

Abstract
Tau is a microtubule associated protein that fulfills several functions critical for neuronal formation and health. Tau discharges its functions by producing multiple isoforms via regulated alternative splicing. These isoforms modulate tau function in normal brain by altering the domains of the protein, thereby influencing its localization, conformation and post-translational modifications and hence its availability and affinity for microtubules and other ligands.
Disturbances in tau expression result in disruption of the neuronal cytoskeleton and formation of tau structures (neurofibrillary tangles) found in brains of dementia sufferers. More specifically, aberrations in tau splicing regulation directly cause several neurodegenerative diseases which lead to dementia. In this review, I present our cumulative knowledge of tau splicing regulation in connection with neurodegeneration and also briefly go over the still-extensive list of questions that are connected to tau (dys)function.
Keywords: MAP tau, Splicing regulation, Dementia
Tau is a microtubule-associated protein (MAP) enriched in axons of growing and mature neurons that is critical for neuronal function. Among its many roles, tau promotes neurite outgrowth, organizes axonal microtubules (MTs) and is involved in kinesin-dependent axonal transport (Wang and Liu, 2007; Morfini et al., 2009). Hyperphosphorylated, MT-dissociated tau is the component of neurofibrillary tangles (NFTs), hallmark structures of many neurodegenerative diseases (Gasparini et al., 2007; Gendron and Petrucelli, 2009).
Despite the ubiquity of NFTs in brains of people with dementia, tau was delegated to the back seat of neurodegeneration research for many years because no mutations had been found in it. This changed after close examination of frontotemporal dementia (FTDP) pedigrees. In many FTDP pedigrees, misregulation of tau exon 10 splicing results in wild-type protein but disturbs the normal isoform ratio and causes neurodegeneration regardless of which isoform becomes prevalent (Gasparini et al., 2007). These findings established not only that tau can cause neurodegeneration by itself in the absence of amyloid plaques, but also that it can do so in the absence of mutations in the protein -- a subtle form of a dosage disease.
Although we have made significant advances in dissecting the mechanisms of tau splicing, many aspects of tau (dys)function are still unclear, including how changes in the ratio of exon 10 cause neurodegeneration.
Alternative splicing, the complex choreographer of complexity
Bioinformatics analysis of the human genome indicates that almost all human genes are alternatively spliced (Pan et al., 2008). Alternative splicing is the primary contributor to proteomic complexity and plays a critical role in controlling differentiation and development (Stamm et al., 2005). Misregulation of alternative splicing is the cause of many life-threatening human diseases (Tazi et al., 2009). Despite the high fidelity of exon recognition in vivo, it is currently impossible to accurately predict alternative exons; it appears that combinatorial control and “weighing” of splice element strength are used to enable precise recognition of the short and degenerate splice sites (Hertel, 2008).
Exonic and intronic enhancers and silencers are involved in splicing regulation (Wang and Burge, 2008). These cis elements are regulated by trans-acting factors that mostly belong to two superfamilies, the SR/SR-like and hnRNP proteins (Long and Cáceres, 2009; Martinez-Contreras et al., 2007). Both families have additional functions beyond their involvement in splicing regulation: the former are also components of the spliceosome, whereas the latter are also involved in pre-mRNA transport, mRNA stability and translational regulation.
Splicing factors bind to the pre-mRNA they regulate or to other splicing factors; whether they act directly or indirectly and as activators or inhibitors of a particular splicing event depends on the specific transcript. This flexibility has complicated the investigations of splicing regulation since it is hard to make a priori assignments.
Several mammalian splicing factors are enhanced in or restricted to neurons. Nevertheless, it appears that the exquisite calibration of mammalian alternative splicing is primarily achieved by spatial and temporal variation in the expression and activity levels of quasi-ubiquitous splicing regulators (Hertel, 2008). This pleiotropy frustrates the prospects of any potential therapy based on modulation of splicing factors or their kinases.
Structure, regulation and functions of tau
The single-copy human tau gene is located on 17q21. The tau transcript undergoes extensive alternative splicing that is regulated spatially and temporally and can give rise to 30 isoforms (Andreadis, 2005). Fig. 1 shows the exon structure and splicing patterns of the tau gene (Fig. 1A), the effects of splicing decisions on the molecule’s function (Fig. 1B) and the tau isoforms that are prevalent in brain (Fig. 1C).
Fig. 1.
