

Abstract
Desmocollin 3 (DSC3) is a desmosomal cadherin that is required for maintaining cell adhesion in the epidermis as demonstrated by the intra-epidermal blistering observed in Dsc3 null skin. Recently, it has been suggested that deregulated expression of DSC3 occurs in certain human tumor types. It is not clear whether DSC3 plays a role in the development or progression of cancers arising in stratified epithelia such as the epidermis. To address this issue, we generated a mouse model in which Dsc3 expression is ablated in K-Ras oncogene-induced skin tumors. Our results demonstrate that loss of Dsc3 leads to an increase in K-Ras induced skin tumors. We hypothesize that acantholysis-induced epidermal hyperplasia in the Dsc3 null epidermis facilitates Ras-induced tumor development. Further, we demonstrate that spontaneous loss of DSC3 expression is a common occurrence during human and mouse skin tumor progression. This loss occurs in tumor cells invading the dermis. Interestingly, other desmosomal proteins are still expressed in tumor cells that lack DSC3, suggesting a specific function of DSC3 loss in tumor progression. While loss of DSC3 on the skin surface leads to epidermal blistering, it does not appear to induce loss of cell-cell adhesion in tumor cells invading the dermis, most likely due to a protection of these cells within the dermis from mechanical stress. We thus hypothesize that DSC3 can contribute to the progression of tumors both by cell adhesion-dependent (skin surface) and likely by cell adhesion-independent (invading tumor cells) mechanisms.
Keywords: Desmosome, Desmocollins, Cell Adhesion, Carcinogenesis, Squamous Cell Carcinoma
INTRODUCTION
Desmocollins (DSC) are type-1 transmembrane glycoproteins localized in desmosomes, cell adhesion junctions that are formed in epithelial cells. The three Dsc genes expressed in mammals show cell- and tissue-type specific expression patterns (e.g. [1]). In mouse epidermis, all three isoforms are expressed; DSC3 is present throughout the epidermis, with higher levels in the suprabasal cell layers. DSC1 is restricted mainly to the granular layer. The distribution of DSC2 is unknown since mouse-specific antibodies do not exist. In humans, weak DSC2 expression is found in the basal layer of the epidermis [2].
DSC3 is a component of the transmembrane core of desmosomes and is thought to engage in homophilic as well as heterophilic adhesive interactions in the intercellular space [3], thus contributing to desmosome-mediated cell-cell adhesion. The cytoplasmic domain of this protein interacts with the armadillo proteins plakoglobin (JUP) and plakophilin(s) (PKP), in particular PKP3 [4]. These proteins link DSC3 to the intermediate filament cytoskeleton (keratin intermediate filaments in keratinocytes) via the adaptor protein desmoplakin (DSP). In addition to their role as cytoskeletal adapter molecules, both PKP and JUP can also function as signal transducing molecules (see references in [5,6]).
DSC3 is crucial for mouse development as demonstrated by the embryonic lethality of mice carrying a germline Dsc3 null mutation [7]. More important for the current study is the observation that DSC3 is essential for maintaining epidermal integrity. We recently generated mice with a conditional Dsc3 null mutation in stratified epithelia [8]. Dsc3 null keratinocytes showed normal proliferation and migration abilities in vitro and developed morphologically normal desmosomes. However, loss of DSC3 led to desmosomes that did not maintain cell adhesion when exposed to mechanical stress. Newborn mutant mice developed skin erosions caused by acantholysis (loss of cell adhesion) in the deep layers of the epidermis. Histologically, these lesions were indistinguishable from those previously observed in Dsg3 null mice [9,10] and were similar to the lesions observed in the skin of pemphigus vulgaris patients. We thus hypothesized that impaired DSC3 function, either caused by mutations or induced by autoantibodies, could lead to blistering skin diseases, a hypothesis supported by several recent studies (see references in [11]).
Down-regulation or loss of Dsc3 gene expression has been linked to breast cancer progression [12–14]. In oral squamous cell carcinoma (SCC), DSC3 down-regulation correlates with loss of differentiation markers and consequently more aggressive tumors [15]. Nevertheless, Kurzen and colleagues did not observe a correlation between DSC3 expression and tumor progression in human skin SCC [16]. However, the authors observed cytoplasmic DSC3 localization in some tumors, which is suggestive of a loss of protein function.
As demonstrated by this short summary of contradictory findings, the role of DSC3 in tumor development is not well understood. Further, the studies published thus far relied on immunostaining of tumor material and did not provide functional data to address the question of whether DSC3 loss can effect tumor development or progression in vivo.
To address this question, we designed a mouse model that allows for the inducible ablation of Dsc3 gene function and simultaneous activation of an oncogenic K-Ras allele (K-RasG12D; [17]) in the epidermis. Our results indicate that Dsc3 loss facilitates Ras-induced tumor development, most likely due to a cutaneous wounding response caused by acantholysis. Nevertheless, the difference in the tumor development between mutant and control mice was smaller than anticipated, prompting us to determine whether DSC3 is spontaneously lost in control tumors during tumor progression (i.e. tumors in which we did not inactivate both copies of the Dsc3 gene). This was indeed the case. Focal loss of DSC3 expression occurred early during skin tumor progression in tumor cells that had penetrated the dermis. These DSC3-negative cells appear to be protected from mechanical stress-induced acantholysis. If DSC3 plays a major role in the progression of invading tumor cells, cell adhesion – independent mechanisms have to be considered. Consequently, we propose that DSC3 might play two different roles in tumor development and progression. Loss of cell adhesion can facilitate tumor progression on the stress-exposed skin surface, while adhesion-independent DSC3 function are likely to play a role in tumor progression in the dermis. We further demonstrate that loss of DSC3 occurs during human tumor progression, indicating that our findings are relevant for the understanding of human skin cancer progression.
MATERIALS AND METHODS
Animal Experimentation
Animal experiments were conducted in compliance with all applicable local and federal requirements, and were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado Denver.
