

Abstract
Hairpin peptides bearing cross-strand Trp-Trp and Tyr-Tyr pairs at non-H-bonded strand sites modulate the aggregation of two unrelated amyloidogenic systems: human pancreatic amylin (hAM) and α synuclein (α-syn), associated with type II diabetes and Parkinson’s disease, respectively. In the case of hAM, we have previously reported that inhibition of amyloidogenesis is observed as an increase in the lag time to amyloid formation and a diminished thioflavin (ThT) fluorescence response. In this study, reduced hAM fibril formation is confirmed by TEM imaging. Several of the hairpins tested were significantly more effective inhibitors than rat amylin. Moreover, a marked inhibitory effect on hAM-associated cytotoxicity by the more potent hairpin peptide is demonstrated. In the case of α-syn, the dominant effect of active hairpins was, besides a decreased ThT fluorescence response, the earlier appearance of insoluble aggregates that do not display amyloid characteristics with the few fibrils observed having abnormal morphology. We attribute the alteration of the α-synuclein aggregation pathway observed to the capture of a preamyloid state and diversion to non-amyloidogenic aggregates. These β hairpins represent a new class of amyloid inhibitors that bear no sequence similarity to the amyloid-producing polypeptides that are inhibited. A mechanistic rationale for these effects is proposed.
Keywords: human pancreatic amylin, α-synuclein, Parkinson’s disease, amyloid fibril formation, cytotoxicity inhibition, circular dichroic detection of β structure, bioactive β–hairpins, time course of thioflavin T fluorescence enhancement
Protein folding diseases are an area of intense interest at present. Many protein folding diseases involve the formation of polypeptide aggregate deposits with a common cross-β-sheet fibrillar geometry and dye staining properties. Such amyloid fibrils are associated with more than 40 human diseases and conditions (1), including type II diabetes (2), Parkinson’s disease (PD) (α-synuclein aggregates in Lewy bodies) (3), and other neurodegenerative conditions (e.g. Alzheimer’s and Huntington’s diseases). Fibril formation kinetics (4, 5) imply a complex multi-stage, autocatalytic nucleation-dependent polymerization process with a lag phase followed by rapid, cooperative fibril formation.
While there are many therapeutic strategies (6, 7) for amyloid-associated diseases, there is a commonly-held expectation that amyloidogenesis inhibition has potential as either a preventative or ameliorating therapy for some of these medical conditions that cause human suffering and exact a tremendous societal burden. Three strategies related to the amyloidogenesis process are given here: 1) interfering with the processing of the proteins that afford the amyloidogenic peptides, 2) diverting preamyloid intermediates prior to the toxic states to non-toxic aggregates, and 3) reducing the steady-state concentration of toxic intermediates (8, 9) in the amyloidogenic pathway by tinkering with the relative rates of the steps in the aggregation pathway. Therapeutic development based on the third strategy requires greater definition of the mechanisms of amyloidogenesis and the determination of the toxic species for each of the disease-related amyloidogenic species. Selective inhibitors of these processes should prove useful in this endeavor.
Numerous inhibitors of amyloid formation have been discovered or designed; these include small molecules, peptides and proteins that affect amyloid formation either by delaying the onset of fibril formation or diverting toxic aggregates to non-toxic aggregates of different morphology. Most of the small molecule amyloidogenesis inhibitors are polyphenols which display inhibition for a wide variety of amyloidogenic sequences and fragments. In the case of (−)- epigallocatechin-3-gallate (EGCG), a green tea component, ‘inhibitory potency’ against at least five diverse amyloidogenic systems has been demonstrated.(10, 11) It has been proposed that EGCG works by diverting poorly folded species to non-amyloidogenic oligomers and eventually, non-toxic aggregates, rather than to amyloid fibrils via toxic preamyloid species.
Most of the peptide amyloidogenesis inhibitors presented in the literature, are solubilized (12, 13) and/or mutated versions (14) of the most amyloidogenic sequence fragments of the polypeptide system of interest. The common strategy is “β assembly disruption” by introducing residues that discourage β strand formation and/or association such as proline, N-methylated or α-disubstituted amino acid residues (14–17).
A report by Ghosh and coworkers (18), which demonstrated that a hyper-stable mutant of the B1 domain of protein G could be ‘evolved’ into a potent inhibitor of the aggregation of Aβ (1–40) peptide, served to focus our attention on Trp/Tyr bearing β hairpin peptides as potential amyloidogenesis inhibitors. The substitutions seen in the inhibitory protein included K→W, G→W, K→Y and E→Y mutations. Seven of the eight mutations that appeared occurred on the exposed face of a single hairpin of the B1 domain. Over the last eight years, the a priori design of β hairpins has been improved (19–23) to the point that 10 – 16 residue constructs that are >85 % folded in water can be prepared routinely. As a result, we became interested in establishing whether designed β hairpins could serve as mini-receptors and pharmacophore display scaffolds for drug lead discovery.
We have previously reported that β-hairpin peptides bearing both Trp and Tyr residues inhibit fibril formation by human pancreatic amylin (hAM) (24). These inhibitory peptides bear no structural resemblance to hAM and expose Trp and Tyr residues at varying positions along the β strands of the hairpin structure. The mechanistic hypothesis behind the use of these hairpins as inhibitors can be stated as follows: that the prestructured strands of the β hairpins bearing Trp and Tyr residues will facilitate intermolecular association and then sheet formation with preamyloid states and thus prevent or delay the self-self recognition associated with β oligomerization and fibril growth.
To probe the generality of our hypothesis regarding hairpin inhibition of amyloidogenesis we tested a selection of aromatic containing hairpins against another unstructured amyloidogenic system, α-synuclein (α-syn), using CD, Thioflavin T (ThT) fluorescence (25, 26), TEM imaging and Congo red (CR) staining: modulation of amyloidogenesis by β hairpins was also observed for α-syn. We also confirm the inhibitory effect of our most potent Trp-containing hairpin inhibitor against hAM by TEM imaging, examine additional controls, and extend the study to cytotoxicity effects. These aromatic-containing hairpins are peptidic aggregation inhibitors that lack any sequence similarity with the amyloidogenic polypeptides yet appear to be among the most potent small peptide inhibitors of amyloidogenesis reported to date.
Experimental Procedures
Materials
α Synuclein. The α synuclein sample was prepared at the University of Padova. Human α-synuclein cDNA was subcloned into the NcoI and XhoI restriction sites of the pET28a plasmid (Novagen). Since ~20% of the expression product from Escherichia coli represents mistranslation such that a cysteine residue is incorporated at position 136 instead of a tyrosine), a site-directed mutagenesis of codon 136 (TAC to TAT) was employed. This results in the 100% expression of α-synuclein with the correct sequence. The protein was expressed in E. coli BL21(DE3) growing the cells in Luria Bertani medium. The over-expression product was recovered from the periplasm by osmotic shock as previously described (27). The cell homogenate is boiled for 15 min and the soluble fraction was treated with a two step (35% and 55%) ammonium sulfate precipitation. The pellet was resuspended, dialyzed against 20 mM Tris-HCl pH 8.0, loaded into a 6 mL Resource Q column (Amersham Biosciences) and eluted with a 0–500 mM gradient of NaCl. After dialysis against milli-Q water, the protein is lyophilized and stored at −20 °C.
hAM
For our preliminary studies, samples of hAM(1–37) remaining from prior studies (28) were re-purified by HPLC (C18). In addition, Amylin Pharmaceuticals provided two lots (25 and 10 mg, respectively) of hAM for the fibrilization inhibition studies. Synthetic hAM (1–37) obtained by Fmoc-solid phase peptide synthesis, as previously described (29), was employed for the cytotoxicity studies.