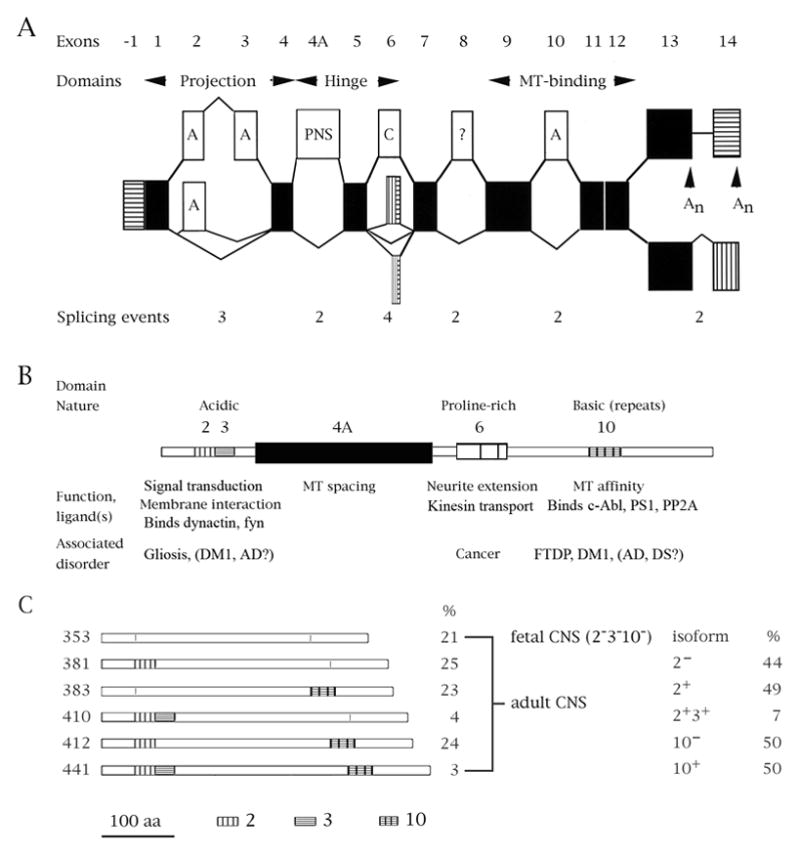
Tau mRNA species and the functions of the ensuing domains. (A) Schematic representation of exons and splicing pathways in the tau gene. Black = constitutive; white = regulated (A = adult-specific, PNS = specific to the peripheral nervous system, C = complex, ? = unknown); horizontal stripes = transcribed, untranslated regions; vertical stripes = alternative/additional reading frames. An indicate polyadenylation sites. The numbers underneath the exons indicate the possible number of outcomes from each alternatively spliced region within the tau transcript. (B) Diagram of the longest tau isoform. Above the diagram the general nature of the domain is noted. Below the diagram is a list of domain functions and of diseases in which the splicing of that particular region is or may be altered. (C) Schematic depictions of tau isoforms abundant in the central nervous system. On the left is the length of each isoform in amino acids, on the right its relative abundance in brain. Below the diagrams is a scale bar (aa=amino acids) and the drawing conventions for exons 2, 3 and 10.
Exons 2, 3 and 10 are adult-specific, but their ratios differ in various central nervous system compartments. All six possible product combinations of the 2/3/10 splicing events have been observed, indicating that separate factors govern their splicing – a conclusion confirmed by extensive studies of splicing factor effects on these exons (Andreadis, 2006).
The tau protein contains three domains. The acidic N-terminal region (“projection domain”), modulated by exons 2 and 3, interacts with the plasma membrane, dynactin and tyrosine kinase fyn (Lee, 2005; Lebouvier et al., 2009; Pooler and Hanger, 2010). The proline-rich middle region (“hinge”) influences MT spacing (by inclusion of exon 4A, specific to the peripheral nervous system) whereas alternative splicing of hinge-region exon 6 gives rise to tau variants that lack the MT binding domain and inhibit kinesin-dependent axonal transport (Andreadis, 2005; La Pointe et al., 2009). The basic C-terminal region, modulated by exon 10, contains either three or four MT binding motifs (changing tau affinity for MTs) and interacts with tyrosine kinase c-Abl (Lebouvier et al., 2009).
Although tau is primarily axonal, it is also found in dendrites (where it may act as a post-synaptic scaffolding protein) and in the nucleus, where it interacts with the nucleolus. It is also present in oligodendrocytes and astrocytes. As already mentioned, tau undergoes phosphorylation on as many as forty serines/threonines and four tyrosines (Dolan and Johnson, 2010; Gendron and Petrucelli, 2009). Like tau splicing, phosphorylation is developmentally regulated and decreases the affinity of tau for MTs. Hyperphosphorylated, MT-dissociated tau is the major component of NFTs (Gasparini et al., 2007).
