+/+
|
Cc1−/−
|
|||
|---|---|---|---|---|
| RD | HF | RD | HF | |
| PPARα | 5.57 ± 0.64 | 9.50 ± 0.33a | 8.50 ± 0.37b | 10.9 ± 0.67a |
| CYP2E1 | 555.0 ± 77.0 | 559.0 ± 70.0 | 500.0 ± 38.4 | 817.0 ± 136.0a |
Notes: Male mice (n > 7; 3 months of age) were fed RD or HF for 3 months. Mice were fasted overnight, the tissues removed and gene expression assessed by semiquantitative real time-PCR analysis. Values are normalized to GAPDH and expressed as mean units ± SEM.
P < 0.05 HF vs RD;
P < 0.05 Cc1−/− vs Cc1+/+ in the same feeding group.
Abbreviations: Cc1−/−, global ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet; PPARα, peroxisome proliferator-activated receptor α; CYP2E1, cytochrome p450 enzyme.
Figure 4.
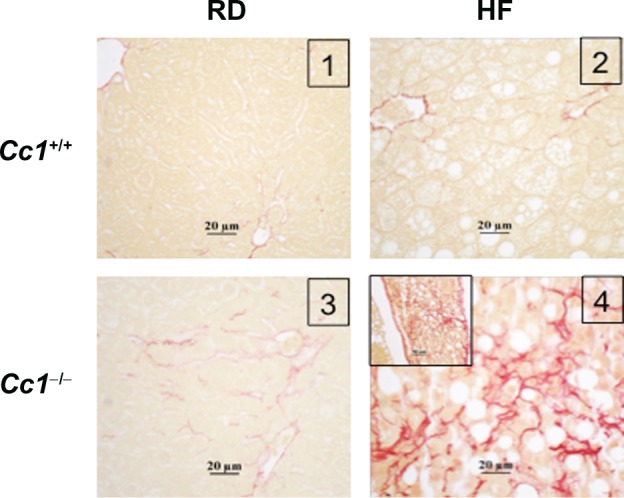
Effect of HF diet and genotype on liver fibrosis. Liver sections from n > 4 mice per feeding group were stained with Sirius red. Panel 2: RD-fed Cc1−/− mice show mild pericellular fibrosis. Panel 4: HF-fed Cc1−/− mice, show extensive pericellular and perivascular fibrosis.
Abbreviations: Cc1−/−, global Ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet.
Evaluating total NAS score from H&E liver sections based on three histological features (steatosis, inflammation and ballooning) following the Kleiner’s scoring system,27 indicated that HF-fed Cc1−/− mice developed NASH (NAS score of ~5), as opposed to the other groups, which scored <2.75 (Table 5).
Table 5.
NAS score of Cc1+/+ and Cc1−/− mice
| Group/number | Steatosis | Inflammation | Ballooning | Total NAS | Designation | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Micro/macro (0–3) | Lobular | Portal (0–3) | Hepatocytes (0–2) | Score (8) | NASHa | ||
| Cc1+/+RD | >4 | 0 | 0.5 | 0 | 0 | 0.5 | No NASH |
| Cc1+/+HF | >4 | 2(/br)1 | 0.625 | 0 | 0 | 2.6 | No NASH |
| Cc1–/–RD | >4 | 0 | 0.625 | 0.125 | 0 | 0.75 | No NASH |
| Cc1–/–HF | >4 | 2.75 | 1.25 | 0.625 | 0 | 4.625a | NASH |
Notes: Male mice (n > 4; 3 months of age) were fed RD or HF for 3 months. Scoring for NAS from H&E sections of liver tissues was evaluated following Kleiner’s scoring system. The criteria included 3 major histological features, comprised of micro and macrosteatosis (0–3), inflammation involving the hepatic lobules as well as portal triads (0–3), and hepatocyte ballooning (0–2).
The ~4.6 score in HF-fed Cc1−/− mice supported the diagnosis of NASH in this group of mice. The other groups of a total NAS score of <3 do not fit into the criteria of NASH.
Abbreviations: NASH, nonalcoholic steatohepatitis; H&E, hematoxylin-eosin; Cc1−/−, global Ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet.