Hexafluoroisopropanol used in this study was a gift (lot # 08012) from Halocarbon Corp., which is hereby acknowledged. All other solvents and chemicals used were reagent or spectroscopic grade commercial materials.
Peptide synthesis
Peptide hairpins and controls were synthesized on an Applied Biosystem 433A peptide synthesizer using standard Fmoc solid-phase peptide synthesis methods. Wang resins preloaded with the C-terminal amino acid were employed. C-terminal amides were prepared similarly but using Rink resins. Peptides are cleaved from the resin using a 95:2.5:2.5 trifluoroacetic acid (TFA): triisopropylsilane: water mixture. The cleaved peptides were purified by reverse phase HPLC on a Varian C18 prep-scale column using gradients of water/acetonitrile (having 0.1% and 0.085% TFA respectively). Collected fractions were lyophilized and their identity and molecular weight confirmed using a Bruker Esquire ion trap mass spectrometer.
NMR Spectroscopy
The derivation of NMR structures for bioactive peptides and controls was carried out using NOESY/TOCSY spectra as previous described (21, 22).
Aggregation and Aggregation Inhibition Assays
hAM assays were performed as previously described (24). Stock solution of hAM (400 μM) were prepared in neat HFIP and vortexed for 5 min. To initiate aggregation/fibril formation, the stock is diluted to 8 μM hAM with 5mM pH 7.2 phosphate buffer, producing a final 2 vol-% HFIP concentration.
In the case of α-synuclein, we prepare 250 μL of 100μM α-syn in 20mM Tris-HCl buffer, pH 7.5 with 1.5 vol-% HFIP in a vial containing a stirring bar with or without added peptide inhibitors and/or ThT as follows. The ThT stock was 800 μM in20mM Tris-HCl buffer (pH 7.5). Peptide inhibitor stocks were 1 mM in the same buffer. The required amount of α-syn was placed in the 1 mL glass vial equipped with a small Teflon stir bar, to which was added either 200 μL of buffer or 150 μL of buffer and 50 μL of inhibitor stock (at 37°C); and aggregation was initiated by adding 50 μL of the HFIP stock solution. The HFIP stock was 7.5 vol-% HFIP in pH 7.5 20mM Tris-HCl buffer. The process was carried out with continuous vigorous stirring while the vial was incubated in a 37°C waterbath, aliquots were removed at different time points (every 2–3 hrs) for assays. To follow the time course of ThT signal development in controls and for inhibitors that resulted in accelerated cloudiness, 7μL of ThT stock was added just prior to HFIP addition and fluorescence measurements were obtained at t = 0 and every 2 – 3 h thereafter as described below.
Circular Dichroism Spectroscopy
Stock peptide solutions for CD experiments are prepared either in 10mM phosphate buffer (or water/HFIP mixtures). The concentrations of the stock solutions are determined by the UV absorption of tyrosine and tryptophan (ε = 1190 M−1 cm−1 and 5580 M−1 cm−1, respectively at 278 nm). CD samples with 6–30μM peptide concentrations are prepared by dilution of the stock solution. Spectra are recorded on a Jasco J720 instrument in cells with a path-length of 0.1 cm. Typical spectral accumulation parameters are a scan rate of 100 nm/min with a 2 nm band-width, and a 0.1 nm step resolution over the wavelength range 185–270 nm with 4–8 scans averaged for each spectrum. For time course experiments this allows the collection of full spectral scans every 10 min. Raw ellipticity data are converted into molar, not residue-molar, ellipticity units (deg.cm2/dmol), using the Jasco software.
For CD experiments on α-syn aggregation samples, 5μL aliquots of assay sample (without added ThT) is removed and diluted with 195 μL of the 1.5% HFIP Tris-HCl buffer (2.5 μM final α-syn concentration). This was done every 2–4 hours in order to monitor transition from random coil to α sheet signal. Correction for the CD contribution of the inhibitor is less significant, but is carried out nonetheless.
ThT Fluorescence Detection of Amyloid Species
For α-syn aggregation assays, 200 μL of assay mixture is employed as described above. For assays that had been monitored by CD, 5 μL of ThT stock is added 5 min prior to the fluorescence measurements, typically at the 16 h point in the assay.
Imaging Fibrils and other Oligomers by Congo Red Staining and TEM
For TEM imaging, a 5μL aliquot of a freshly agitated aggregation assay sample was adsorbed onto formvar/carbon-coated 400 mesh copper grids and negatively stained with 2% uranyl acetate. Images were acquired using a Philips CM100 transmission electron microscope. For hAM assays, TEM images were typically obtained at 1, 2, 4, and/or 16 hrs. In the case of α-syn, images were obtained after 16–24 hrs.
Congo red staining solution was prepared according to Nilsson (2004) (30). After incubation for 16 – 48 hrs, an aggregation assay mixture is agitated just prior to withdrawing a 10 μL sample which is placed onto a glass slide, air-dried, and 200μL of Congo red solution is placed onto the dried polypeptide sample. After a few minutes, the excess dye solution is blotted away and the sample is observed in a light microscope (Olympus BX 60) equipped with crossed polarized filters to observe birefringence.
Cell viability assays
The rat insulinoma cell line RIN5fm was employed; the cells were cultured and platted as previously described (31). Freshly made solutions of hAM (5 μM) alone or mixtures of hAM (5 μM) with various amounts of WW2 or μPro1 as indicated were made in 10 mM sodium phosphate buffer, pH 7.4 containing 1% HFIP and incubated at room temperature for ~20 h. Thereafter, the solutions were diluted with cell culture medium and added to the cells at the indicated final hAM concentrations. Following incubations with the cells for 20 h, cell damage was assessed by measuring the cellular reduction of MTT as previously described (32). Of note, under the experimental conditions employed, hAM was in a nonfibrillar state at the beginning of the pre-incubation (15).
Results
Hairpin Peptides Examined as Modulators of Amyloidogenesis
The hairpins tested in this study were previously characterized by NMR and CD, and are least 50% folded under the assay conditions. None of the hairpins or non-hairpin controls aggregated or enhanced thioflavin T (ThT) fluorescence, even at 2 mM concentrations after 5 days. The hairpins used in this study are shown in scheme 1. The list includes controls as well as μPro1, a β-capped microprotein (21).
Scheme 1.

a The peptide with no citation is analogous to the others of the same class (HP7)(18), prepared specifically for this study and was synthesized and characterized by the methods given in the citation.
b The peptides are classified by whether they have a Trp-flanked reversing loop(17, 18) and whether they are derived from a hairpin originally based on the dimer interface of the Met repressor (MrH) (60, 61). Bold lower case, p = D-Pro; aromatic residues are also highlighted in bold.