Paths from tau splicing to neurodegeneration
NFTs are insoluble tau aggregates found within the neurons and glia of people with sporadic and familial Alzheimer’s disease (AD), sporadic and familial tangle-only dementias (“tauopathies”, exemplified by frontotemporal dementia with Parkinsonism -- FTDP-17), Down syndrome (DS; trisomy 21) and myotonic dystrophy type 1 (DM1). NFT numbers correlate with dementia severity. Additionally, tau null mice and human pedigrees that contain microdeletions and microduplications in the tau locus show developmental defects and learning disabilities (Pennisi, 2008).
The genetic and clinical connections between tau and dementia were long known and tantalizing, yet a direct cause-and-effect link had proved elusive. Finally, in 1998, the characterization of several FTDP-17 pedigrees firmly placed tau and, specifically, its splicing directly upstream of the process that causes dementia (Andreadis, 2006; Liu and Gong, 2008). Tangle-only tauopathies primarily affect the frontal and temporal cortex and their associated executive and cognitive functions (empathy, affect, social behavior, language use and comprehension). Collectively, they are the second most common type of sporadic dementia after AD and the familial cases show a far earlier age of onset than AD.
Although these tauopathies show such clinical variability that they have often been misdiagnosed, their molecular causes are remarkably uniform: the afflicted pedigrees predominantly show mutations in tau exon 10, although several carry missense mutations in tau exons 1, 9, 11, 12 and 13 which influence MT binding or tau conformation (Liu and Gong, 2008). Some of the exon 10 mutations are missense which influence MT binding. However, the majority are silent at the protein level but alter the ratio of exon 10 isoforms.
A few years after the FTDP discoveries, a second connection was discovered between tau and an odd kind of neurodegeneration. Myotonic dystrophy 1 (DM1) is the most common disease of its kind in adults, a multisystemic dominantly inherited disorder whose outcome includes dementia classified as an atypical tauopathy. DM1 brains show tau hyperphosphorylation, formation of intraneuronal aggregates, and significant reduction of tau isoforms containing exon 2 and 10 (Jiang et al., 2004, Sergeant et al. 2001).
The disease arises from titration of splicing regulators (the CELF and MBNL proteins) which explains its pleiotropic phenotype (Llamusí and Artero, 2008). Among their other targets, these regulators modulate splicing of the two affected tau exons (Andreadis, 2006). Additionally, analysis of AD cases has shown that exon 2 decreases and exon 10 increases in AD (Glatz et al., 2005; Conrad et al., 2007).
There is no direct connection (yet) between exon 2 and brain pathology, unlike the clear-cut link between exon 10 and FTDP. As a result, in a repetition of the earlier pattern for tau research in general, very little has been done on either the splicing regulation or the specific function of exon 2 in the last six years. Given the dearth of new data on that portion of tau, I will focus the rest of this review on exon 10 with a brief detour into a new and mysterious arrival, saitohin.
The type and behavior of the tau mutations makes it a unique system for two reasons. Tau belongs to a tiny category of genes in which disturbances of alternative splicing cause disease despite production of wild-type protein. Additionally, tau is the only system documented so far in which changes in isoform ratios arising from splicing misregulation can cause neurodegeneration both by cis and trans mechanisms: respectively, mutations in tau exon 10 (FTDP) and variations in levels of trans factors that regulate tau exons 2 and 10 (DM1 and perhaps AD and DS).
Notorious exon 10 and its confirmed link to FTDP
Tau exon 10 is adult-specific, encodes the second of four imperfect repeats that bind MTs and shows a species-specific difference crucial to neurodegeneration: in adult rodents, exon 10 becomes constitutive. In contrast, in adult humans exon 10 remains regulated in the central nervous system where the 10+ (4-repeat, 4R) and 10− (3-repeat, 3R) isoforms are present in a 1:1 ratio (Andreadis, 2005; Gasparini et al., 2007).
Splicing of tau exon 10 splicing is affected by exonic and intronic enhancers and silencers as well as by several trans factors (Andreadis, 2006; Liu and Gong, 2008). Investigations of FTDP pedigrees established that the proximal downstream intron of exon 10 is a hotspot for tauopathy mutations. This is particularly interesting in view of the fact that this region diverges between humans and rodents past intron position +8, whereas exon 10 and its proximal upstream intron are essentially 100% conserved.
The exon 10 mutations found in tauopathy pedigrees are shown in Fig. 2A. The mutations clustering around the 5′ splice site of exon 10 engendered two hypotheses. One is that its splicing is partly modulated by a putative hairpin loop which hinders interaction with the U1 snRNP (the first step of spliceosome formation); the other is that this region has two splicing elements: an intronic silencer followed by an intronic enhancer (Andreadis, 2006; Wolfe, 2009).
Fig. 2.