Generation of Dsc3fl/fl; K-RasG12D; K5.Cre*PR1 Mice
We generated trigenic mice carrying a conditional Dsc3 null allele (Dsc3tm2PKo; referred to as Dsc3fl/fl; [8]), an inducible K-Ras oncogene (LSL K-Ras G12D, referred to as K-RasG12D; [17]) and an RU486-inducible version of Cre (Cre*PR1). The Cre transgene is expressed under the control of the keratin 5 promoter (K5.Cre*PR1); [18]), which is transcriptionally active in the basal layer of stratified epithelia. We will refer to Dsc3fl/f; K-RasG12D; K5.Cre*PR1 mice as mutants and to Dsc3fl/+; K-RasG12D; K5.Cre*PR1 mice as controls.
Induction of Skin Tumors
The back skins of newborn mutant and control mice were treated with 1mg/ml of RU486 dissolved in 50% ethanol and 50% DMSO once a day for five consecutive days. At the age of four weeks, we started to treat the back skin of the RU486-induced mice with the tumor promoter TPA (12-O-tetradecanoylphorbol-13-acetate; Sigma; 10ug/200ul in acetone). The TPA treatment was repeated once a week for 25 weeks. A total of 34 mutant and 25 control mice were used in this experiment.
Immunofluorescence Microscopy, Histology, In Situ Hybridizations and Quantification of Immunofluorescence Signals in Tissue Sections
Immunofluorescence microscopy and histology (H&E staining) were done following standard protocols. In situ hybridizations with Dsc3 sense and antisense mRNA probes (covering positions 1–500 of the cDNA; ATG, position 1–3) were done essentially as described [19]. Antibody staining and histology were documented with a Nikon Eclipse 90I microscope equipped with a Coolsnap HQ2 and a DS-Fi1 camera using the NIS Elements 3.10 imaging software package from Nikon. The following antibodies were used: Gp2280 (DSC3; [7,8]), U114 (human DSC3; Fitzgerald), DSG3 [9,10], ITGA6 (α6 integrin; Chemicon), collagen IV (Progen, Germany), keratin 13, keratin 14, keratin 5 (KRT13, KRT14, KRT5; generous gifts from Dennis Roop, University of Colorado Denver), PKP3 (a generous gift from James Wahl III, UNMC College of Dentistry, Lincoln, NE), desmoplakin (DSP, Research Diagnostics), plakoglobin (JUP, clone PG5.1; Research Diagnostics), and PCNA (Santa Cruz Biotechnology). Secondary (Alexa Fluorochrome-labeled) antibodies for immunofluorescence microscopy were purchased from Invitrogen.
Immunofluorescence microscopy signals were quantified using the ZEN 2009 software package (Carl Zeiss MicroImaging GmbH, Germany). Seven areas of approximately 6000 pixels in size were selected in both the hyperplastic epidermis and in the tumor tissue (within the same tissue section) to determine fluorescence signal strengths (i.e. 14 measurements were done in each tumor section shown in Figure 4). Averages, standard deviations and p values were determined based on these measurements.
Figure 4.


Loss of DSC3 expression in control mouse skin tumors. (A-G) Immunofluorescence microscopy staining of sections through a well-differentiated control (Dsc3fl/+; K-RasG12D; K5.Cre*PR1) tumor. The tumor sections were stained with the antibodies indicated on the left. (A) Note that DSC3 is present at cell-cell borders in the hyperplastic epithelium (HE) overlaying the tumor tissue (T). The tumor has lost DSC3 staining at the cell-cell borders. The remaining weak signal in the cytoplasm most likely represents background staining. (B-E) Other desmosomal markers, such as desmogleins 3 (DSG3), plakophilin 3 (PKP3; a cytoplasmic binding partner for DSC3), desmoplakin (DSP) and plakoglobin (JUP) show normal expression patterns in the tumor tissues. Note that desmosomal proteins normally show a cell type specific expression patterns. For example, DSG3 expression is restricted to the basal and first suprabasal cell layers in normal epidermis and in the tumor tissue (arrow in (B)). (F) The tumor progression marker KRT13 is expressed in tumor tissue that has lost DSC3 expression (compare consecutive tissue sections in (A) and (F)]. (G) Staining with normal guinea pig IgG (negative control staining for section (A)). Counterstaining with an antibody against α6 integrin (ITGA6) demarcates the basement membrane (red). Bar, 50μm. (H) Histological section through the tumor shown in (A) – (G). (I) Histological section through the tumor shown in (J-K). In situ hybridization with Dsc3 (J) anti-sense and (K) sense probes on control tumor sections. The basement membrane zone of the overlaying HE is marked with a dashed line. Note that the hyperplastic epithelium (HE) is positive for Dsc3 mRNA while part of the tumor tissue (T) is negative (arrow in J), indicating loss of Dsc3 gene expression. The sense probe did not hybridize to the tissue section, demonstrating specificity of the ani-sense hybridization for Dsc3 mRNA. Bar, 50μm. (L) Quantification of the immunofluorescence signals obtained with the antibodies shown in A-E. As outlined in MATERIAL AND METHODS, the fluorescence signals in hyperplastic and tumor areas of the same tumor were determine and compared to each other. Expression levels of DSC3 were significantly reduced in the tumor tissue. Note that due to the diffuse background staining in the tumor portion of A, we are likely to overestimate residual DSC3 staining. Note the small but statistically significant reduction in PKP3 expression in the tumor tissue when compared to the hyperplastic epithelium.
Human Tissue Arrays
The human skin cancer tissue array was purchased from US Biomax (Rockville, MD. Cat#: SK208) and stained with antibodies against human DSC3 (U114) and against KRT5 (see above). The tumor classification as indicated in Figure 5 was provided by the manufacturer of the array (Grade1, well-differentiated; Grade 2, moderately-differentiated). Grade 1–2 tumors were counted as being grade 2. Three independent observers evaluated antibody staining. The results shown in Figure 5E are based on the consensus findings of three observers. Only KRT5-positive cells were evaluated for DSC3 expression. Tumors were classified as either DSC3-positive or as showing reduced or absent staining.
Figure 5.