Assay Conditions and Methods
Most previously reported α-syn aggregation assays have reported long lag times (days to many weeks) even at high protein concentrations (150–250 μM) with warming and aggressive agitation (33, 34). We chose to design an assay with a shorter lag time that would allow for ready monitoring of the course of amyloid formation by employing media containing 1.5 – 2 vol-% hexafluoroisopropanol (HFIP). The incorporation of small amounts of HFIP is a common feature in many fibrilization assays (34). The specific assay which we developed for hAM, 8 μM hAM in pH 7.2 phosphate buffer containing 2 vol-% HFIP, gave results consistent with data of Miranker and coworkers (35–37). The hAM fibrilization kinetics, as monitored by the ThT binding assay, were very similar to prior studies by some of us (15, 31). The Raleigh group (38) has adopted a similar assay but with constant stirring which also accelerates aggregation. In the case of α-synuclein, the assay that gave reproducible aggregation time courses employed 100 μM α-syn, with HFIP addition (to 1.5 vol-% final concentration) to the stirred solution as the aggregation-initiating event. Munishkina (39) has also reported shorter lag times in buffers with added HFIP. For both amyloidogenic systems, lag times were measured using both CD spectroscopy and a ThT fluorescence assay. CR-stained and/or TEM images were obtained at the time corresponding to complete fibrilization in uninhibited assays (16 h for α-syn, 40 – 60 min for hAM) and at later time points.
hAM aggregation inhibition (TEM imaging confirms the CD and ThT assay results)
Our previously reported results indicated that Trp containing hairpins were among the more potent hairpins against hAM fibrillization (24). The effects of selected hairpin peptides on hAM aggregation lag times appear in Table 1. The most potent inhibitor (WW2) is a Trp-flanked turn species; as a result, we added additional W-loop-W peptides to our screen. In light of the recent report (38) on the inhibition of hAM aggregation by rat amylin (rAM) we also included this non-amyloidogenic analog as a ‘control’ amylin-like species. The extent of inhibition observed (see also Table 3, vide infra) and the effects of selected hairpin peptides on hAM aggregation lag times appear in Table 1; representative time courses of ThT fluorescence in our assays appear in Figure 1.
Table 1.
Hairpin inhibition of hAM aggregation (lag times observed) a -- data for ThT data with errors are from triplicate determinations; data from comparable CD assaysb is given in parentheses.
| Peptide | Lag time in minutes | |||
|---|---|---|---|---|
| Molar equivalents added | ||||
| 1 eq | 2eq | 4 eq | 6eq | |
| rAM | 50 (45–50) | 50 | 93 | |
| MrH3b | -- | -- | 55 (45) | 65 (95) |
| WW2 | 151±42 (167±25) | 253±52 (120–300) | 344±28 | -- |
| WW3 | (60) | (80) | 98±10 (180) | 107±33 |
| WW4 | (114) | 82±33 (105–120) | 155±11 | 333 |
| YY2 | 139 | 201±42 (70–80) | 190 | 303±37 |
| YY3 | -- | 58 | 52 | 44 (40) |
| Other W-loop-W hairpins | ||||
| HP7AAA | -- | 70 | -- | -- |
| μPro1 | -- | 30 (35) | 50 | 60 |
The lag time for hAM aggregation is defined as the time at which the ThT signal reaches ca. 15% of its final value, does vary, 61±14 min. (n > 15), this also appears to be the point at which CD β minimum (217 nm) in the CD spectrum becomes distinct (35–70 min.).
The extreme sensitivity to the precise amount of HFIP is likely a source of lag time variability since it is difficult to control HFIP content to 1.5±0.1 vol-% of the medium. Since the two measurements are always obtained in separate experiments, we do not know whether the measures are coincident in any specific run, but it appears that either can be used to monitor the aggregation time course.
Table 3.
Summary of hairpin peptide effects on hAM and Synuclein amyloid formation. The ThT fluorescence (as % of uninhibited control) observed with 2 molar equivalents of added hairpin appears as the first entry, followed by other observations for each species.
| Hairpin peptide | hAM a | Synuclein b |
|---|---|---|
| WW2 | < 5 % c 4-fold extended lag phase c inhibits cytotoxicity |
19±5% A few fibrils with alternate morphology non-amyloid (CR) aggregate precipitation |
| WW3 | 80–100 % slight delay in onset at 6 equiv. |
20% fibrils with alternate morphology |
| YY2 | 42 ± 6 % delayed onset d |
18±7% spherical aggregates non-amyloid (CR) aggregate precipitation |
| MrH3b | No effect even at 4 equiv. | 32% |
| μPro1 | 123 ± 7 % Shorter lag phase greater yield of fibrils by TEM no effect on hAM cytotoxicity |
69% normal fibril morphology and staining |
| HP7AAA | 90 % No effects, confirmed by TEM |
80% Slight delay in onset |
ThT fluorescence measured at 2.5 hours, the full reference response is observed at 1.2 hours for uninhibited controls with little or no loss in signal over the next 2 hours.
ThT fluorescence measured at 16 hours for α-syn assays. CR refers to Congo red staining and the observation of birefringence.
ThT fluorescence measured at 5 hours was 43% of that observed in a control.
A 5-fold extension of the lag phase is observed when 6 equivalents of YY2 are added to hAM, with modest levels of ThT fluorescence observed only after 5 hours.
Figure 1.

Human amylin aggregation assays. a, the time course of ThT fluorescence development in a control and in the presence of selected peptides. b, TEM image of hAM (uninhibited control) after one hour; the scale bar represents 500 nm.
In our assay, rAM was only a modest inhibitor requiring 6 molar equivalents to effect a significant inhibition or display a lag time increase. Of the additional W-loop-W peptides, one was essentially inactive and the other (μPro1) appeared to accelerate and enhance (an increased ThT signal) aggregation, see Figure 1. In a recent study, the Raleigh group (38) cautioned against relying on ThT fluorescence assays and employed TEM imaging for validation. In the case of uninhibited assay, Figure 1B indicates that fibrils are indeed being formed. These fibrils are morphologically comparable to those in previously reported hAM studies with and without HFIP (35, 36, 40). We chose our most active inhibitor (WW 2, which increases the aggregation lag time by at least a factor of 3 when present in a 1-fold molar excess1 and the aggregation-enhancing μPro1 for TEM validation of hairpin effects on hAM aggregation (Figure 2) and cytotoxicity testing.
Figure 2.

TEM images from hAM aggregation assays in the presence Trp-bearing hairpin peptides. Panel a, b, c are representative panels from hAM plus 2 molar equivalents of WW2 at 2h (a), 5h (b) and 20 h (c). The scale bar for a, b and d represent 500nm; the scale bar for panel c = 100nm. Panel c was the grid displaying the greatest density of fibrils. Panel d was recorded after 40 min in the presence of 2 molar equivalents of μPro1; a denser fibrillar network is observed at an early time point.
In the presence of WW2 at 2 molar equivalent, the hAM sample shows, in agreement with the CD and ThT assays (24), no trace of fibrils after 2 hours (Figure 2, panel A). After 5 hours, a few amorphous aggregates are observed (panel B), as well as occasional fibrils (none appear in most grids). After 20 hours, some fibrils were found in most grids, panel C illustrates a particularly fibril-rich grid, but significant inhibition of fibril formation extended to the longest times examined. The TEM image of hAM in the presence of 2 molar equivalents of μPro1 (Figure 2, Panel D) shows a denser web of fibrils after only 40 min. The fibril yield appears to be significantly greater than that observed for the uninhibited control after 60 min, which corroborates the ThT and CD assays for this species (Table 1).