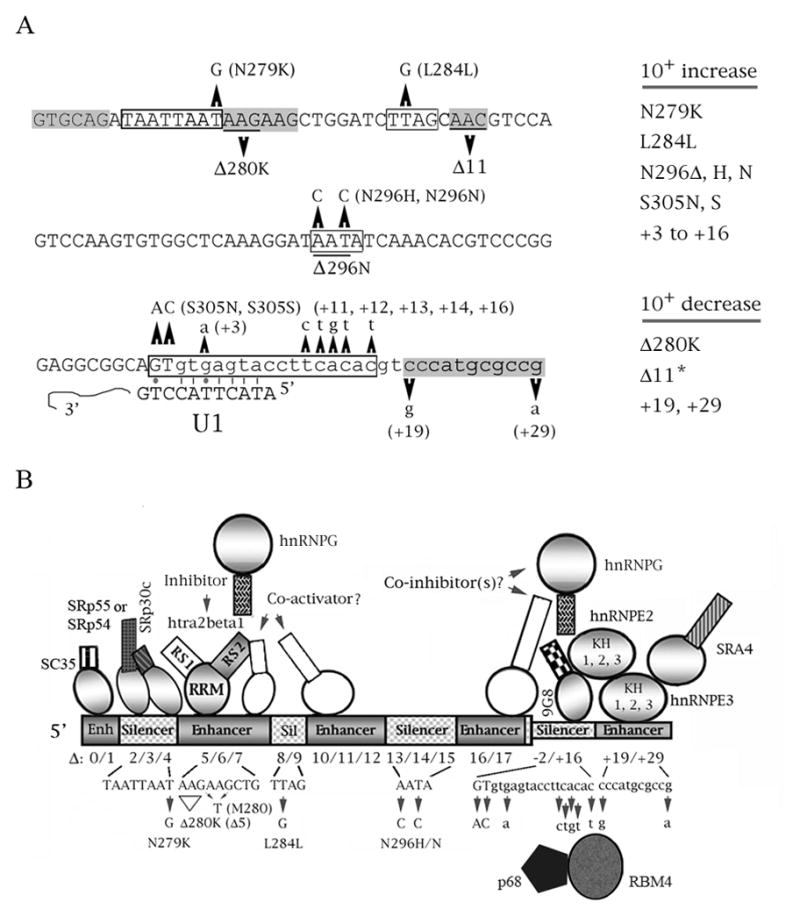
Cis elements and trans factors involved in the splicing regulation of tau exon 10. (A) Mutations of exon 10 found in pedigrees of neurodegenerative diseases (the exception is Delta11, designated by an asterisk, which was defined by the behavior of deletion constructs). The sequence of exon 10 (uppercse) and its proximal downstream intron (lowercase) is shown. Point mutations are indicated, as well as their resulting missense mutations, if any. Deletions are underlined. The only mutation not shown is P301L/S, which does not affect splicing. The boxed regions define enhancers (gray) or silencers (white). Also shown is the complementarity of the 5′ splice site with the U1 snRNA. Lines are Crick/Watson pairs, dots are G-T pairs. The effect of each mutation on exon 10 splicing is listed on the right. (B) Cumulative model of splicing interactions for tau exon 10. Cis elements that act as silencers (gray) and enhancers (striped) are indicated (not to scale). For the trans factors, binding to an enhancer implies that the factor activates splicing, whereas binding to a silencer implies that the factor inhibits it. For the factors, circles depict RNA recognition (RRM) domains, rectangles protein interaction (RS) domains. Factors whose identity and mechanisms of action have been confirmed are shades of gray, whereas factors whose details of action are speculative are white. Deletions and FTDP mutations are indicated as well as a double mutation (M280) which weakens the purine-rich enhancer.
The two hypotheses are not mutually exclusive, but thermodynamic considerations and specific results from the tau system make the linear theory likelier. All mutations that weaken the putative stem also increase complementarity to the 5′ end of U1, so the regulation can be explained by a suboptimal 5′ splice site without invoking a hairpin loop. Additionally, compensatory mutations that restore stem strength do not restore wild-type splicing regulation. The proposed stem/loop structures differ by research group and are unstable by the Tinoco rules. Furthermore, it is known that such structures do not form in vivo (Andreadis, 2006; Caffrey and Wade-Martins, 2007).
Indeed, all the work that supports formation of the stem-loop structure has been done in vitro. The sole exception to this is a recent result that shows helicase p68 is involved in the splicing of tau exon 10 at this region (Kar et al., 2011). However, p68 is part of the spliceosome and helicases are integral to the obligatory unwinding of the pre-mRNA prior to splicing.
