

Abstract
Background
General anesthesia has been likened to a state in which anesthetized subjects are locked out of access to both rapid eye movement (REM) sleep and wakefulness. Were this true for all anesthetics, one might expect a significant REM rebound following anesthetic exposure. However, for the intravenous anesthetic propofol, studies demonstrate that no sleep debt accrues. Moreover, pre-existing sleep debts dissipate during propofol anesthesia. To determine whether these effects are specific to propofol or are typical of volatile anesthetics we tested the hypothesis that REM sleep debt would accrue in rodents anesthetized with volatile anesthetics.
Methods
Electroencephalographic and electromyographic electrodes were implanted in 10 mice. After 9–11 days of recovery and habituation to a 12h:12h light:dark cycle, baseline states of wakefulness, non-rapid eye movement sleep, and REM sleep were recorded in mice exposed to 6 hours of an oxygen control and on separate days to 6 hours of isoflurane, sevoflurane, or halothane in oxygen. All exposures were conducted at the onset of light.
Results
Mice in all three anesthetized groups exhibited a significant doubling of REM sleep during the first six-hours of the dark phase of the circadian schedule while only mice exposed to halothane displayed a significant increase in non-rapid eye movement sleep that peaked at 152% of baseline.
Conclusion
REM sleep rebound following exposure to volatile anesthetics suggests that these volatile anesthetics do not fully substitute for natural sleep. This result contrasts with the published actions of propofol for which no REM sleep rebound occurred.
Introduction
Nearly two decades ago, the call was issued for multidisciplinary studies to address the mechanisms through which anesthetics produce a state of unconsciousness.1,2 The state of general anesthesia produced by many drugs shares striking similarities with that of non-rapid eye movement (NREM) sleep.3 During unconsciousness accompanying both states there is a breakdown in effective cortical communication,4,5 the appearance of slow waves that share a similar origin and directionality,6 and asymmetry of directional information transfer.7–9 Functional imaging studies demonstrate that activity in the default mode network is changed with the unconsciousness accompanying both NREM sleep and general anesthesia, although the specific adaptations in default mode network are admittedly not identical,10,11 leading to questions about the true degree of convergence between sleep and anesthetic-induced hypnosis.12 Nonetheless, neurophysiologic similarities extend beyond the cortex to subcortical systems as well, where imaging studies have revealed a deactivation of the thalamus and midbrain reticular formation coincident with loss of consciousness.13–16 Moreover, animal studies show that sleep deprivation potentiates the hypnotic potency of anesthetics17,18 as does administration of endogenous somnogens.19,20 Although the states of anesthesia and NREM sleep are clearly not the same, anesthetics impair the neuronal circuits that promote endogenous arousal.21–24 This has led to the speculation that the brains of anesthetized subjects may be locked into a NREM-like state while being mutually excluded from accessing rapid eye movement (REM) sleep and wakefulness.13,25 If true, then time spent in anesthesia might fulfill NREM sleep requirements, but one might expect a REM rebound upon termination of the anesthetic exposure as the homeostatic drive for REM sleep accrues.26
However, seminal studies conducted with the intravenous anesthetic propofol refute the preceding hypothesis. In rats, pre-existing sleep debts accumulated over the previous 24 hours dissipate identically under propofol-anesthesia as they would have during recovery sleep.27 Moreover, unlike true sleep deprivation, when propofol anesthesia is induced at the onset of the rest cycle in rodents and maintained for 12 hours, neither REM nor NREM sleep debts accrue.28 With respect to subsequent sleep, the degree to which the state of anesthesia produced by propofol may be reflective of other anesthetics remains unknown. It has become increasingly clear that many agent-specific differences exist amongst anesthetics, including diversity in the neuronal mechanisms through which individual drugs produce the anesthetic state.23,29–34
Contradictory evidence with volatile anesthetics in multiple species, including humans, suggests that anesthetics may not substitute for natural NREM and REM sleep.35–39 However, detailed studies of sleep and wakefulness following anesthetic exposure as defined by incorporating electroencephalographic-based analysis of sleep and its microarchitecture are currently lacking. Moreover, comparative studies examining the isolated effects of volatile anesthetics on sleep in the absence of surgical insults are also needed. We therefore conducted the following studies in mice to test the hypothesis that REM but not NREM sleep debts would accrue during exposure to volatile anesthetics and that unlike propofol, the volatile anesthetics would not fully substitute for endogenous REM and NREM sleep. In addition to their potential clinical application, these studies also suggest that important differences exist in the ways that volatile anesthetics and propofol interact with endogenous arousal circuits.
Materials and Methods
Animals
All studies were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania (Philadelphia, PA) and were conducted in accordance with the National Institutes of Health guidelines.Ψ Adult C57BL6/J male mice (Jackson Labs, Bar Harbor, ME) aged 8–12 weeks were placed on a 12hr light:dark cycle and given food and water ad libidum. Lights were turned on at 7am—Zeitgeber time (ZT) 0 and turned off at 7pm, ZT12.