Development of early fibrosis in Cc1−/− null mice
In NASH, fibrosis usually starts in zone 3 as perivenular and/or pericellular fibrosis, a pattern referred to as “chicken wire” fibrosis. Sirius red stain revealed a “chicken wire” pattern of collagen deposition in Cc1−/− (stage 2),28,29 but not Cc1+/+ wild-type mice on the regular diet (Figure 5, panel 3 versus 1), in spite of elevated levels of the proinflammatory IFNγ cytokine, which decreases collagen synthesis.30 Consistent with the profibrogenic effect of IL-6 cytokine and TGFβ,30 the mRNA content was higher in the liver of Cc1−/− than Cc1+/+ mice on the regular diet (Table 3). Sustained HF feeding exacerbated these fibrogenic changes in Cc1−/− mice (panel 4 versus 3) without causing further changes in IL-6 and TGFβ mRNA levels. Instead, HF induced serum leptin (Table 2) and hepatic TNFα mRNA levels (Table 3), which together could mediate progression of fibrosis and inflammation in NASH.31
Figure 5.
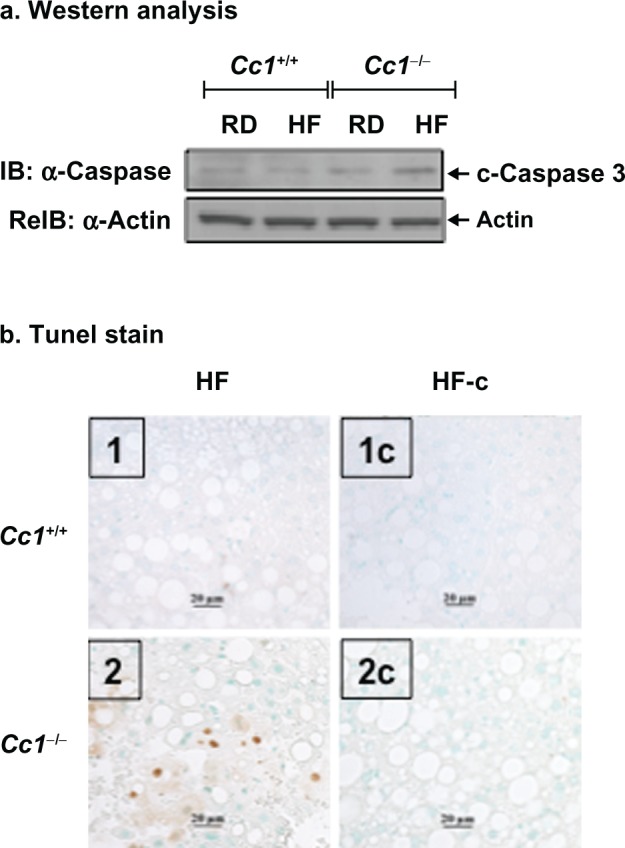
Effect of HF diet and genotype on liver cell apoptosis. a) Liver lysates were analyzed by sequential immunoblotting (IB) with antibodies against caspase 3 (upper gel), followed by α-actin (lower gel) to detect cleaved caspase 3 (17 kdaltons) as an index of apoptosis. The gel represents at least three mice from each feeding group. b) Liver sections from n > 4 mice per feeding group were analyzed by TUNEL staining for apoptotic cells, as described in Materials and Methods. Representative sections are shown. Panel 1: HF-fed Cc1+/+; Panel 2, HF-Fed Cc1−/−, Apoptotic cells are observed only in Cc1−/− livers. Panels 1-c and 2-c are negative controls without TDT enzyme following digestion with proteinase K to account for nonspecific binding of enzyme conjugate. Sections from mice on the regular diet are not shown because there was little evidence of apoptosis in either genotype.
Abbreviations: Cc1−/−, global Ceacam1 null mouse; Cc1+/+, wild-type mouse from the same genetic background as Cc1−/− mice; RD, regular diet; HF, high-fat diet; IB, immunoblotting; reIB, reimmunoblotting.
In Cc1+/+ wild-type mice, HF elevated leptin (Table 2) and hepatic TGFβ and TNFα (Table 3), without altering hepatic IL-6 mRNA levels (Table 3). HF induced a small increase in collagen production, as shown by Sirius red staining (Figure 5, panel 2 versus 1).