Besides regulation by p68, work from my laboratory (Gao et al., 2007; confirmed by Ding et al., 2011) demonstrated that SR protein 9G8, the most potent inhibitor of exon 10 splicing, interacts directly with the intronic silencer at position +14 (defined by an FTDP mutation). Further work in our laboratory established that the intronic enhancer downstream of the silencer is regulated by a complex consisting of hnRNPE3, hnRNPE2 and SRA4 (Wang et al., 2010).
The action of several factors that regulate exon 10 is modulated by nuclear and shuttling kinases, including DYRK1A (Ding et al., 2011; Shi et al., 2008). The involvement of hnRNPE3, SRA4 and DYRK1A is intriguing because their genes are located on chromosome 21. This ties tau splicing regulation and isoform ratio balance into the early-onset of cognitive impairment in DS.
Besides the involvement of the 5′ splice site, exon 10 splicing is affected by additional exonic silencers and enhancers identified by FTDP mutations and/or systematic deletions (Andreadis, 2006). The most extensively studied of these elements is a purine-rich enhancer that overlaps mutants N279K and Delta280K (Fig. 2A). This element binds SR-like protein htra2beta1, which moderately activates splicing of exon 10. Its action is antagonized by hnRNPG, which sequesters htra2beta1 by binding to it, and by SR proteins SRp54 and SRp55/SRp30c that sterically interfere with htra2beta1 by binding to a silencer upstream of the purine-rich enhancer (Fig. 2B).
Work from several laboratories has matched additional cis elements with trans factors, but others are still orphans waiting for their partners. Fig. 2B shows a cumulative picture of the various splicing interactions discovered for exon 10. As I mentioned earlier, neurodegeneration occurs regardless of whether the ratio shifts towards 10− (mutations Delta280K, +19, +29) or 10+ (all the others) and regardless of how much the ratio changes. Interestingly, the tangles that form differ in terms of exactly how the tau in them aggregates, depending on which isoform (10− or 10+) is overexpressed (Gasparini et al., 2007).
Enigmatic saitohin and the unique human susceptibility to dementia
A decade ago, another potential player entered the already crowded neurodegeneration stage. Saitohin (STH) is an intronless gene located in the intron between exons 9 and 10 of the human tau gene that encodes an open reading frame of 128 amino acids (Conrad et al., 2002). STH expression is highly congruent with that of tau in human tissues. The DNA sequences homologous to the human STH gene reveal an intact, highly conserved open reading frame in the primates most closely related to humans (chimpanzee, bonobo and gorilla) but not in other primates or rodents (Holzer et al., 2004).
A single nucleotide polymorphism of human STH has been identified that changes glutamine residue 7 to arginine (Q7R). This polymorphism is associated with the two non-recombining tau gene haplotypes: the Q allele with H1, the R allele with H2 (Conrad et al., 2004). Although our primate cousins show a mix of the H1 and H2 haplotypes, they are all homozygous for the R allele (Holzer et al., 2004). So the Q allele is a human-specific marker that came into existence after the hominin lineages separated from the ancestor we shared with bonobos and chimpanzees. This wrinkle is interesting beyond evolutionary relationships: the STH Q allele is over-represented in several tangle-only tauopathies, as well as Parkinson’s disease (Tobin et al., 2008).
STH does not have obvious motifs and domains, which has hampered studies of its function. My laboratory found that it interacts in allele-specific fashion with peroxiredoxin 6, the sole member of that family with a unique phospholipase function whose levels also increase in Pick’s disease, a tangle-only tauopathy (Gao et al., 2005). This is an intriguing observation in view of the fact that mitochondrial dysfunction appears to be a component of neurodegeneration (Eckert et al., 2010). We are continuing to search for additional STH ligands, because we suspect that the protein’s Q allele may be connected to the uniquely human susceptibility to neurodegeneration.
The never-shortening list of unanswered questions
Despite progress along several fronts, many crucial questions around tau remain unanswered. Some may appear literally of academic interest, but their elucidation will inform diagnostic and therapeutic approaches to tauopathies. From molecule to organism, here is a partial list that does not include equally important questions connected to tau post-translational modifications (de/phosphorylation, nitration, glycosylation, truncation) or degradation pathways:
Which model of the splicing regulation at the 5′ splice site of exon 10 is correct? The two theories (stem-loop versus linear) are not mutually exclusive but resolution is important as an aid to designs of future RNA-based interventions, even after we have solved the thorny issues of how to deliver and calibrate the expression of such constructs.
How does the imbalance of tau isoforms cause neurodegeneration? Since neurodegeneration occurs regardless of which way the ratio of exon 10 tilts, the inevitable conclusion is that balance between 3R and 4R isoforms must remain within a narrow window to ensure normal neuronal function. This correlates with the finding that mice which overexpress tau develop severe neuropathies or gliopathies regardless of transgene details (Brandt et al., 2005; LaFerla, 2010).