Inhibition of hAM Cytotoxicity
Inhibition of hAM fibrillogenesis has been previously shown to correlate with suppression of formation of cytotoxic hAM assemblies (e.g. citation 41). As a result, we sought to establish whether WW2 (the best of the here-identified hAM fibrillogenesis inhibitors) would also suppress hAM cytotoxicity. For comparison, the effect of μPro1 on hAM cytotoxicity was also studied as this WW-peptide has no inhibitory effect on hAM fibrillogenesis. For these studies, 24 h aged incubations of hAM and mixtures of hAM with WW2 or μPro1 at different hAM/inhibitor molar ratios in 10 mM sodium phosphate buffer (1% HFIP) were added to RIN5fm cells (Figure 3). Following incubation for 20 h, cell viabilities were assessed by the MTT reduction assay. In the presence of 1 equivalent of WW2, no inhibition of formation of cytotoxic assemblies was observed. However, a marked inhibition of hAM cytotoxicity was observed when a two-fold excess of WW2 was applied which was consistent with the results of the ThT binding assay (Figures. 1a and 2). A clear concentration dependence of the inhibitory effect of WW2 was found, with a 6- and 10-fold excess of this peptide strongly suppressing formation of cytotoxic hAM assemblies (Figure 3). In contrast, no reduced hAM cytotoxicity was observed for hAM solutions containing μPro1 even at a hAM:μPro1 1:10 molar ratio. These results were consistent with the results of the ThT assay and demonstrated that WW2, but not the control peptide (μPro1) with a similar Trp/Trp interaction geometry, when applied in a two to tenfold molar excess to hAM, is able to either delay and suppress formation of cytotoxic assemblies derived from hAM or blunt their cytotoxic effect on rat insulinoma cells.
Figure 3.

Effects of WW2 and μPro1 on cytotoxic self-assembly of hAM. Aliquots of 24 h aged incubations of hAM and mixtures of hAM with WW2 or μPro1 at hAM/inhibitor molar ratios of 1/1, 1,2, 1/6, and 1/10 (in the case of WW2) and 1/1 or 1/10 (in the case of μPro1) in 10 mM sodium phoshate buffer (1% HFIP) were diluted with cell medium and added at the indicated final hAM concentrations to RIN5fm cells. Following incubation for 20 h, cell viabilities (% of control) were assessed by the MTT reduction assay. Data are means (±SEM) from 3 assays (n=3 each).
α-Synuclein Studies
The α-syn species employed in our studies is the full-length 140-residue construct produced by over expression in E. coli BL21 (DE3) as described in the Materials & Methods section. The top panels of Figure 4 illustrate α-syn aggregation as monitored by the CD and ThT assays. Uninhibited control solutions become somewhat cloudy 4 – 7 hours2 after HFIP addition (see Figure. 1S); this is also the time point at which ThT fluorescence and the CD β signature are in their growth phase. The aggregation of α-syn results in complete fibril formation over a 16 hour time course. Amyloid fibril formation was also confirmed by CR staining (panels C, D) and TEM imaging (E). The α-syn fibrils produced in the 1.5% HFIP medium display morphology comparable to previous reports for media lacking co-solvent addition (33) and CR staining results in very bright apple-green birefringence, a hallmark of amyloid (26, 42).
Figure 4.
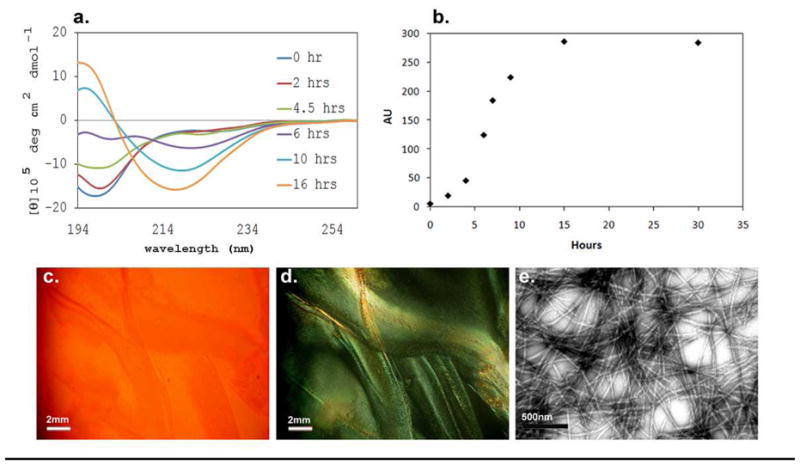
a) CD, molar ellipticity versus wavelength and b) ThT assays monitoring α-syn aggregation over a period of 16–30 hours. The lower panels illustrate CR-stained images (at 48 hours) under non polarized (c) and polarized (d) light, bright green birefringence is observed in panel d; scale bars represent 2mm. A TEM images of the α-syn assay after 16 hours of incubation is shown in panel e; scale bar represents 500nm.
We selected both potent and inactive hairpins from our study of hAM for screening in the α-syn aggregation assay. The inclusion of two molar equivalents of potentially inhibiting peptides in α-syn fibrilization experiments resulted in a variety of responses (Table 2). Although modest delays in lag time as assayed by CD were observed in some cases, the more common observation was an earlier cloud point or immediate precipitation of non-amyloid aggregates. Even MrH3b, with only a single aromatic residue, which was without effect on hAM aggregation, produced accelerated precipitation. Other MrH hairpin sequences (data not shown), even ones with greatly reduced hairpin populations, also displayed some acceleration of precipitation. When the MrH hairpins bearing two aromatic sidechains were present, immediate cloudiness was observed upon HFIP addition2. CD data could not be collected for α-syn assay solution containing these hairpins since the aggregates that formed reduced the observed CD intensities and precluded accurate determination of the CD difference spectrum. The cloudiness that appears 4 – 7 hours after initiation with α-syn alone or in the presence of peptides that do not affect the lag-time did not have this effect: reproducible CD difference spectra could be generated for at least 20 hours after HFIP addition. The enhancement of ThT fluorescence at 16 h was strongly inhibited by hairpins that produced immediate cloudiness.
Table 2.
The effects of added peptides on α-syn aggregation and fibril formation.
| Peptide Inhibitor | Visible Cloudiness | β CD signal | ThT Fl a | Fibril Morphology b | Congo Red staining c |
|---|---|---|---|---|---|
| WW2 | Immediate | n/a | 19±5% | Short thick fibrils + AA. | Little/no BiR |
|
| |||||
| WW3 | Immediate | n/a | 20 % | Short thick fibrils | n.d. |
|
| |||||
| YY2 | Immediate | n/a | 18±7% | Spherical aggregates | Little/no BiR |
|
| |||||
| MrH3b | within 1 hour | CD intensity implies ppt’n, 10% β | 32±7% | n.d | n.d |
|
| |||||
| HP6AYW | Slight after 4–6 hours | β signal within 6 hours, 93% 16 h | 93±12% | Normal | bright green BiRc |
|
| |||||
| μPro1 | Slight after 8 hours | β signal delayed, but 100% β at 16 h | 69±9% | Normal | green BiRc |
|
| |||||
| HP7AAA | after 7 hours | Delayed but full β signal appears (100% at 16 h) | 81±10% | ||
| 4 equivalents | delayed | Delayed. (84% β at 16h. 100% β at day 3). | 67 % | Normal morphology and yield at 5 days | |
|
| |||||
| Non-hairpin controls | |||||
|
| |||||
| ssMrH | Slight after 4–6 hours | β signal within 6 h, 100% 16 h | ≥90% | Normal | n.d. |
|
| |||||
| ssW | after 6 hours | Helical at 4–6 hours 80 % β at 16 h | 61±7% | Normal + AA | Some/green BiRc |
ThT fluorescence is reported as % of the uninhibited control value at 16 h and is given with the experimental error for triplicate experiments. Typical measurements at zero time were 5 – 26 AU units. A greater than 11-fold enhancement in fluorescence at 482 nm is observed on fibril formation. The control ThT fluorescence value at 16 h was well reproduced, 291 ± 9 AU.