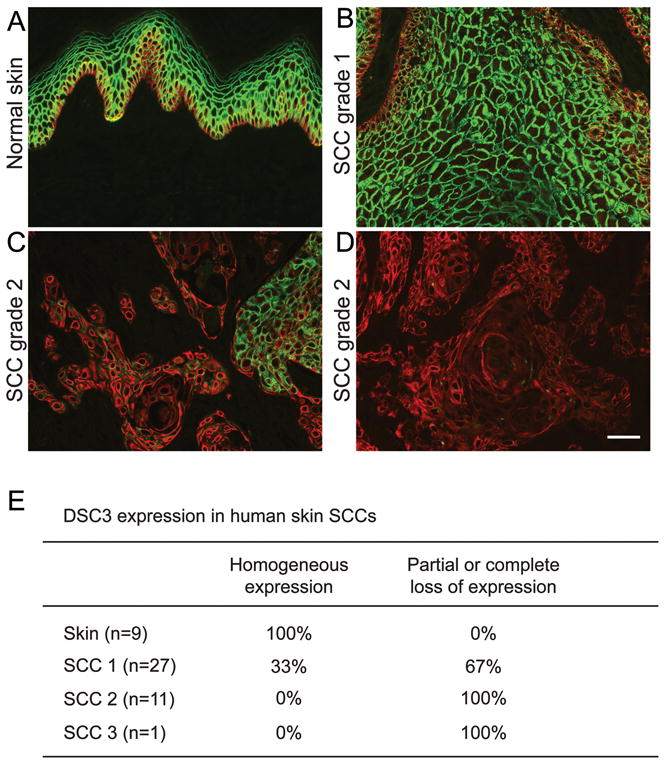
DSC3 expression in human skin SCCs. A human skin tumor tissue array (see MATERIAL AND METHODS) was co-stained with antibodies against keratin 5 (KRT5, red) and DSC3 (green). (A) As expected, both antibodies stained normal human skin. (B-D) Skin SCC sections showed a range of DSC3 expression patterns including (B) strong homogeneous expression (Grade 1 SCC), (C) patchy expression pattern (DSC3-positive and DSC3-negative tumor areas within the same section; Grade 2 SCC), (D) a complete loss of DSC3 expression in KRT5-positive cells (Grade 2 SCC). Note that the DSC3-stained sections were photographed with the same exposure time. Tumor tissues were classified by the provider of the array (see MATERIAL AND METHODS). Bar, 50μm. (E) Percentage of tissue sections showing either homogeneous or reduced/absent staining for DSC3 in KRT5-positive cells.
PCR Analysis
Recombination of the Dsc3 and K-RasG12D alleles was detected by PCR as described [8,18].
RESULTS
Generation of a New Skin Tumor Model
To test whether loss of the Dsc3 gene can facilitate tumor development or progression, we utilized a mouse model that carries a conditional Dsc3 null allele (Dsc3fl/fl). We have previously shown that upon Dsc3 gene ablation in the skin, these mice develop skin blisters with a pemphigus vulgaris-like histo-pathology [8]. To assess the role of Dsc3 in skin cancer, we generated mice that carry the conditional Dsc3fl/fl allele as well as a conditional oncogenic K-Ras allele (K-Ras G12D; [17]). Upon activation of a Cre recombinase, exon 1 and the proximal Dsc3 promoter are deleted thus rendering the gene inactive (Dsc3−/− allele; Figure 1A,B). Simultaneously, Cre activation also removes a floxed stop-cassette from the K-RasG12D allele thus activating oncogenic K-RasG12D expression (Figure 1C,D). We created trigenic mice (Dsc3fl/fl; K-Ras G12D; K5.Cre*PR1; referred to as mutant mice) that expressed an inducible version of Cre under the control of the keratin 5 promoter (K5.Cre*PR1; [18]), which directs transgene expression to the basal layer of stratified epithelia. Mice heterozygous for the conditional null Dsc3 allele were used as controls in the following experiments (Dsc3fl/+; K-Ras G12D; K5.Cre*PR1). The Cre transgene was activated in newborn mice by topical application of RU486 to the back skin. Once the animals reached four weeks of age, we began to treat the back skin once a week with TPA (12-O-tetradecanoylphorpbol-13-acetate), a phorbol ester, that is commonly used to facilitate skin tumorigenesis in the mouse. Both mutant and control mice developed skin tumors in this protocol. Mutant tumors showed both Dsc3 gene inactivation and K-RasG12D activation as demonstrated by PCR (Figure 1B, D). By immunofluorescence microscopy, we observed mutant tumors that had completely lost Dsc3 expression (Figure 2B, 2D; see Figure 2A for an example of normal DSC3 expression in mouse skin). Loss of Dsc3 was often associated with induction of keratin 13 (KRT13) expression, an early marker for tumor progression [20] (Fig. 2C). DSC3-negative tumor tissue maintained expression of other desmosomal proteins, such as desmoplakin (DSP; data not shown), suggesting that DSC3 is not required for forming or maintaining desmosomes, which is consistent with previously published results [8]. We also observed superficial skin blistering and epidermal hyperplasia in mutant tumors (Figure 2F). This observation is consistent with our previous finding that inactivation of the Dsc3 gene in the epidermis of mice leads to acantholysis and hyperplasia ([8]; and Supplemental Figure S1). We did not observe acantholysis in mutant tumor tissue that penetrated the deep dermis, i.e. acantholysis was always restricted to the epidermal tissue on the body surface. Of note is the fact that the Cre transgene showed mosaic activation. Only 20% of the tumors examined by immunostaining showed homogeneous loss of DSC3 throughout the tumor tissue, an indication of successful gene inactivation. Most mutant tumors (80%) showed mosaic Dsc3 gene inactivation (data not shown).
Figure 1.