Surgery
Wild-type (n = 10) mice were surgically implanted with four electroencephalographic and two electromyographic electrodes according to previous published protocols40–42 with the following modifications. Briefly, anesthesia was induced with 2.5% isoflurane and maintained with 1.5–2.0% isoflurane, titrated to the absence of response following tail and foot pinch. Animals were placed into a digital stereotactic frame (David Kopf Instruments, Tujunga, CA) and warmed on a heating pad to maintain core body temperature at 36 ± 1C. Aseptic technique was undertaken. The skin was cleaned with betadine. A midline incision over the scalp exposed the skull to permit drilling of four burr holes at ±2mm lateral to Bregma and 1mm rostral overlying primary motor cortex and ±2mm lateral to Bregma and 3mm caudal to Bregma overlying primary visual cortex. A lightweight socket headpiece, created by soldering 28-gauge Teflon insulted silver wires (AM systems, Sequim, WA) onto a 6-pin straight dual-row personal computer board connector (Allied Electronics, Ft Worth, TX), was inserted with one wire cemented into each burr hole to create four epidural electroencephalographic leads. The remaining two wires were placed into the nuchal musculature to create electromyographic leads. The entire headpiece was secured to the skull with dental cement (AM systems).
Mice were allowed a minimum of 7 days of recovery following surgery. During the recovery period, mice were singly housed and acclimated to the recording chamber, a custom designed cylindrical cage with a diameter of 7.625″, a height of 6.5″, and a volume of 4.9L, which also served as their new home cage. The recording chamber could be sealed to become gas tight, had fresh gas inlet and outlet ports, along with two luer adaptors at the base for sampling and returning the recording chamber gas mixture. Temperature in the cages was kept at 25 ± 2C. Following recovery, the implanted socket headpieces were connected to the recording apparatus via a flexible tethering cable (Plastics One, Roanoke, VA) allowing the mice to habituate for another 2–4 days prior to baseline sleep-wake recordings.
Physiological Recording of Behavioral State
Polysomnographic recordings of murine electroencephalographic and electromyographic waveforms were amplified and sampled at 200Hz using an MP150 recordings system (Biopac Systems Inc, Goleta, CA). High-and low-pass filters were set at 1.0 Hz and 100Hz for electroencephalogram and 1.0 Hz and 500Hz for electromyogram recordings, respectively. Data were sent to an eMac computer running AcqKnowledge 3.9.1 (Biopac Systems Inc). Data were converted for analysis with Somnologica Studio version 3.2.1 with Rodent Sleep Scoring Module (EMBLA, Broomfield, CO). States of REM, NREM, and wakefulness in mice were determined by an experienced scorer and verified by a board certified sleep medicine physician (E.B.F.) on the basis of the predominant state within each 10 second epoch. Both scorers were blinded to the experimental condition. There was a 91.7% overall agreement between double-scored recordings of sleep and wakefulness in keeping with published studies of both human-human and human-computer concordance.43 Wakefulness was recognized by the presence of low amplitude, fast frequency, desynchronized electroencephalogram combined with elevated motor tone. NREM sleep was recognized by the presence of high amplitude, slow frequency electroencephalographic activity with reduced motor tone relative to wake. REM sleep was recognized by the presence of low-amplitude, fast frequency desynchronized electroencephalogram with a peak in theta frequency, together with minimal to absent motor tone.40,44
Sleep microstructure was distinguished by determining the number of wake bouts, NREM bouts, and REM bouts as well as the average bout length for each sleep state. Bout numbers were counted as each continuous episode of the sleep state. Bout length was calculated by the amount of time spent in separate bouts. Spectral analysis was performed using the Somnologica Science software application. Electroencephalographic data were fast Fourier transformed in 10 second epochs. The delta power (1.0 – 4.0 Hz) was computed for each NREM epoch and averaged over all NREM sleep epochs within three temporal periods: L2 corresponding to the 2nd portion of the light cycle (ZT7-12), D1 corresponding to the 1st portion of the dark cycle (ZT12-18), and D2 corresponding to the 2nd portion of the dark cycle (ZT18-24). Delta power is reported as a percentage of raw NREM delta power at baseline to raw NREM delta power following anesthetic exposure. Since each mouse was exposed to all conditions (baseline, sevoflurane, isoflurane, and halothane), a decision was made not to normalize total power, but rather to score and report absolute power at each frequency in 0.2Hz bins.