Perhaps the 1:1 3R:4R ratio is critical for correct MT dynamics in specific contexts within the various cell types of the human brain, a balancing act between fluidity and stability. Alternatively or additionally, ratio imbalances of the domains encoded by alternatively spliced exons can influence tau subcellular localization and interactions with other cytoskeletal or membrane components, including regulatory kinases and phosphatases (Gendron and Petrucelli, 2009).
Which is the neurotoxic tau species? Results from animal, cellular and in vitro models give inconsistent results: some support the long-held view that tau aggregated in NFTs is the culprit, while others indicate that NFTs are inert, safe “warehouses” of otherwise toxic soluble tau species (Götz et al., 2008; Sahara et al., 2008; Jellinger, 2009). The latter theory gains support from both invertebrate and vertebrate animal models that overexpress tau, in which neurodegeneration and cognitive impairment occur without tangle formation.
This paradigm shift, according to which neurons are fated to degenerate once toxic tau oligomers accumulate, highlights the fact that prevention of neuronal death may require intervention at a stage earlier than NFT formation -- and that a reagent which dissolves NFTs may be in fact be terribly harmful if it leads to re-formation of toxic oligomers.
Which function of tau is crucial to neuronal health and/or compromised in neurodegeneration? Although tau was originally defined as an organizer of axonal MTs, its functions continue to expand. Tau is now known to be involved in kinesin-dependent axonal transport (Morfini et al., 2009) and in signal transduction in dendritic spines, in connection with the interaction of kinase fyn (which phosphorylates tau on Tyr18) with NMDA receptors (Ittner and Götz, 2011). Synapse loss and disruption of axonal transport are both early events in neurodegeneration that occur in advance or in the absence of NFT formation. Association of tau with the membrane appears to be equally crucial during neuronal development, as tau is then found in the growth cones of extending neurites which exclude MTs via a complex actin-based “shield” (Gordon-Weeks, 1993).
The prevailing paradigm of tau conformation has also changed, from the traditional view of it as a coil-coil protein to increasing evidence that it normally exists in a paperclip configuration that unravels during neurodegeneration (Jeganathan et al., 2006). This places tau in the lengthening list of proteins that may cause neuronal damage by accumulation of misfolded aggregates, from amyloid to prions.
Is tau inherently toxic in late life? Between the mild phenotype of tau null mice (although they would clearly fare poorly in the wild) and the decrease of amyloid-mediated toxicity upon tau removal (Denk and Wade-Martins, 2009; LaFerla, 2010), some researchers are starting to argue that we might be better off without tau in late life. However, the deficits in human pedigrees with tau microdeletions and the human-specific aspects of tau (discussed in the next section) should give us pause before we contemplate large-scale alterations to our brain/mind.
Of mice and (wo)men
Mouse AD and FTDP models have been very helpful in shedding light on specific aspects of the neurodegenerative cascade (Denk and Wade-Martins, 2009; Ashe and Zahs, 2010). However, they do not fully recapitulate either disease and often show disparate or even contradictory outcomes, in part due to differences in epigenetic or environmental details. In particular, no mouse model so far reproduces the regulatory profile of tau exon 10. This is not surprising, since the FTDP hotspot region is not conserved between rodents and humans (Andreadis, 2006) and different sets of genes are affected by age in the two species, often in completely opposite ways (Bishop et al., 2010).
Furthermore, the intrinsic nature of these types of dementia makes it certain that their species specificity needs to be taken into account to address them effectively. FTDP primarily affects the frontal cortex, a brain compartment uniquely enlarged in humans, and tau disturbances in either early or late life affect higher executive functions (social affect, empathy, decision-taking, judgment, language skills) that are also unique to humans.
This is mirrored at the molecular level: human tau differs from its rodent counterpart in several small but crucial ways. In addition to the human-specific splicing of exon 10 and the human-specific Q allele of saitohin, human exon 1 has a small insertion that may affect fyn binding and tau folding, and human exon 4A is longer than its mouse counterpart. In biology, some aspects are conserved across species (molecular functions foremost among them) but the devil is in the details.
It is a sad irony that we humans have become susceptible to neurodegeneration due to our greatly increased lifespan, made possible by clean water, antibiotics and vaccines. In this connection, tau may be a case of Medawar and Williams’ antagonistic pleiotropy: beneficent in development, detrimental in advancing age. It is possible that tau dysfunction contributes to neurodegeneration by many mechanisms and at different stages of the process.
At the cellular level, toxicity is defined by the final outcome of neuronal death. If enough neurons die in a brain compartment, the decreasingly plastic adult brain can no longer rewire and reroute local functions, eventually resulting in the clinical presentations of dementia. Short-lived mammals such as rodents appear largely immune to such diseases, which require chronic imbalance, progressive loss of normal function and/or accumulation of toxic species.