Generation of an animal model to study the role of Dsc3 in skin tumorigenesis. A conditional Dsc3 null allele (Dsc3 fl/fl; [8]) was introduced into a mouse line carrying a conditional oncogenic K-RasG12D allele [17]. Inducible expression of Cre recombinase (K5.Cre*PR1 transgene; [18]) in the basal layer of mutant (Dsc3fl/f; K-RasG12D; K5.Cre*PR1) epidermis leads to Dsc3 ablation (Dsc3−/− alleles) and K-RasG12D activation. Note that the Cre recombinase is only active after topical application of the inducer RU486. (A) Schematic representation of the 5′ end of the Dsc3 gene. LoxP sites (triangles) were inserted in the proximal promoter and in the first intron of the gene (ATG demarcates the start codon in exon 1). Cre-mediated recombination leads to the excision of promoter elements and exon 1, rendering the gene nonfunctional. Successful recombination between the loxP sites can be detected with primers P1 and P2, which amplify a DNA fragment only after successful deletion of the floxed sequence (see [8] for details). (B) PCR analysis of genomic DNA isolated from two RU486-induced Dsc3fl/f; K-RasG12D; K5.Cre*PR1 tumors (Tumor 1, Tumor 2). Note that both tumors demonstrated successful recombination at the Dsc3 gene locus as demonstrated by the presence of the P1/P2 PCR product. Tail DNA (Tail) from a mutant mouse served as a negative control. Since the tail was not treated with the inducer RU486, recombination did not occur. (C) Activation of the conditional K-RasG12D allele. Cre-mediated recombination removes a stop-flox sequence thus allowing expression of the oncogene from the endogenous promoter. Primers P3 and P4 are used in PCR to distinguish between the activated oncogene and the K-Ras wild type allele. The star demarcates the position of the mutant sequence in Exon 1 (modified from [38]). The arrow with hatched lines symbolizes a silent K-Ras locus, while the arrows without the hatched lines symbolize transcriptionally active loci. (D) The genomic DNA samples used in (B) were subjected to PCR using primers P3 and P4. As expected, the mutant tail DNA only showed the wild type allele whereas the tumor samples showed both the wild type and the recombined (activated) oncogene allele. The size difference between the two products is due to the presence of the LoxP sequence in the product amplified from the recombined locus.
Figure 2.

Tumor induction and Dsc3 ablation in mutant mice. Immunofluorescence microscopy (A-D) using antibodies against DSC3, keratin 13 (KRT13) and the basement membrane markers α6 integrin (ITGA6) and collagen IV (COLIV). (A) DSC3 (green) and COLIV (red) staining of newborn epidermis. Bar, 50μm (B) DSC3 (green) and ITGA6 (red) staining of a tumor from a RU486-treated mutant mouse. Note the strong DSC3 staining in the epidermis (E) and the absence of DSC3 staining in the tumor tissue (T) that has penetrated the dermis. Bar, 100μm (C) KRT13, an early marker for tumor expression [20], was frequently induced in DSC3-negative tumor tissues (Bar, 50μm). (D) DSC3 staining of the section shown in (C) and in the boxed area of panel (B). Note the complete absence of DSC3 staining in the tumor cells. (E) Histological staining of the tumor section shown in (B and C). (F) Histological section of a mutant tumor. Note the suprabasal acantholysis (*) in the tumor tissue. Bar, 100μm
Dsc3 Ablation Increases the Incidence of Skin Tumors in Mice Expressing an Oncogenic K-Ras Allele in the Epidermis
As shown in Figure 3A, mutant mice showed an increase in tumor incidence. The difference between mutants and controls was noticeable after 15 weeks of TPA promotion and reached statistical significance (p<0.05) by 20 weeks of treatment. At 20 weeks, 91% of the mutant mice had developed tumors whereas only 68% of the controls carried tumors. Mutants also showed a slight increase in the tumor load (number of tumors per tumor-bearing mouse). This phenomenon was transient with a significant difference observed only until 15 weeks of treatment (Figure 3B). We did not observe a statistically significant difference in the volume of mutant and control tumors (data not shown). We analyzed tumors from 71 mutant and 32 control mice by histology. 41% of the mutant mice developed tumors classified as moderate SCC, low grade SCC or SPCC. In the control group, only 27% of the mice developed tumors that fell into these categories suggesting accelerated de-differentiation of tumors developed by mutant mice (data not shown). Nevertheless, we did not observe an increased rate of metastasis in mutant mice. In fact, we identified only two mice (one mutant and one control) that developed metastases. This resistance to metastasis in both genotypes might be due to the C57Bl genetic background of our lines, a strain that is resistant to carcinogenesis (e.g. [21]).
Figure 3.

Comparison of tumor development in mutant and control mice. (A) Kaplan-Meier graph showing the percentage of tumor-free mice over time (weeks of TPA treatment). Note that mutant mice showed a significant increase in tumor incidence which reached statistical significance at the 20 week time point (p<0.05). At this time point, 32% of the control mice were free of tumors while only 9% of the mutants had not developed visible tumors. (B) Average number of tumors in tumor-bearing mice. Note that mutants showed a slightly but statistically significant higher number of tumors by 15 weeks of TPA treatment. Nevertheless, control animals eventually caught up and both genotypes yielded similar average tumor loads at the end of the observation period (25 weeks of TPA treatment).
DSC3 Loss is an Early Marker for Tumor Progression in Skin Tumorigenesis
Mutant mice developed a higher percentage of low-grade tumors than control mice did. However, the difference between the two was smaller than anticipated. This led to the question of whether DSC3 was spontaneously lost in control animals during tumor progression, effectively generating Dsc3 null tumors in the control group. To address this question, we analyzed control tumors (Dsc3fl/+; KrasG12D; K5.Cre*PR1; n=23) by immunofluorescence microscopy for the expression of DSC3 and other desmosomal markers such as desmoglein 3 (DSG3), plakophilin 3 (PKP3), desmoplakin (DSP), plakoglobin (JUP), adherens junction markers (E-cadherin (CDH1), β-catenin (CTNB1); data not shown) and marker proteins for the basement membrane zone (α6 integrin (ITGA6), collagen IV (COLIV)). We noticed that DSC3 expression disappeared, often at the transition from papillomas to SCC. Of the 23 control tumors analyzed, 17 (74%) showed loss or reduced expression of DSC3 while 6 tumor samples (26%) maintained DSC3 expression as judged by immunofluorescence microscopy. The chimeric nature of the tumors analyzed (areas with and without DSC3 expression) prevented us from assessing DSC3 loss by Western Blotting.