Discussion
Farrell10 and others11 have shown that similar to L-SACC1 mice with liver-specific inactivation of CEACAM1, mice homozygous for null mutation of the Ceacam1 gene on the C57BL/6 (BL6) genetic background manifest impairment of insulin clearance and hyperinsulinemia. This leads to insulin resistance and increases transcription of lipogenic enzymes (including FAS) to promote de novo hepatic lipid production.19 On the BL6 genetic background, Ceacam1 null mutation causes preferential lipid redistribution to white adipose tissue and an increase in visceral obesity.10
The current studies show that sustained HF feeding of Cc1−/− mice caused lipid accumulation in hepatocytes despite increase in fatty acid β-oxidation. Similar changes are seen in L-SACC1 mice13 and human NASH.32,33Cc1−/− mice also developed NASH-characteristic fibrosis when fed a HF diet; even on a regular chow diet, they exhibited low levels of pericellular fibrosis. In response to a HF diet, which triggers inflammation,34–36Cc1−/− mice exhibited other key features of progressive NAFLD, including diffuse macrosteatosis, inflammation and apoptosis, and possibly oxidative changes, as suggested by increased hepatic CYP2E1 protein levels. These data provide further in vivo evidence that altered CEACAM1-dependent insulin clearance pathways can provide a link between insulin resistance and NASH development, at least in mice with a BL6 genetic component background.37 They also confirm that male BL6 mice are susceptible to developing a NASH-like phenotype.38
Ceacam1 null mutation did not affect total and free cholesterol content in the liver. However, reduction of NPC1 protein level in HF-fed Cc1−/− mice suggests increased partitioning of free cholesterol from cytosolic lipid droplets to mitochondria.39 This could reduce mitochondrial GSH stores40 and increase sensitivity to the cytotoxic effect of the proinflammatory cytokine, TNFα, the level of which was high in Cc1−/− livers, resulting partly from steatosis-induced changes in the inflammatory milieu.41,42 Consistent with a role for TNFα-dependent activation of IKK-β in oxidative stress and inflammation,43 HF diet activated NF-κB pathways to elicit a more robust inflammatory response in Cc1−/− than Cc1+/+ mice.
The population of CD4+ T cells was basally higher in Cc1−/− than Cc1+/+ mice fed a regular diet. This could, at least in part, be due to Ceacam1 deletion in T cells, as suggested by increased CD4+ T pool in mice with conditional deletion of Ceacam1 in T cells,44 but not in regular chow-fed L-SACC1 mice with functional inactivation of Ceacam1, specifically in hepatocytes.13 The increase in CD4+ T cell pool was accompanied by elevation in hepatic mRNA content of IFNγ without changes in IL-4/IL-13 levels, suggesting increased production of CD4+ Th1, but not CD4+ Th2 cytokine in Cc1−/− livers.45 Elevated basal leptin level and its inducing effect on TNFα could contribute to the increase in CD4+ Th1 response.46 Given that Cc1−/− mice lack the anti-inflammatory effect of CEACAM1 in T47 and B48 cells, it is likely that deletion of Ceacam1 in lymphocytes contributed to the more robust inflammatory response to HF diet in mutant mice.