Parting words
Despite the harnessing of enormous intellectual and physical resources, the frustrating fact remains that since I began working on tau in 1989, we have not come a single step closer toward preventing or curing dementia.
When I entered the tau field and became familiar enough with the intricacies of the molecule to hazard educated guesses, I made two predictions: mis-splicing of tau would be found to directly cause neurodegeneration; and the tau splicing variants I discovered that lack the microtubule-binding domain would moderate a tau function in the manner of graphite rods in a nuclear reactor. Both predictions have been confirmed, although I was wrong about the target of the latter: my prime suspect was neurite extension, but it appears to be axonal transport.
I will now make a few more predictions, based on what we have started to glimpse from recent investigations into tau and dementia that veered off heavily trodden paths: signal transduction will turn out to be a major tau function; the human-unique STH Q allele will affect a tau function relevant to neurodegeneration; and the means of preventing and/or curing tauopathies (conceptually, if not technologically) will come from an unexpected finding, far from the street lamp of the obvious.
Discovery of the specific functions of the tau isoforms arising from alternative splicing has given and will continue to give significant insights into the cooperative networks that establish and maintain neuronal function. Additionally, work on species-specific regulation will keep us alert to the need for subtlety, a crucial attribute for devising effective therapies targeted to chronic diseases that do not fit into the heroic medicine model. Results from such work may reveal the processes common to dementia in which NFTs are the sole or major pathological manifestation and in the long term give us a handle for ameliorating, preventing or even reversing dementia – a specter that looms ever darker as the human lifespan lengthens.
Acknowledgments
I apologize to colleagues whose work I could not cite due to space constraints. I want to thank NSF and NIH for making my contributions to tau research possible.
Literature cited
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochem Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Andreadis A. Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog Mol Subcell Biol. 2006;44:89–107. doi: 10.1007/978-3-540-34449-0_5. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta. 2005;1739:331–54. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Caffrey TM, Wade-Martins R. Functional MAPT haplotypes: bridging the gap between genotype and neuropathology. Neurobiol Dis. 2007;27:1–10. doi: 10.1016/j.nbd.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Vianna C, Freeman M, Davies P. A polymorphic gene nested within an intron of the tau gene: implications for Alzheimer’s disease. Proc Natl Acad Sci USA. 2002;99:7751–7756. doi: 10.1073/pnas.112194599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Vianna C, Schultz C, Thal DR, Ghebremedhin E, Lenz J, Braak H, Davies P. Molecular evolution and genetics of the Saitohin gene and tau haplotype in Alzheimer’s disease and argyrophilic grain disease. J Neurochem. 2004;89:179–188. doi: 10.1046/j.1471-4159.2004.02320.x. [DOI] [PubMed] [Google Scholar]
- Conrad C, Zhu J, Conrad C, Schoenfeld D, Fang Z, Ingelsson M, Stamm S, Church G, Hyman BT. Single molecule profiling of tau gene expression in Alzheimer’s disease. J Neurochem. 2007;103:1228–1236. doi: 10.1111/j.1471-4159.2007.04857.x. [DOI] [PubMed] [Google Scholar]
- Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging. 2009;30:1–13. doi: 10.1016/j.neurobiolaging.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Shi J, Qian W, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F. Regulation of alternative splicing of tau exon 10 by 9G8 and Dyrk1A. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2010.11.021. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan PJ, Johnson GV. The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Devel. 2010;13:595–603. [PMC free article] [PubMed] [Google Scholar]
- Eckert A, Schulz KL, Rhein V, Götz J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107–14. doi: 10.1007/s12035-010-8109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Tse SW, Conrad C, Andreadis A. Saitohin, which is nested in the tau locus and confers allele-specific susceptibility to several neurodegenerative diseases, interacts with peroxiredoxin 6. J Biol Chem. 2005;280:39268–39272. doi: 10.1074/jbc.M506116200. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang J, Wang Y, Andreadis A. SR protein 9G8 modulates splicing of tau exon 10 via its proximal downstream intron, a clustering region for frontotemporal dementia mutations. Mol Cell Neurosci. 2007;34:48–58. doi: 10.1016/j.mcn.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Terni B, Spillantini MG. Frontotemporal dementia with tau pathology. Neurodegen Dis. 2007;4:236–253. doi: 10.1159/000101848. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13–31. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, Rosenberg C, Hulette C, Jellinger K, Hampel H, Rieder P, Moeller HJ, Andreadis A, Henkel K, Stamm S. The alternative splicing of tau exon 10 and its regulatory proteins clk2 and tra2-beta1 changes in sporadic Alzheimer’s disease. J Neurochem. 2005;96:635–644. doi: 10.1111/j.1471-4159.2005.03552.x. [DOI] [PubMed] [Google Scholar]