Nevertheless, we measured and compared the immunofluorescence signals generated from hyperplastic epidermis and tumor tissue in the same tissue sections (see MATERIAL AND METHODS) to assess expression levels of DSC3 and other epithelial marker proteins (see below). Hyperplastic epidermis adjacent to tumor tissue showed normal expression of DSC3 while tumor tissue invading the dermis gradually lost expression, often showing a patchy DSC3 expression pattern (see example in Figure 4A, L). Interestingly, loss of DSC3 often coincided with the onset of keratin 13 (KRT13) expression, an early marker for tumor progression (Figure 4F; [20]). In fact, 82% of the control tumors with loss of DSC3 expression showed concomitant induction of KRT13. Cells with loss or a reduction in DSC3 staining were morphologically indistinguishable from surrounding keratinocytes expressing normal levels of DSC3. Our immunofluorescence signal quantification shown in Figure 4L suggested a twofold reduction of DSC3 staining in tumor tissues when compared to hyperplastic epidermis. Nevertheless, this most likely represents an underestimate due to the presence of unspecific diffuse cytoplasmic and perinuclear staining when labeling tumors with the DSC3 antibody, suggesting that most cells within the tumor tissue were effectively Dsc3 null (Figure 4A). This conclusion is consistent with the observed loss of Dsc3 mRNA expression in control tumor tissue as demonstrated by in situ hybridizations (Figure 4J, K). We also observed a slight reduction in the expression of PKP3 in the tumor tissue, a structural and signaling protein that directly binds to DSC3 (Figure 4C)[4,5]. This observation might reflect the reduced availability of the transmembrane protein (DSC3) that serves as a membrane anchor for PKP3. The expression of DSG3, a transmembrane receptor co-expressed with DSC3, and of JUP were not significantly affected in tumor tissue (Figure 4).
To ensure that the observed loss of DSC3 in control tumors was not caused by the fact that we used Dsc3fl/+ heterozygous mice as controls, we also stained archival tumors (papilloma and SCC) sections derived from wild type mice that had been subjected to a chemical carcinogenesis protocol (DMBA/TPA protocol; see [22] for references and protocols). As expected, these tumors, which are usually caused by H-Ras mutations, showed focal loss of DSC3 in tumor cells invading the dermis (data not shown).
DSC3 is an Early Marker for Tumor Progression in Human Skin SCCs
Next, we tested whether loss of DSC3 expression is also a feature of human skin SCCs. To that end, we co-stained a human skin tumor tissue array with antibodies against DSC3 and keratin 5 (KRT5; Figure 5). KRT5 is expressed in the basal layer of stratified epithelia, such as the epidermis, and can thus be used as a marker for keratinocytes. Only KRT5-positive cells were evaluated for the presence or absence of DSC3. As expected, all normal human skin samples in the array (n=9) stained homogenously for DSC3 (Figure 5A). We observed a gradual loss of DSC3 expression concomitant with tumor cell dedifferentiation. The percentage of SCCs that showed patchy, weak or absent DSC3 expression (see examples in Figure 5B-D) increased significantly in tumors that showed impaired keratinocyte differentiation (Grade 2 tumors). None of the grade 2 SCC showed homogeneous DSC3 expression (Figure 5E). These results suggest that loss of DSC3 expression occurs early during tumor progression and becomes more prevalent in tumors with lower differentiation grade.
DISCUSSION
The role of desmosomal proteins in cancer development and progression is not well understood. Both tumor-promoting as well as tumor-suppressing functions have been assigned to desmosomal proteins [23], often to the same protein. Loss of DSC3, for example, has been observed in a high percentage of breast cancer cell lines [12–14]. In this system, promoter silencing appears to cause loss of Dsc3 expression. Changes in DSC3 synthesis have also been observed in oral SCC, where reduced expression is predominantly observed in mid- to low-grade tumors [15]. Nevertheless, Wang et al. also reported reduced expression of other desmosomal proteins (e.g. DSG3), suggesting the possibility that a general desmosomal defect occurred in the tumors analyzed. Conversely, increased DSC3 expression has been observed in certain types of lung cancer [24], and ectopic DSC3 expression was observed in colorectal adenocarcinomas [25].
The hypothesis emerging from the data summarized above is that desmosomal genes have cell type-specific functions that determine whether deregulated expression promotes or prevents tumor progression. These effects might not always be related to altered cell adhesion. Ectopic expression of DSC3 in colon cancer cells, for example, is not likely to impair cell adhesion. It is thus likely that abnormal cell signaling triggered by changes in DSC3 expression might contribute to carcinogenesis. DSC3 binds the cytoplasmic armadillo proteins plakoglobin and plakophilin at the plasma membrane. These molecules have been shown to modulate key cell biological properties important for cancer development and progression, such as cell migration and the cellular response to stress (e.g. [5,26–29]). It is thus tempting to speculate that DSC3 loss might affect the signaling pool of these molecules, thus facilitating cancer progression. In this context it is noteworthy that we observed reduced expression of PKP3 in DSC3-negative tumor cells invading the dermis (Figure 4L). Nevertheless, further experiments are required to establish a mechanistic link between the loss of DSC3 and changes in the signaling pool of armadillo proteins in tumor cells.
In the present study, we focused on a possible role of DSC3 in skin cancer. Previous studies did not provide evidence in support of a causal link between changes in DSC3 expression and skin cancer development; although a correlative link between DSC3 loss and oral SCC progression has been described. To test whether loss of DSC3 would affect cancer development or progression in the skin, we designed a trigenic mouse system that allows for the inducible Cre-mediated inactivation of the Dsc3 gene and the simultaneous activation of a K-Ras oncogene. We have previously shown that Dsc3 ablation in the skin leads to intra-epidermal blistering with pemphigus vulgaris like histopathology [8]. To avoid extensive skin blistering in our trigenic system, which would have made long-term cancer studies impossible to conduct, we utilized an RU486-inducible Cre transgene (K5.Cre*PR1) with weak activity in the basal layer of the epidermis [18]. As expected, our trigenic mice did not develop extensive skin blistering, although we did observe micro-blisters as shown in Figure 2F. Of note is the observation that we did not detect acantholysis in the tumor tissue that penetrated deep into the dermis. It is likely that this failure to observe loss of cell-cell adhesion might be due to a protection of this tissue from mechanical forces, which contribute to blistering on the body surface. Nevertheless, our histological analysis did not exclude the possibility of subtle cell adhesion defects at the submicroscopic level.