Consistent with insulin resistance being an independent predictor for fibrosis in NASH,49 chow-fed Cc1−/− null mice developed a low level of pericellular fibrosis. This could be attributed to elevated hepatic content of the profibrogenic factors, IL-6 and TGFβ,30,50 In addition to IL-6, leptin increases transcription of TNFα,46,51 which, together with IFNγ, reduces collagen synthesis.30 The combined effect of TNFα and IFNγ inflammatory cytokines could limit the profibrogenic effect of IL-6 and TGFβ in RD-fed null mice. HF feeding caused progressive fibrosis in parallel to preferentially inducing TNFα without changing IFNγ levels. TNFα triggers apoptosis, which in turn, leads to fibrosis and inflammation.28,50,52 Thus, it is possible that further induction in TNFα by HF diet activated caspase 3 to cause apoptosis and subsequently, promote fibrosis more preferentially in the null mouse. The increase in serum leptin levels could exacerbate the fibrogenic effect of TNFα31,53
In contrast to the null mouse, fat feeding elevated hepatic TGFβ, but not IL-6 mRNA content in Cc1+/+ wild-type mice. It also failed to produce NASH-like fibrogenic changes in these mice. IL-6 could be secreted basally from macrophages, but also from B cells as part of the adaptive immune response.30 Given that IL-6 release from B cells induces differentiation of hepatic stellate cells to myofibroblasts to mediate hepatic fibrosis in response to CCl4 in mice, independently of T cell or antibody stimulation,54 it is possible that changes in hepatic IL-6 levels contributed to the differential development of hepatic fibrosis in Cc1−/−, but not wild-type mice. This is in concordance with the positive correlation between the severity of the disease in NASH patients and hepatic IL-6 levels.55,56
Our data emphasize that loss of CEACAM1 causes insulin resistance, hepatic steatosis and visceral obesity, with a subsequent increase in leptin production and release from white adipose tissue. This, in turn, promotes production of IL-6 and TNFα, which in addition to other adipokines, trigger a proinflammatory state. In the absence of CEACAM1, sustained HF feeding exacerbates the inflammatory response and elevates hepatic TNFα content and its proapoptotic and fibrogenic effect. This model illustrates that null mutation of Ceacam1 integrates metabolic, apoptotic, inflammatory, and fibrogenic signals leading to progressive NAFLD. Given that murine models rarely develop spontaneous fibrosis,1 the Cc1−/− mouse provides a unique tool to investigate the effect of environmental factors on genetic predisposition to NAFLD progression and sheds light on the pathogenesis of NASH. The significance of this finding to human disease is highlighted by the strong conservation of CEACAM1 functional domains in humans and rodents and by the reduction of hepatic Ceacam1 mRNA levels in low aerobic capacity running rats that exhibit features of metabolic syndrome57 and NASH.58
Acknowledgments
We thank Dr Larisa Fedorova for advice on TUNEL assays and Dr Cara Gatto-Weis for guidance on pathological evaluations. We also thank Jehnan Liu and Garrett Heinrich (Najjar laboratory) for continuous discussion and manuscript preparation, as well as Jennifer Kalisz (Najjar laboratory) and Steven Lear (Erickson laboratory) for excellent technical assistance.
This work was supported by grants from the National Institutes of Health (DK054254 and DK083850 to SM Najjar and DK072187 to SK Erickson), the United States Department of Agriculture (USDA 38903-02315) to SM Najjar, a Merit Award from the Department of Veteran’s Affairs to SK Erickson, and by institutional funds to RE Bourey.
Footnotes
Disclosures
The authors declare that no conflicts of interest exist in this work.
References
- 1.Erickson SK. Nonalcoholic fatty liver disease. J Lipid Res. 2009;50(Suppl):S412–S416. doi: 10.1194/jlr.R800089-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis. 2008;28:339–350. doi: 10.1055/s-0028-1091978. [DOI] [PubMed] [Google Scholar]
- 3.Ong JP, Younossi ZM. Epidemiology and natural history of NAFLD and NASH. Clin Liver Dis. 2007;11:1–16. doi: 10.1016/j.cld.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Marrero JA, Fontana RJ, Su GL, Conjeevaram HS, Emick DM, Lok AS. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology. 2002;36:1349–1354. doi: 10.1053/jhep.2002.36939. [DOI] [PubMed] [Google Scholar]