- Gordon-Weeks PR. Organization of microtubules in axonal growth cones: a role for microtubule-associated protein MAP 1B. J Neurocytol. 1993;22:717–725. doi: 10.1007/BF01181317. [DOI] [PubMed] [Google Scholar]
- Götz J, Ittner LM, Fändrich M, Schonrock N. Is tau aggregation toxic or protective: a sensible question in the absence of sensitive methods? J Alzheimers Dis. 2008;14:423–429. doi: 10.3233/jad-2008-14410. [DOI] [PubMed] [Google Scholar]
- Hertel KJ. Combinatorial control of exon recognition. J Biol Chem. 2008;283:1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- Holzer M, Craxton M, Jakes R, Arendt T, Goedert M. Tau gene (MAPT) sequence variation among primates. Gene. 2004;341:313–22. doi: 10.1016/j.gene.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Götz J. Amyloid-β and tau--a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Recent advances in our understanding of neurodegeneration. J Neural Transm. 2009;116:1111–1162. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- Kar A, Fushimi K, Zhou X, Ray P, Shi C, Chen X, Liu Z, Chen S, Wu JY. RNA Helicase p68 (DDX5) regulates tau exon 10 splicing by modulating a stem-loop structure at the 5′ Splice Site. Mol Cell Biol. 2011;31:1812–1821. doi: 10.1128/MCB.01149-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Pathways linking Abeta and tau pathologies. Biochem Soc Trans. 2010;38:993–995. doi: 10.1042/BST0380993. [DOI] [PubMed] [Google Scholar]
- Llamusí B, Artero R. Molecular Effects of the CTG Repeats in Mutant Dystrophia Myotonica Protein Kinase Gene. Curr Genomics. 2008;9:509–16. doi: 10.2174/138920208786847944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–51. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebouvier T, Scales TM, Williamson R, Noble W, Duyckaerts C, Hanger DP, Reynolds CH, Anderton BH, Derkinderen P. The microtubule-associated protein tau is also phosphorylated on tyrosine. J Alzheimers Dis. 2009;18:1–9. doi: 10.3233/JAD-2009-1116. [DOI] [PubMed] [Google Scholar]
- Lee G. Tau and src family tyrosine kinases. Biochim Biophys Acta. 2005;1739:323–30. doi: 10.1016/j.bbadis.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Long JC, Cáceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008;3:8–17. doi: 10.1186/1750-1326-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. HnRNP proteins and splicing control. Adv Exp Med Biol. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Pennisi E. 17q21.31: not your average genomic address. Science. 2008;322:842–845. doi: 10.1126/science.322.5903.842. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Hanger DP. Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans. 2010 Aug;38(4):1012–5. doi: 10.1042/BST0381012. [DOI] [PubMed] [Google Scholar]
- Sergeant N, Sablonnière B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, Vermersch P, Delacourte A. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10:2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Takashima A. Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr Alzheimer Res. 2008;5:591–598. doi: 10.2174/156720508786898442. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, Zhou J, Hwang YW, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem. 2008;283:28660–28669. doi: 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochem Biophys Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin JE, Latourelle JC, Lew MF, Klein C, Suchowersky O, Shill HA, Golbe LI, Mark MH, Growdon JH, Wooten GF, Racette BA, Perlmutter JS, Watts R, Guttman M, Baker KB, Goldwurm S, Pezzoli G, Singer C, Saint-Hilaire MH, Hendricks AE, Williamson S, Nagle MW, Wilk JB, Massood T, Laramie JM, DeStefano AL, Litvan I, Nicholson G, Corbett A, Isaacson S, Burn DJ, Chinnery PF, Pramstaller PP, Sherman S, Al-hinti J, Drasby E, Nance M, Moller AT, Ostergaard K, Roxburgh R, Snow B, Slevin JT, Cambi F, Gusella JF, Myers RH. Haplotypes and gene expression implicate the MAPT region for Parkinson disease: the GenePD Study. Neurology. 2008;71:28–34. doi: 10.1212/01.wnl.0000304051.01650.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gao L, Tse SW, Andreadis A. Heterogeneous nuclear ribonucleoprotein E3 modestly activates splicing of tau exon 10 via its proximal downstream intron, a hotspot for frontotemporal dementia mutations. Gene. 2010;451:23–31. doi: 10.1016/j.gene.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS. Tau mutations in neurodegenerative diseases. J Biol Chem. 2009;284:6021–6025. doi: 10.1074/jbc.R800013200. [DOI] [PubMed] [Google Scholar]