Unfortunately, the weak Cre activity also resulted in mosaic recombination activity, i.e. the development of tumors that expressed the K-Ras oncogene but also maintained Dsc3 expression. Tumors were defined as Dsc3 null only if the tumor tissue was homogenously negative for DSC3 expression. We concluded that only 20% of the RU486-treated Dsc3fl/fl; K-RasG12D; K5.Cre*PR1 tumors were truly Dsc3 null.
Mutant mice showed an increased tumor incidence with 97% of these mice developing tumors within the 25 weeks observation period. Only 68% of the control mice developed tumors in this period, pointing towards a tumor promoting effect of the Dsc3 null mutation. As indicated above, due to the mosaic activity of the Cre recombinase used, it is likely that the true difference between mutant and control mice in terms of tumor susceptibility is even greater than what we have observed. It is noteworthy that more aggressive tumors (mid to low grade SCCs and SPCC) were more prevalent in the mutant group. Based on published evidence summarized below, we hypothesize that the increased frequency of tumor initiation and the acceleration of tumor tissue dedifferentiation in mutants are due to a cutaneous wound healing response (epidermal hyperplasia) induced by intraepidermal blistering.
Argyris and colleagues have shown that DMBA treated mice develop tumors at sites of skin injury [30]. DMBA is a chemical carcinogen that induces Ras mutations, suggesting that Ras initiation and skin injury can act synergistically to promote tumor induction. Similarly, Leder et al showed that transgenic mice expressing a v-HA-Ras oncogene develop skin tumors at sites of abrasion [31]. Furthermore, it has recently been shown that activation of the Ras pathway in mice expressing RASGRP1 can induce tumors in injured skin, even in the absence of Ras mutations [32]. All of these studies point towards skin wound-induced epidermal hyperplasia as a trigger for tumor initiation in cells with an activated Ras pathway. Epidermal hyperplasia is a typical consequence of acantholysis in Dsc3 null skin as shown in Supplemental Figure 1 [8,11] and is thus predicted to act as a tumor promoter.
Given the increase in tumor incidence that we observed, it was surprising that we observed a higher tumor load of mutant mice (average number of tumors per mice) only 15 weeks after TPA promotion (Figure 3B). At later time points, in particular at the conclusion of our experiments at 25 weeks of tumor promotion, mutants and controls showed similar tumor loads. We hypothesize that TPA-induced inflammation and hyperproliferation, a necessary step in the skin tumor protocol used, might have covered up the more subtle effects of acantholysis-induced hyperplasia. Nevertheless, the higher tumor incidence documented in Figure 3A combined with the higher initial tumor load shown in Figure 3B strongly support our hypothesis that loss of DSC3 drives tumor development in our experimental system.
We also observed spontaneous loss of Dsc3 expression in control tumors, generally in papillomas or well-differentiated SCCs. If Dsc3 is eventually lost in both genotypes, why do we observe a difference in the tumor incidence between mutants and controls? Induced loss of Dsc3 occurs early during tumor development in the epidermis, leading to acantholysis and regenerative hyperplasia. The spontaneous loss of Dsc3 in control tumors on the other hand was detected at later stages of tumor progression, usually in well-differentiated SCCs that had begun to grow into the dermis. This tissue did not show acantholysis by histological analysis, probably because this tissue is protected from mechanical forces prevalent on the body surface, and thus would not be predicted to induce hyperplasia and accelerated tumor development.
Future experiments, designed to prevent spontaneous loss of Dsc3 gene expression are required to elucidate its role during tumor progression.
Does epidermal hyperplasia, triggered by acantholysis, play a role in human tumor development and progression? A subgroup of SCC (ASCC, acantholytic squamous cell carcinoma) and actinic keratosis (acantholytic actinic keratosis) of the skin is associated with suprabasal acantholysis (see examples in Supplemental Figure S2; e.g. [33–37]). The mechanisms that trigger acantholysis in these tumors are not known. However, a survey of a small number of ASCC indicated that loss of desmosomal gene expression occurs in these tumors (own unpublished observations). Based on our animal study, we would predict that acantholysis-induced epidermal hyperplasia might contribute to tumor progression in ASCC.
In summary, we have shown that Dsc3 gene ablation increases the incidence of Ras-induced skin tumors in mice. The synergistic effects of intra-epidermal blistering and oncogene activations might be the underlying mechanism driving tumor development in a subclass of human tumors, namely ASCC. Further, the spontaneous loss of DSC3 expression in tumor cells invading the dermis might also contribute to tumor progression. This loss of DSC3 does not appear to be the result of a general desmosomal defect, since other desmosomal marker proteins are still expressed in these cells. It is tempting to speculate that loss of DSC3 might affect adhesion-independent tumor cell functions, such as signaling. Further experiments will be required to elucidate this point.
Supplementary Material
Supplemental Figure 1. Loss of DSC3 induces suprabasal acantholysis and epidermal hyperplasia in conditional Dsc3 null skin. (A-F) The back skin of adult Dsc3 mutant mice (Dsc3fl/fl; K14-Cre; [8]) and (G, H) age-matched wild type control skin are shown. Sections A, C, E and G were stained with antibodies against the proliferation marker PCNA (green) and antibodies against keratin 5 (KRT5, red). Sections B, D, F and H represent matching histological sections. Note that the mutant skin shows prominent acantholysis (filled arrows) and epidermal hyperplasia. The lesion shown in C and E represent an area in which epidermal wound healing occurs. Lesional mutant skin shows a higher proliferation index as determined by the percentage of PCNA-positive nuclei (regenerative hyperplasia; e.g. please compare panels A and G). Note the presence of suprabasal proliferating cells (open arrows) in the mutant sample shown in (E). On average, we found a more than twofold increase in the proliferation index (percentage of PCNA-positive nuclei in the basal cell layer) in acantholytic epidermis when compared to age-and strain-matched normal control skin. Bars (E, F), 50μm; Bars (C, D, G, H), 100μm.