- 5.Bullock RE, Zaitoun AM, Aithal GP, Ryder SD, Beckingham IJ, Lobo DN. Association of non-alcoholic steatohepatitis without significant fibrosis with hepatocellular carcinoma. J Hepatol. 2004;41:685–686. doi: 10.1016/j.jhep.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Ariz U, Mato JM, Lu SC, Martinez Chantar ML. Nonalcoholic steatohepatitis, animal models, and biomarkers: what is new? Methods Mol Biol. 2010;593:109–136. doi: 10.1007/978-1-60327-194-3_6. [DOI] [PubMed] [Google Scholar]
- 7.Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 9.Green RM. NASH – Hepatic metabolism and not simply the metabolic syndrome. Hepatology. 2003;38:14–17. doi: 10.1053/jhep.2003.50325. [DOI] [PubMed] [Google Scholar]
- 10.Farrell GC. The liver and the waistline: fifty years of growth. J Gastroenterol Hepatol. 2009;24(Suppl 3):S105–S118. doi: 10.1111/j.1440-1746.2009.06080.x. [DOI] [PubMed] [Google Scholar]
- 11.Formisano P, Najjar SM, Gross CN, et al. Receptor-mediated internalization of insulin. Potential role of pp120/HA4, a substrate of the insulin receptor kinase. J Biol Chem. 1995;270:24073–24077. doi: 10.1074/jbc.270.41.24073. [DOI] [PubMed] [Google Scholar]
- 12.Poy MN, Yang Y, Rezaei K, et al. CEACAM1 regulates insulin clearance in liver. Nature Genet. 2002;30:270–276. doi: 10.1038/ng840. [DOI] [PubMed] [Google Scholar]
- 13.Lee SJ, Heinrich G, Fedorova L, et al. Development of non-alcoholic steatohepatitis in insulin resistant L-SACC1 mice. Gastroenterology. 2008;135:2084–2095. doi: 10.1053/j.gastro.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung N, Turbide C, Olson M, Marcus V, Jothy S, Beauchemin N. Deletion of the carcinoembryonic antigen-related cell adhesion molecule 1 (Ceacam1) gene contributes to colon tumor progression in a murine model of carcinogenesis. Oncogene. 2006;25:5527–5536. doi: 10.1038/sj.onc.1209541. [DOI] [PubMed] [Google Scholar]
- 15.Erickson SK, Lear SR, Dean S, et al. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female cyp7A1-deficient mice. J Lipid Res. 2003;44:1001–1009. doi: 10.1194/jlr.M200489-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Park SY, Cho YR, Kim HJ, et al. Mechanism of glucose intolerance in mice with dominant negative mutation of CEACAM1. Am J Physiol Endocrinol Metab. 2006;291:E517–E524. doi: 10.1152/ajpendo.00077.2006. [DOI] [PubMed] [Google Scholar]
- 17.Bondini S, Kleiner DE, Goodman ZD, Gramlich T, Younossi ZM. Pathologic assessment of non-alcoholic fatty liver disease. Clin Liver Dis. 2007;11:17–23. doi: 10.1016/j.cld.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Najjar SM, Yang Y, Fernstrom MA, et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005;2:43–53. doi: 10.1016/j.cmet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 19.DeAngelis AM, Heinrich G, Dai T, et al. CEACAM1: A link between insulin and lipid metabolism. Diabetes. 2008;57:2296–2303. doi: 10.2337/db08-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu E, Dubois MJ, Leung N, et al. Targeted disruption of carcinoembryonic antigen-related cell adhesion molecule 1 promotes diet-induced hepatic steatosis and insulin resistance. Endocrinology. 2009;150:3503–3512. doi: 10.1210/en.2008-1439. [DOI] [PubMed] [Google Scholar]
- 21.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 22.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel R, Ferrante AWJ. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavin KD, Yang S, Lin HZ, et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem. 1999;274:5692–5700. doi: 10.1074/jbc.274.9.5692. [DOI] [PubMed] [Google Scholar]
- 24.Surwit RS, Wang S, Petro AE, et al. Diet-induced changes in uncoupling proteins in obesity-prone and obesity-resistant strains of mice. Proc Natl Acad Sci U S A. 1998;95:4061–4065. doi: 10.1073/pnas.95.7.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1135–G1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 26.McClain CJ, Mokshagundam SP, Barve SS, et al. Mechanisms of nonalcoholic steatohepatitis. Alcohol. 2004;34:67–79. doi: 10.1016/j.alcohol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 28.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Dig Dis Sci. 2010;55:560–578. doi: 10.1007/s10620-009-1081-0. [DOI] [PubMed] [Google Scholar]