AA designates amorphous aggregates,
Congo red staining leads to bright green birefringence (BiR) for amyloid species.
In contrast, a hairpin with shorter strands (HP6AYW) but retaining a biaryl-flanked turn was essentially inactive (see Figure. S2). Peptides μPro1 and HP7AAA have even shorter β strand segments. HP7AAA, a stable hairpin with an EtF W/W interaction displayed dose-dependent inhibition: the β CD signal development was still incomplete after 48 h at a 4:1 molar ratio (inhibitor/α-syn), see Table 2. After 5 days, fibrils of control morphology are present by TEM. Peptide μPro1 was also modestly inhibiting with no evidence of accelerated precipitation.
The precipitation phenomenon, not observed in the hAM studies, lead us to use CR-staining as an additional assay. Although this assay is not quantitative, it does visualize and characterize the aggregates formed. Since the complete assay mixture, dispersed by agitation just prior to slide preparation, is assayed, precipitation does not present problems in the TEM imaging and CR staining assays. Representative images for WW2 and YY2 inhibited runs appear in Figure 5. According to TEM and CR staining, few, if any, fibrils of normal morphology were observed for α-syn in the presence of WW2 and YY2. Rather WW2 produced thicker short fibrils, while YY2 resulted in what appears to be spherical aggregates. For both “inhibitors”, CR staining was less extensive and no trace of bright green birefringence was observed, see Figure 5.
Figure 5.
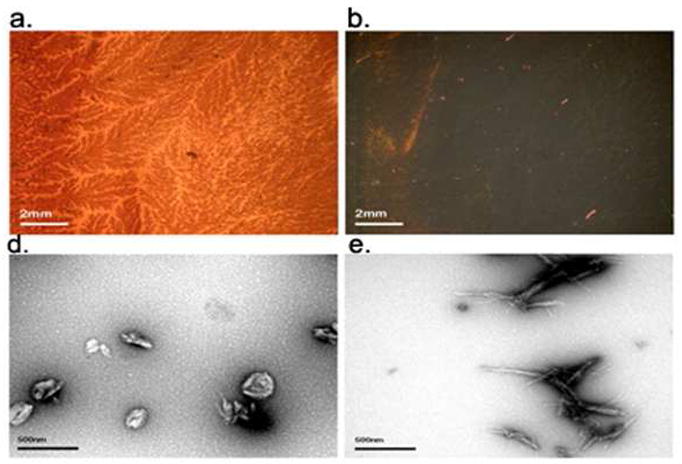
CR stained and TEM images of α-syn in the presence of beta hairpin peptides. Panels a and b illustrate CR stained images with and without polarized light respectively for α-syn aggregation in the presence of 2 molar equivalents of YY2. TEM images of α-syn with 2 molar eq. of YY2 (c) and WW2 (d) are also shown. Congo red images of WW2 are almost identical to those seen for YY2 (data not shown).
Control peptides
Two specific single strand controls for hairpins (ssMrH and ssW) were included in our study. Ac-KIVTSAK (ssMrH) represents one strand of MrH3b and KKLTVWI (ssW) serves as the single strand control for the WW2 series of hairpin peptides. Like a number of short peptides tested (data not shown), ssMrH had no effects on the course of α-syn aggregation. However, ssW (the Trp-containing control) delays aggregation (as monitored by CD) and displayed a significant reduction in ThT fluorescence at the 16 h read time. Unlike non-inhibited control assays, which shift cleanly from random coil to β by CD, in the presence of ssW, a helical CD spectrum (minima at 207 and 222nm) is present at the 4 and 6 h points in the assay (Figure 6).
Figure 6.
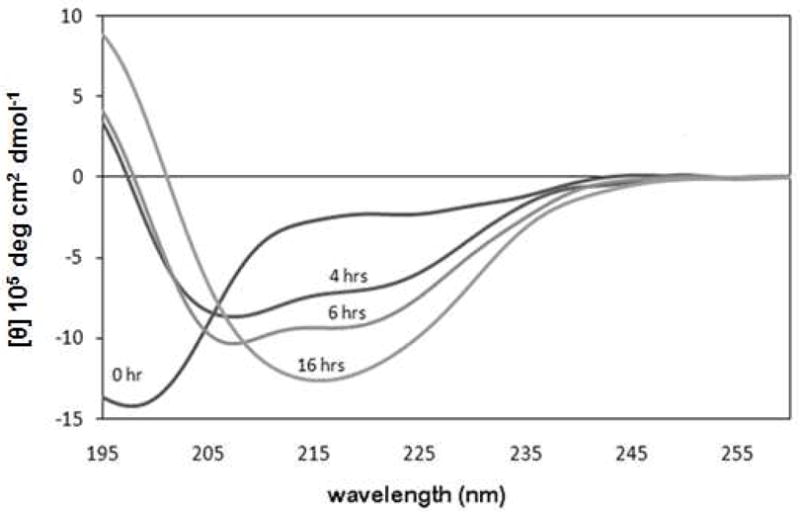
CD difference spectra recorded for α-syn in the presence of peptide ssW and 1.5 vol-% HFIP. The ellipticity [θ] scale is in molar not residue molar units. The zero time point is immediately after HFIP addition.
Discussion
The two amyloidogenic systems examined in this study (hAM and α-syn), although having no sequence similarities, are both random coil peptides that convert to amyloid fibrils over a period of days at the concentrations examined in the aqueous buffers emplyed. Including 1.5 – 2 vol-% HFIP in the assay buffer, resulted in complete amyloid formation over a period of hours rather than days. Under these conditions, lag times were reproducible as was the extent of ThT fluorescence enhancement observed. The TEM images and CR staining characteristics of the amyloid fibrils produced correspond to those observed in the absence of HFIP by other researchers. This is also the case for the amplitude of the β CD signal that is observed 3.
Our previous report on hAM inhibition indicated that stable MrH hairpins with aromatic sidechains at non-H-bonded sites were effective at retarding β structure formation (CD) and delaying fibril formation. Inhibition of hAM aggregation was observed as a dose dependent increase in the lag time to amyloid nucleation coupled, in most cases, with a decreased yield of fibrils as quantitated by ThT fluorescence (24), see also Figure 1. The decreased yield of fibrils has now been confirmed by TEM (Figure 2) which also verifies the aggregation acceleration and enhancement produced by μPro1. Rat amylin (rAM) has recently (38) been reported as an effective inhibitor of WT hAM amyloid formation though prior reports (15) indicated that rAM does not significantly inhibit the formation of cytotoxic hAM aggregates.. Hairpins WW2, YY2 and WW4 are significantly more potent than rAM in our assay. Control peptides, including hairpins lacking aromatic residues, and at least one stable hairpin with a W-loop-W unit (HP7AAA) had no significant effect on hAM aggregation. A summary of the observed effects of added hairpins on amyloid fibril formation assays for both hAM and α-syn appears in Table 3.