Supplemental Figure 2. Histology of malignant and pre-malignant skin lesions with acantholysis. Acantholysis occurs in a subset of (A) human acantholytic actinic keratosis and (B) human acantholytic squamous cell carcinoma. Stars demarcate areas of cell-cell separation. Bars, A), 50μm; (B), 100μm
Acknowledgments
This work has been supported by grants from the National Institutes of Health (NIH/NIAMS) to PJ Koch (RO1 AR050439; RO1 AR053892).
We would like to thank Bryan McNair (Colorado Biostatistics Consortium, University of Colorado Denver) for conducting the statistical analysis for our tumor experiments and Dr. Radu Moldovan (Advanced Light Microscopy Core Facility; UC Denver) for assistance with quantifying immunofluorescence signals in tissue sections. We are also grateful to Dr. Radhika Ganeshan for critical reading of this manuscript. This work has been supported by grants from the National Institutes of Health (NIH/NIAMS) to PJ Koch (RO1 AR050439; RO1 AR053892).
Abbreviations
- DSC
Desmocollin
- DSG
Desmoglein
- DSP
Desmoplakin
- JUP
Plakogobin
- KRT
Keratin
- CDH1
E-cadherin
- ITGA6
α6 integrin
- COLIV
Collagen IV
- SCC
Squamous Cell Carcinoma
References
- 1.Nuber UA, Schafer S, Stehr S, Rackwitz HR, Franke WW. Patterns of desmocollin synthesis in human epithelia: immunolocalization of desmocollins 1 and 3 in special epithelia and in cultured cells. Eur J Cell Biol. 1996;71(1):1–13. [PubMed] [Google Scholar]
- 2.Nuber UA, Schafer S, Schmidt A, Koch PJ, Franke WW. The widespread human desmocollin Dsc2 and tissue-specific patterns of synthesis of various desmocollin subtypes. Eur J Cell Biol. 1995;66(1):69–74. [PubMed] [Google Scholar]
- 3.Spindler V, Heupel WM, Efthymiadis A, et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J Biol Chem. 2009;284(44):30556–30564. doi: 10.1074/jbc.M109.024810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonne S, Gilbert B, Hatzfeld M, Chen X, Green KJ, van RF. Defining desmosomal plakophilin-3 interactions. JCell Biol. 2003;161(2):403–416. doi: 10.1083/jcb.200303036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neuber S, Muhmer M, Wratten D, Koch PJ, Moll R, Schmidt A. The desmosomal plaque proteins of the plakophilin family. Dermatol Res Pract. 2010;2010:101452. doi: 10.1155/2010/101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt A, Koch PJ. Desmosomes: just cell adhesion or is there more? Cell Adh Migr. 2007;1(1):28–32. doi: 10.4161/cam.1.1.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Den Z, Cheng X, Merched-Sauvage M, Koch PJ. Desmocollin 3 is required for pre-implantation development of the mouse embryo. J Cell Sci. 2006;119(Pt 3):482–489. doi: 10.1242/jcs.02769. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 2008;121(Pt 17):2844–2849. doi: 10.1242/jcs.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch PJ, Mahoney MG, Ishikawa H, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137(5):1091–1102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch PJ, Mahoney MG, Cotsarelis G, Rothenberger K, Lavker RM, Stanley JR. Desmoglein 3 anchors telogen hair in the follicle. J Cell Sci. 1998;111 ( Pt 17):2529–2537. doi: 10.1242/jcs.111.17.2529. [DOI] [PubMed] [Google Scholar]
- 11.Ganeshan R, Chen J, Koch PJ. Mouse models for blistering skin disorders. Dermatol Res Pract. 2010;2010:584353. doi: 10.1155/2010/584353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klus GT, Rokaeus N, Bittner ML, et al. Down-regulation of the desmosomal cadherin desmocollin 3 in human breast cancer. Int J Oncol. 2001;19(1):169–174. doi: 10.3892/ijo.19.1.169. [DOI] [PubMed] [Google Scholar]
- 13.Oshiro MM, Kim CJ, Wozniak RJ, et al. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 2005;7(5):R669–R680. doi: 10.1186/bcr1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshiro MM, Watts GS, Wozniak RJ, et al. Mutant p53 and aberrant cytosine methylation cooperate to silence gene expression. Oncogene. 2003;22(23):3624–3634. doi: 10.1038/sj.onc.1206545. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Liu T, Wang Y, et al. Altered expression of desmocollin 3, desmoglein 3, and beta-catenin in oral squamous cell carcinoma: correlation with lymph node metastasis and cell proliferation. Virchows Arch. 2007;451(5):959–966. doi: 10.1007/s00428-007-0485-5. [DOI] [PubMed] [Google Scholar]
- 16.Kurzen H, Munzing I, Hartschuh W. Expression of desmosomal proteins in squamous cell carcinomas of the skin. J Cutan Pathol. 2003;30(10):621–630. doi: 10.1034/j.1600-0560.2003.00122.x. [DOI] [PubMed] [Google Scholar]
- 17.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 18.Caulin C, Nguyen T, Longley MA, Zhou Z, Wang XJ, Roop DR. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004;64(15):5054–5058. doi: 10.1158/0008-5472.CAN-04-1488. [DOI] [PubMed] [Google Scholar]
- 19.Cheng X, Mihindukulasuriya K, Den Z, et al. Assessment of splice variant-specific functions of desmocollin 1 in the skin. Mol Cell Biol. 2004;24(1):154–163. doi: 10.1128/MCB.24.1.154-163.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nischt R, Roop DR, Mehrel T, et al. Aberrant expression during two-stage mouse skin carcinogenesis of a type I 47-kDa keratin, K13, normally associated with terminal differentiation of internal stratified epithelia. Mol Carcinog. 1988;1(2):96–108. doi: 10.1002/mc.2940010205. [DOI] [PubMed] [Google Scholar]