- 29.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 30.Bhogal RK, Bona CA. B cells: no longer bystanders in liver fibrosis. J Clin Invest. 2005;115:2962–2965. doi: 10.1172/JCI26845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carter-Kent C, Zein NN, Feldstein AE. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am J Gastroenterol. 2008;103:1036–1042. doi: 10.1111/j.1572-0241.2007.01709.x. [DOI] [PubMed] [Google Scholar]
- 32.Charlton M, Sreekumar R, Rasmussen D, Lindor K, Nair KS. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology. 2002;35:898–904. doi: 10.1053/jhep.2002.32527. [DOI] [PubMed] [Google Scholar]
- 33.Abdelmalek MF, Diehl AM. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med Clin North Am. 2007;91:1125–1149. doi: 10.1016/j.mcna.2007.06.001. ix. [DOI] [PubMed] [Google Scholar]
- 34.Chiang SH, Bazuine M, Lumeng CN, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138:961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim F, Pham M, Maloney E, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–1988. doi: 10.1161/ATVBAHA.108.169722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill-Baskin AE, Markiewski MM, Buchner DA, et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum Mol Genet. 2009;18:2975–2988. doi: 10.1093/hmg/ddp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirsch R, Clarkson V, Shephard EG, et al. Rodent nutritional model of non-alcoholic steatohepatitis: species, strain and sex difference studies. J Gastroenterol Hepatol. 2003;18:1272–1282. doi: 10.1046/j.1440-1746.2003.03198.x. [DOI] [PubMed] [Google Scholar]
- 39.Shen WJ, Patel S, Natu V, et al. Interaction of hormone-sensitive lipase with steroidogenic acute regulatory protein: facilitation of cholesterol transfer in adrenal. J Biol Chem. 2003;278:43870–43876. doi: 10.1074/jbc.M303934200. [DOI] [PubMed] [Google Scholar]
- 40.Mari M, Caballero F, Colell A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 41.Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Ann Intern Med. 1997;126:137–145. doi: 10.7326/0003-4819-126-2-199701150-00008. [DOI] [PubMed] [Google Scholar]
- 42.Bigorgne AE, Bouchet-Delbos L, Naveau S, et al. Obesity-induced lymphocyte hyperresponsiveness to chemokines: a new mechanism of fatty liver inflammation in obese mice. Gastroenterology. 2008;134:1459–1469. doi: 10.1053/j.gastro.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 43.Crespo J, Cayon A, Fernandez-Gil P, et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 44.Nagaishi T, Pao L, Lin SH, et al. SHP1 phosphatase-dependent T cell inhibition by CEACAM1 adhesion molecule isoforms. Immunity. 2006;25:769–781. doi: 10.1016/j.immuni.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 45.Aarsland A, Chinkes D, Wolfe RR. Contributions of de novo synthesis of fatty acids to total VLDL-triglyceride secretion during prolonged hyperglycemia/hyperinsulinemia in normal man. J Clin Invest. 1996;98:2008–2017. doi: 10.1172/JCI119005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 47.Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- 48.Lobo EO, Zhang Z, Shively JE. Pivotal advance: CEACAM1 is a negative coreceptor for the B cell receptor and promotes CD19-mediated adhesion of B cells in a PI3K-dependent manner. J Leukoc Biol. 2009;86:205–218. doi: 10.1189/jlb.0109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 50.Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13:565–580. doi: 10.1016/j.cld.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 52.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 53.Manco M, Marcellini M, Giannone G, Nobili V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;127:954–960. doi: 10.1309/6VJ4DWGYDU0XYJ8Q. [DOI] [PubMed] [Google Scholar]
- 54.Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115:3072–3082. doi: 10.1172/JCI24798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103:1372–1379. doi: 10.1111/j.1572-0241.2007.01774.x. [DOI] [PubMed] [Google Scholar]
- 56.Lemoine M, Ratziu V, Kim M, et al. Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int. 2009;29:1431–1438. doi: 10.1111/j.1478-3231.2009.02022.x. [DOI] [PubMed] [Google Scholar]
- 57.Wisloff U, Najjar SM, Ellingsen O, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005;307:418–420. doi: 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]
- 58.Thyfault JP, Rector RS, Uptergrove GM, et al. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J Physiol. 2009;587:1805–1816. doi: 10.1113/jphysiol.2009.169060. [DOI] [PMC free article] [PubMed] [Google Scholar]