A mechanistic rationale that includes the original basis for these studies, hairpin association with a preamyloid patch retarding self-self recognition, and can be used to discuss the results, appears in Figure 7. In Figure 7, the course of hAM amyloid fibril formation is modeled after the recent report by Martin Zanni and co-workers (43) and incorporates the fibril cross-section geometry of Luca et al. (44). The preamyloid conformation can be viewed as a partially-formed β arch (38, 45) According to Shim et al. (43), the hAM oligomerization process begins near the HSSNN reversing loop of the β arch. The asterisk on the initiating, partially-formed β arch indicates a hydrophobic site, which includes V17, the site that achieves a 13C=18O coupled β state earlier than any other in oligomerization studies of hAM isotopomers (43). We view inhibitor binding at such a site as the initial driving force for complex formation with β-sheet formation serving to mask the self-self recognition region required for oligomerization. This model predicts the dose dependent increase in lag-time that is observed. Given that cytotoxicity is generally attributed to oligomers (7–9), the observation of hAM cytotoxicity inhibition by peptide WW2 supports the hypothesis that the hairpins bind to a preamyloid state. It has been established (15) that hAM is in a nonfibrillar state at the beginning of the pre-incubation used in the cytotoxicity assay. In the uninhibited pre-incubations of hAM both fibril and cytotoxic assemblies form, with significant rat insulinoma cell toxicity evident at concentrations as low as 1 nM (Figure. 3). Two molar equivalents of peptide WW2 provide nearly complete protection from this cytoxic challenge.
Figure 7.

A mechanistic model for the inhibition of amyloid formation by a hairpin bearing an exposed hydrophobic site (shown as a tryptophan sidechain). The β arch geometry of the preamyloid state is modeled after the proposal of Shim et al. (43) for hAM aggregation. The asterisk on the starting structure indicates a hydrophobic binding site. The alternative route, the formation of non-amyloid aggregates is included to extend the model to α-syn, for which this pathway appears to be the dominant one in the presence of the hairpin examined in this study.
While the specific model is based on hAM, the general features are likely to apply to other amyloidogenic systems as well. In the case of α-syn, there are several suspect amyloidogenic patches (12,46) with hydrophobic binding sites in the NAC region. However, the preamyloid conformation is likely to be a more complex superpleated β-structure (45). The alternative pathway to non-amyloid aggregates was added to Figure 7 since this mechanism is suggested for α-syn amyloid fibril inhibition.
The course of α-syn amyloidogenesis was more sensitive to hairpin structures. To date, all hairpin peptides with 5-residue long β-strands, even those with less stable folds, have resulted in some inhibition as assessed by ThT fluorescence at 16 hours. The interpretation of the ThT assay data has a potential caveat: the reduced signal may reflect the formation of aggregates that have precipitated and are thus not observed in the fluorimetric assay. As a result, we rely to a greater extent on visual inspection (appearance of precipitates), TEM, and CR-stained imaging of the complete assay mixtures for the analyses. For peptides WW2, WW3 and YY2, TEM imaging indicated either no fibrils, or a greatly reduced yield of fibrils of abnormal morphology. These assay mixtures displayed diminished CR staining with little or no green birefringence. The common feature of these inhibitors was immediate or accelerated (versus non-inhibited controls) cloudiness and the production and precipitation of amorphous aggregates. In the case of peptides WW2 and YY2 amorphous aggregates were also observed in the absence of added HFIP. Whether these non-amyloid aggregates contain the added hairpin peptides has not been determined to date, Figure 7 recognizes both possibilities.
In one case (with peptide YY2 present), we observed what appear to be spherical aggregates (Figure 5). Tubular and annular structures, designated as protofibrils, have been the subject of significant study (12, 47–49). However, the protofibrils reported are much smaller (10–24 nm diameter) than the structures seen in the present case (160–260 nm diameter). 70–90 nm diameter annular oligomers have been observed for α-syn under quite different conditions with their formation accelerated in the presence of 100 μM Ca+2 ions (48). We view the larger spherical species observed in the presence of YY2 as off-path, non-amyloidogenic aggregates resulting from the diversion of a preamyloid state present at a low equilibrium concentration from the onset to an alternative aggregation pathway. This mechanism of amyloidogenesis inhibition, has been suggested for EGCG, a polyphenol which also affords spherical aggregates with α-syn (10), that has high-profile citations as a potential therapeutic strategy (7). We observe non-amyloid precipitates of α-syn with as little as 0.5 molar equivalents of peptide WW2; the similar effect with EGCG requires a 5-fold excess of the “inhibitor”. A similar diversion of a pre-amyloid state has also been observed with IAPP-GI, a designed hAM mimic (15): we also found spherical oligomers and suggested the sequestering of hAM and Aβ from their cytotoxic self-association pathways in the form of nonfibrillar heterocomplexes with IAPP-GI (29).
Stable hairpins retaining similar W/W pairs (e.g. μPro1 and HP7AAA), but with shorter β strands, appear to have diminished inhibitory activity for both amyloid systems. In the case of hAM, μPro1 was the only species to accelerate aggregation and afford an increased yield of fibrils (Figure 2 panel d) while HP7AAA had no significant effects on hAM aggregation. Consistent with this observation, μPro1 had no effect on hAM-induced cytotoxicity (Figure 3). In the case of α-syn, μPro1 had, if any effect, a slight lag time prolongation. HP7AAA consistently prolonged the α-syn lag time. While further screening, including other classes of hairpins, will be needed for a confirmation, our tentative conclusion is that a minimum β strand length is required to compete with the oligomerization that leads to α-syn fibril growth.
CD assays of amyloid formation for both hAM and α-syn imply, with one exception, a direct conversion of a random coil signal to a α structure signal with no observable intermediate state. The exception was the observation of a helical signal for α-syn at intermediate times in the presence of a control peptide (ssW) that corresponds to a single strand of an active hairpin inhibitor (Figure 6). Helix conformations have recently been implicated in the fibril formation pathways for Aβ and hAM (50, 51). α-Syn has been reported to favor helix formation in SDS-containing and organic solvent media (52–54). Some reports note that membrane surfaces can accelerate α-syn fibrillization (55); others (56), however, suggest that lipid binding and helical conformations result in fibrillization inhibition. Since we never observed helical features in CD spectra recorded at intermediate times for non-inhibited α-syn control assays, we attribute the helical CD signals observed at intermediate points when the ssW peptide is present to oligomerization inhibition which allows the observation of a helical preamyloid state.
The present studies suggest that aromatic residue display is involved in the initial formation of preamyloid/inhibitor complexes but this is, in the case of α-syn, not sufficient to prevent self-self recognition. In the case of hAM, the aromatic moieties play a deciding role; the exposed β-strand edges of hairpins are not sufficient for establishing inhibitory interactions with hAM amyloidogenesis intermediates. The MrH hairpins with multiple aromatic residues are, in both systems, amyloid formation inhibitors. They are mostly effective when the hairpins include Trp residues. This observation is not that surprising given that Trp residues are often observed at peptide/protein interfaces (57), form favorable cross-strand interactions in β sheet systems, and occur frequently in prior peptide modulators of amyloidogenesis (18, 58, 59).
In the present case, we have demonstrated both cross-reactivity with hAM and α-syn and the inhibition of hAM-induced cytotoxicity by a hairpin peptide that bears no sequence similarity to either amyloidogenic system. Whether designed hairpins can serve as leads for therapeutic development remains to be determined, however, the modulation of amyloidogenesis pathways by these hairpins should provide probes for NMR studies of the early stages of the processes by adjusting conditions under which lag time of non-inhibited assay may be further delayed to allow for NMR measurements. We anticipate that preamyloid/inhibitor complex formation will occur in the absence of added HFIP and that these complexes will have sufficient lifetimes for NMR characterization to provide both insights into the biorecognition phenomena along the pathway to cytotoxic assemblies and guidance for the design of more effective inhibitors that could serve as leads for therapeutic development.