- 21.Woodworth CD, Michael E, Smith L, et al. Strain-dependent differences in malignant conversion of mouse skin tumors is an inherent property of the epidermal keratinocyte. Carcinogenesis. 2004;25(9):1771–1778. doi: 10.1093/carcin/bgh170. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Cheng X, Merched-Sauvage M, Caulin C, Roop DR, Koch PJ. An unexpected role for keratin 10 end domains in susceptibility to skin cancer. J Cell Sci. 2006;119(Pt 24):5067–5076. doi: 10.1242/jcs.03298. [DOI] [PubMed] [Google Scholar]
- 23.Chidgey M, Dawson C. Desmosomes: a role in cancer? Br J Cancer. 2007;96(12):1783–1787. doi: 10.1038/sj.bjc.6603808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boelens MC, van den Berg A, Vogelzang I, et al. Differential expression and distribution of epithelial adhesion molecules in non-small cell lung cancer and normal bronchus. J Clin Pathol. 2007;60(6):608–614. doi: 10.1136/jcp.2005.031443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan K, Hardy R, Haq A, Ogunbiyi O, Morton D, Chidgey M. Desmocollin switching in colorectal cancer. Br J Cancer. 2006;95(10):1367–1370. doi: 10.1038/sj.bjc.6603453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kundu ST, Gosavi P, Khapare N, et al. Plakophilin3 downregulation leads to a decrease in cell adhesion and promotes metastasis. Int J Cancer. 2008;123(10):2303–2314. doi: 10.1002/ijc.23797. [DOI] [PubMed] [Google Scholar]
- 27.Papagerakis S, Shabana AH, Depondt J, Pibouin L, Blin-Wakkach C, Berdal A. Altered plakoglobin expression at mRNA and protein levels correlates with clinical outcome in patients with oropharynx squamous carcinomas. Hum Pathol. 2004;35(1):75–85. doi: 10.1016/j.humpath.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 28.Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann I, Casella M, Schnolzer M, Schlechter T, Spring H, Franke WW. Identification of the junctional plaque protein plakophilin 3 in cytoplasmic particles containing RNA-binding proteins and the recruitment of plakophilins 1 and 3 to stress granules. Mol Biol Cell. 2006;17(3):1388–1398. doi: 10.1091/mbc.E05-08-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Argyris TS. Tumor promotion by abrasion induced epidermal hyperplasia in the skin of mice. JInvest Dermatol. 1980;75(4):360–362. doi: 10.1111/1523-1747.ep12531153. [DOI] [PubMed] [Google Scholar]
- 31.Leder A, Kuo A, Cardiff RD, Sinn E, Leder P. v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesis: effects of phorbol esters and retinoic acid. Proc Natl Acad Sci U S A. 1990;87(23):9178–9182. doi: 10.1073/pnas.87.23.9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diez FR, Garrido AA, Sharma A, et al. RasGRP1 transgenic mice develop cutaneous squamous cell carcinomas in response to skin wounding: potential role of granulocyte colony-stimulating factor. Am J Pathol. 2009;175(1):392–399. doi: 10.2353/ajpath.2009.090036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerawala CJ. Acantholytic squamous cell carcinoma of the oral cavity: a more aggressive entity? Br J Oral Maxillofac Surg. 2009;47(2):123–125. doi: 10.1016/j.bjoms.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Nappi O, Pettinato G, Wick MR. Adenoid (acantholytic) squamous cell carcinoma of the skin. J Cutan Pathol. 1989;16(3):114–121. doi: 10.1111/j.1600-0560.1989.tb00024.x. [DOI] [PubMed] [Google Scholar]
- 35.Cherpelis BS, Marcusen C, Lang PG. Prognostic factors for metastasis in squamous cell carcinoma of the skin. Dermatol Surg. 2002;28(3):268–273. doi: 10.1046/j.1524-4725.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- 36.Cassarino DS, Derienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. Part one. J Cutan Pathol. 2006;33(3):191–206. doi: 10.1111/j.0303-6987.2006.00516_1.x. [DOI] [PubMed] [Google Scholar]
- 37.Papadopoulou E, Tosios KI, Nikitakis N, Papadogeorgakis N, Sklavounou-Andrikopoulou A. Acantholytic squamous cell carcinoma of the gingiva: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109(6):e67–71. doi: 10.1016/j.tripleo.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 38.Caulin C, Nguyen T, Lang GA, et al. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest. 2007;117(7):1893–1901. doi: 10.1172/JCI31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Loss of DSC3 induces suprabasal acantholysis and epidermal hyperplasia in conditional Dsc3 null skin. (A-F) The back skin of adult Dsc3 mutant mice (Dsc3fl/fl; K14-Cre; [8]) and (G, H) age-matched wild type control skin are shown. Sections A, C, E and G were stained with antibodies against the proliferation marker PCNA (green) and antibodies against keratin 5 (KRT5, red). Sections B, D, F and H represent matching histological sections. Note that the mutant skin shows prominent acantholysis (filled arrows) and epidermal hyperplasia. The lesion shown in C and E represent an area in which epidermal wound healing occurs. Lesional mutant skin shows a higher proliferation index as determined by the percentage of PCNA-positive nuclei (regenerative hyperplasia; e.g. please compare panels A and G). Note the presence of suprabasal proliferating cells (open arrows) in the mutant sample shown in (E). On average, we found a more than twofold increase in the proliferation index (percentage of PCNA-positive nuclei in the basal cell layer) in acantholytic epidermis when compared to age-and strain-matched normal control skin. Bars (E, F), 50μm; Bars (C, D, G, H), 100μm.
Supplemental Figure 2. Histology of malignant and pre-malignant skin lesions with acantholysis. Acantholysis occurs in a subset of (A) human acantholytic actinic keratosis and (B) human acantholytic squamous cell carcinoma. Stars demarcate areas of cell-cell separation. Bars, A), 50μm; (B), 100μm