Supplementary Material
Acknowledgments
NMR studies were carried out on instruments that were purchased or upgraded from facility grants from the NIH and NSF. TEM training was provided by W. Chan (Department of Biology UW) and images were collected at The Biology Imaging Facility at the University of Washington. Congo red images were taken with polarized light microscope provided by the Electron Microscopy Center at the University of Washington. We thank J. Bernhagen RWTH Aachen University for help with the cell viability assays.
Abbreviations
- hAM
human amylin
- MTT
3-4,5-Dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide
- rAM
rat amylin
- TEM
transmission electron microscopy
- ThT
thioflavin T
- CR
Congo red
Footnotes
Funding by NSF (CHE0650318) and NIH grants (3R01-GM059658-08S1)
With 4 molar equivalents of WW2, the lagtime increases to 5.7 h (a 5-fold increase), and ~50 % inhibition of the ThT fluorescence signal is still observed at 7 h (24).
In the absence of HFIP addition, α-syn solutions remain fully transparent for many days. When aromatic-bearing hairpins that result in immediate α-syn precipitation upon HFIP addition are added to α-syn in the absence of HFIP, immediate precipitation is not observed. The solutions do become cloudy over a period of hours: 2 h for 2 molar equivalents of peptide WW2, 4–6 h for the same conditions with YY2. For both peptides, precipitation occurs immediately upon adjusting the medium to 1.5 vol-% HFIP content. In the case of WW2/α-syn mixtures, non-amyloid precipitates are obtained with as little as 0.5 equivalents of the added peptide. It is apparent that the interaction of α-syn with these peptides favors aggregate formation, and that this process, like amyloidogenesis, is greatly accelerated by the presence of 1.5 vol-% HFIP. Control experiments show that the hairpin conformational equilibrium is not altered upon addition of these low levels of HFIP co-solvent.
Our prior study of β CD characteristics (62) can be used to estimate the fractional contributions of β strand conformations in the fibril states. The mature fibril (50) [θ]216 values for hAM ranged from −16,000° to −20,000°, indicating that virtually all of the residues are in β strand conformations in the fibril state. Similar values have been reported for hAM aggregation studies in media without added co-solvents (40, 63) The β minimum for α-syn in our assay corresponds to −11,300°/residue, in excellent agreement with values reported by other labs (26, 33) for assays conducted in the absence of co-solvents. This ellipticity value implies that ca. 60% of the synuclein sequence is in β strand conformations in the fibril state.
Supporting information available illustrating the precipitation effect of hairpin peptides on a-syn aggregation as well as TEM and CR images of an additional control hairpin peptide in the presence of α-syn. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nature Chemical Biology. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 2.Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001:2. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 4.Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymology. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 5.Ruschak AM, Miranker AD. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc Natl Acad Sci USA. 2007;104:12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lansbury PT. Inhibition of amyloid formation: A strategy to delay the onset of Alzheimer’s disease. Curr Opin Chem Biol. 1997;1:260–267. doi: 10.1016/s1367-5931(97)80018-x. [DOI] [PubMed] [Google Scholar]
- 7.Roberts BE, Shorter J. Escaping amyloid fate. Nat Struct Mol Biol. 2008;15:544–546. doi: 10.1038/nsmb0608-544. [DOI] [PubMed] [Google Scholar]
- 8.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid [beta]-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 9.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastire A, Wanker EE. EGCG redirects amyloidogenic polypeptides into unsctructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 11.Hudson SA, Ecroyd H, Dehle FC, Musgrave IF, Carver JA. (−)-Epigallocatechin-3-Gallate (EGCG) Maintains κ-Casein in its pre-fibrillar state without redirecting its aggregation pathway. J Mol Biol. 2009;392:689–700. doi: 10.1016/j.jmb.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 12.El-Agnaf OMA, Paleologou KE, Greer B, Abogrein AM, King JE, Salem SA, Fullwood NJ, Benson FE, Hewitt R, Ford KJ, Martin FL, Harriott P, Cookson MR, Allsop D. A strategy for designing inhibitos of alpha-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004;18:1315–1317. doi: 10.1096/fj.03-1346fje. [DOI] [PubMed] [Google Scholar]
- 13.Austen BM, Paleologou KE, Ali SAE, Qureshi MM, Allsop D, El-Agnaf OMA. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s β-amyloid peptide. Biochemistry. 2008;47:1984–1992. doi: 10.1021/bi701415b. [DOI] [PubMed] [Google Scholar]
- 14.Kapurniotu A, Schmauder A, Tenidis K. Structure-based design and study of nonamyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J Mol Biol. 2002;315:339–350. doi: 10.1006/jmbi.2001.5244. [DOI] [PubMed] [Google Scholar]
- 15.Yan LM, Tararek-Nossol M, Velkova A, Kazantzis A, Kapumiotu A. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity of IAPP cytotoxic fibrillogenesis. Proc Natl Acad Sci USA. 2006;103:2046–2051. doi: 10.1073/pnas.0507471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilead S, Gazit E. Inhibition of amyloid fibril formation by peptide analogues modified with α-aminoisobutyric acid. Angew Chem Inl Ed. 2004;43:4041–4044. doi: 10.1002/anie.200353565. [DOI] [PubMed] [Google Scholar]
- 17.Etienne MA, Aucoin JP, Fu Y, McCarley RL, Hammer RP. Stoichiometric inhibition of amyloid β-protein aggregation with peptides containing alternating α,α-disubstituted amino acids. J Am Chem Soc. 2006;128:3522–3523. doi: 10.1021/ja0600678. [DOI] [PubMed] [Google Scholar]
- 18.Smith TJ, Stains CI, Meyer SC, Ghosh I. Inhibition of β-amyloid fibrillization by directed evolution of a β-sheet presenting miniature protein. J Am Chem Soc. 2006;128:14456–14457. doi: 10.1021/ja065557e. [DOI] [PubMed] [Google Scholar]
- 19.Cochran AG, Skelton NJ, Starovasnik MA. Tryptophan zippers: Stable, monomeric β-hairpins. Proc Natl Acad Sci USA. 2001;98:5578–5583. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen NH, Olsen KA, Fesinmeyer RM, Tan Xu, Hudson FM, Eidenschink LA, Farazi SR. Minimization and optimization of designed β-hairpin folds. J Am Chem Soc. 2006;128:6101–6110. doi: 10.1021/ja054971w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kier BL, Andersen NH. Probing the lower size limit for protein-like fold stability: ten-Residue Microproteins with specific, Rigid Structures in Water. Journal of the American Chemical Society. 2008;130:14675–14683. doi: 10.1021/ja804656h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eidenschink LA, Kier BL, Huggins KNL, Anderse NH. Very short peptides with stable folds: Building on the interrelationship of Trp/Trp, Trp/cation, and Trp/backbone-amide interaction geometries. Prot Struct, Funct Bioinform. 2009;75:308–322. doi: 10.1002/prot.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kier BL, Shu I, Eidenschink LA, Andersen NH. Stabilizing capping motif for beta-hairpins and sheets. Proceedings of the National Academy of Sciences. 2010;107:10466–10471. doi: 10.1073/pnas.0913534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huggins KNL, Andersen NH. Hairpin peptide inhibitors of Amyloid fibril formation. In: Lankinen H, editor. Chemistry of Peptides in Life Science, Technology and Medicine; Peptides 2008 (Proceedings of the 30th European Peptide Symposium); 2010. pp. 590–591. [Google Scholar]
- 25.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 26.Conway KA, Harper JD, Lansbury PT. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 27.Huang C, Ren G, Zhou H, Wang CC. A new method for purification of recombinant human alpha-synuclein in Escherichia coli. Protein Express Purif. 2005;42:173–177. doi: 10.1016/j.pep.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 28.Cort JR, Liu Z, Lee GM, Huggins KNL, Janes S, Prickett K, Andersen NH. Solution state structures of human pancreatic amylin and pramlintide. Protein Eng Des Sel. 2009;22:497–513. doi: 10.1093/protein/gzp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan LM, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A. Designed IAPP Mimic Blocks Aβ Cytotoxic Self-Assembly: Cross-Suppression of Amyloid Toxicity of Aβ and IAPP Suggests a Molecular Link between Alzheimer’s Disease and Type 2 Diabetes. Angew Chem Int Ed Engl. 2007;46:1246–1252. doi: 10.1002/anie.200604056. [DOI] [PubMed] [Google Scholar]
- 30.Nilsson MR. Techniques to study amyloid fibril formation in vitro. Methods. 2004;34:151–160. doi: 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Tatarek-Nossol M, Yan LM, Schmauder A, Tenidis K, Westermark G, Kapurniotu A. Inhibition of hIAPP amyloid-fibril formation and apoptotic cell death by a designed hIAPP amyloid-core-containing hexapeptide. Chem Biol. 2005;12:797–809. doi: 10.1016/j.chembiol.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, Merkle ML, Voelter W, Brinner H, Kapurniotu A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J Mol Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 33.Hoyer W, Antony T, Cherny D, Heim G, Jovin TM, Subramaniam V. Dependence of α-synuclein aggregate morphology on solution conditions. J Mol Biol. 2002;322:383–393. doi: 10.1016/s0022-2836(02)00775-1. [DOI] [PubMed] [Google Scholar]
- 34.Fink AL. The aggregation and fibrillation of α-synuclein. Acc Chem Res. 2006;39:628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 35.Miranker AD, Padrick SB. Islet amyloid polypeptide: identification of long-range contacts and local order on the fibrillogenesis pathway. J Mol Biol. 2001;308:783–794. doi: 10.1006/jmbi.2001.4608. [DOI] [PubMed] [Google Scholar]
- 36.Padrick SB, Miranker AD. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41:4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- 37.Koo BW, Miranker AD. Contribution of the intrinsic disulfide to the assembly mechanism of islet amyloid. Protein Sci. 2005;14:231–239. doi: 10.1110/ps.041051205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao P, Meng F, Abedini A, Raleigh DP. The ability of rodent islet amyloid polypeptide to inhibit amyloid formation by human islet amyloid polypeptide has important implications for the mechanism of amyloid formation and the design of inhibitors. Biochemistry. 2009;49:872–881. doi: 10.1021/bi901751b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munishkina LA, Phelan C, Uversy VN, Fink AL. Conformational behaviour and aggregation of α-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry. 2003;42:2720–2730. doi: 10.1021/bi027166s. [DOI] [PubMed] [Google Scholar]
- 40.Goldsbury C, Goldie K, Pellaud J, Seeling J, Frey P, Muller SA, Kistler J, Cooper JS, Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J Struct Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 42.Puchtler H, Sweat F, Levine M. On the binding of congo red by amyloid. J Histochem Cytochem. 1962;10:355–364. [Google Scholar]
- 43.Shim SH, Gupta R, Ling YL, Strasfeld DB, Raleigh DP, Zanni MT. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. PNAS. 2009;106:6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolacular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kajava AV, Baxam U, Stevem AC. Beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du HN, Li HT, Zhang F, Lin XJ, Shi JH, Shi YH, Ji L-Na, Hu J, Lin DH, Hu HY. Acceleration of alpha-synuclein aggregation by homologous peptides. FEBS letters. 2006;580:3657–3664. doi: 10.1016/j.febslet.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 47.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-Synuclein, especially the Parkinson’s diseas-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 48.Lowe R, Pountney DL, Jensen HP, Gair WP, Voelcker NH. Calcium (II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004;13:3245–3252. doi: 10.1110/ps.04879704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rochet JC, Conway KA, Lansbury PT. Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse α-synuclein. Biochemistry. 2000;39:10619–10626. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- 50.Abedini A, Raleigh DP. A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Engin, Des Sel. 2009;22:453–459. doi: 10.1093/protein/gzp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williamson JA, Loria JP, Miranker AD. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J Mol Biol. 2009;393:383–396. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bisaglia M, Tessari I, Pinato L, Bellanda M, Giraudo S, Fasano M, Bergantino E, Bubacco L, Mammi S. A topological model of the interaction between alpha-synuclein and sodium dodecyl sulfate micelles. Biochemistry. 2005;44:329–339. doi: 10.1021/bi048448q. [DOI] [PubMed] [Google Scholar]
- 53.Bisaglia M, Trolio A, Bellanda M, Bergantino E, Bubacco L, Mammi S. Structure and topology of human alpha-synuclein bound to micelles: Implications for the aggregation process. Protein Sci. 2006;15:1408–1416. doi: 10.1110/ps.052048706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munishkina LA, Henriques J, Uversky VN, Fink AL. Role of protein-water interactions and electrostatics in alpha-synuclein fibril formation. Biochemistry. 2004;43:3289–3300. doi: 10.1021/bi034938r. [DOI] [PubMed] [Google Scholar]
- 55.Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 56.Zhu M, Fink AL. Lipid Binding Inhibits alpha-Synuclein Fibril Formation. J Biol Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 57.Ma B, Elkayam T, Wolfson H, Nussinov R. Protein-protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc Natl Acad Sci USA. 2003;100:5772–5777. doi: 10.1073/pnas.1030237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sato T, Kienlen-Campard P, Ahmed M, Liu W, Li H, Elliott JI, Aimoto S, Constantinescu SN, Octave JN, Smith SO. Inhibitors of amyloid toxicity based on β-sheet Packing of Aβ40 and Aβ42. Biochemistry. 2006;45:5503–5516. doi: 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, Goto Y, Naiki H, Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 53.Meier JJ, Kayed R, Lin CY, Gurlo T, Haataja L, Jayasinghe S, Langen R, Glabe CG, Butler PC. Inhibition of human IAPP fibril formation does not prevent beta-cell death: evidence for distinct actions of oligomers and fibrils of human IAPP. Am J Physiol-Endoc M. 2006;291:E1317–E1324. doi: 10.1152/ajpendo.00082.2006. [DOI] [PubMed] [Google Scholar]
- 60.Maynard AJ, Sharman GJ, Searle MS. Origin of β-hairpin stability in solution: Structural and thermodynamic analysis of the folding of model peptide supports hydrophobic stabilization in water. J Am Chem Soc. 1998;120:1996–2007. [Google Scholar]
- 61.Dyer RB, Maness SJ, Franzen S, Fesinmeyer RM, Olsen KA, Andersen NH. Hairpin Folding Dynamics: The cold-denatured state is predisposed for rapid refolding. Biochemistry. 2005;44:10406–10415. doi: 10.1021/bi050698z. [DOI] [PubMed] [Google Scholar]
- 62.Cort JR, et al. β-Structure in human amylin and two designer β-peptides: CD and NMR spectroscopic comparisons suggest soluble β-oligomers and the absence of significant populations of β-strand dimers. Biochem Biophys Res Commun. 1994;204:1088–1095. doi: 10.1006/bbrc.1994.2574. [DOI] [PubMed] [Google Scholar]
- 63.Porat Y, Mazor Y, Efrat S, Gazit E. Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry. 2004;43:14454–14462. doi: 10.1021/bi048582a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.