Experimental Protocol
Baseline recordings of wakefulness, NREM, and REM sleep in instrumented mice were conducted in all mice prior to anesthetic exposure while flowing 100% oxygen through the gas-tight chambers for 6 hours beginning at the onset of the light cycle, ZT0, to simulate conditions on the anesthetic day. Three days after obtaining baseline data, mice were exposed to a volatile anesthetic for 6 hours beginning at ZT0, which is coincident with their rest period. During anesthetic challenges fresh gas flows were set to 1 liter per minute with sevoflurane 2.4%, isoflurane 1.2%, or halothane 1.0%, corresponding to 1.2–1.3 times the ED50 for loss of righting reflex in C57BL/6 mice, respectively yielding burst suppression ratios of 4.59% ± 4.19%, 11.74% ± 8.80%, and 2.29% ± 3.85% as compared with a baseline burst suppression ratio of 0.02% ± 0.03% in the absence of any anesthetic. Burst suppression ratios were determined using epoch length of 60 seconds, a voltage cutoff of 15μV, and an isoelectric duration of 500ms. Burst suppression ratios were averaged across the entire 6 hour exposure for each animal and then averaged across animals. To promote euthermia, each recording chamber was actively heated by partially submerging it inside of a water bath set to 37C, which we have previously demonstrated prevents development of hypothermia in mice exposed to anesthetics.45,46 At the end of the six-hour anesthetic exposure, the anesthetic was discontinued, but fresh oxygen gas continued to flow for an additional thirty minutes. Fresh food and water were then returned to the mice and the chamber lids were reopened to room air. Scoring of NREM sleep, REM sleep, and wakefulness began during the second portion of the light cycle at ZT7 and continued to ZT24 (Figure 1). Mice were given 3–5 days between repeated anesthetic exposures. The first cohort of mice (n=6) was exposed to isoflurane, sevoflurane, and then halothane. The second cohort of mice (n=4) was exposed to sevoflurane, isoflurane, and then halothane. Halothane was deliberately chosen to be last anesthetic based upon a published report of cross-tolerance.47
Fig. 1.

States of Wakefulness, NREM sleep and REM sleep at baseline and following anesthetic exposure. (A) Experimental scheme in which general anesthesia is induced at the beginning of the light cycle and arousal states are recorded over the 2nd portion of the light cycle (L2), along with the 1st (D1) and 2nd (D2) portions of the dark cycle. Gray bar indicates the time given for emergence prior to recordings of sleep and wakefulness. Percentage of time spent in (B) REM sleep, (D) NREM sleep, and (F) wakefulness each hour as a function of time at baseline (black) and following exposure to isoflurane (purple), sevoflurane (yellow), or halothane (red). Total times spent in (C) REM sleep, (E) NREM sleep, or (G) wakefulness over L2, D1, and D2. **, *** respectively indicate p < 0.001 or p < 0.001 relative to corresponding baseline totals by Bonferroni post-hoc analysis of two-way ANOVA. NREM signifies non rapid eye movement whereas REM signifies rapid eye movement. ZT stands for zeitgeber time.
Statistical analysis
The amount of time spent in wake, NREM, and REM sleep was calculated in three blocks covering the second portion of the light cycle (L2) and both six-hour portions of the dark cycle (D1 and D2). Data are presented as mean ± standard error. The data were analyzed with a two-way mixed model repeated measures ANOVA of arousal state as a function of Treatment (baseline, sevoflurane, isoflurane, and halothane) and Time (L2, D1, and D2) using a random effects model for subjects with Prism 4.0c (GraphPad Software, Inc, San Diego, CA) and JMP version 8.0 (SAS Institute, Cary, NC). A Bonferroni correction was applied to all post-hoc comparisons. Interactions with Treatment and Time were considered and included only when statistically significant. Residuals were analyzed and checked for within subjects correlations and for constant variation among groups. We observed no significant departures from these assumptions, except where explicitly noted in the text.
The bout lengths for wakefulness, NREM sleep, and REM sleep were averaged to yield the mean bout length of each state in the given time frame. All values are shown as mean ± standard error. Data were analyzed with a mixed model repeat measure two-way ANOVA with Bonferroni correction applied for post-hoc comparisons. In all cases two-tailed P values less than 0.05 were considered to be statistically significant.
Results
Exposure to volatile anesthetics during the rest period caused a delayed REM rebound
Using continuous recordings of the electroencephalogram and electromyogram, we measured levels of wakefulness, NREM, and REM sleep in wild type C57BL6/J mice exposed to a general anesthetic for six-hours during the first half of the rest period. Mice exposed to hypnotic doses of sevoflurane, isoflurane, and halothane all exhibited an increase in REM sleep in comparison to baseline pre-exposure levels (Figure 1B, E; F3,99 = 8.39, p < 0.0001). This rebound was time dependent and did not manifest until D1, the onset of the dark cycle (F2,99 = 18.44, p < 0.0001). Moreover, the repeated measure two-way ANOVA confirmed a significant interaction between the two factors: treatment × time (F6,99 = 3.36, p = 0.0046). Following halothane exposure, the REM rebound persisted throughout the entire 2nd half of the dark cycle (Figure 1E).
Exposure to sevoflurane or isoflurane did not alter subsequent levels of wakefulness or NREM sleep, but exposure to halothane decreased wakefulness and increased NREM sleep
Previous exposure to isoflurane or sevoflurane did not significantly alter ensuing amounts of NREM sleep or wakefulness. However, previous exposure to halothane was associated with a significant increase in NREM sleep during both the light and first half of the dark periods (Figure 1C, F). Repeated measure two-way ANOVA confirmed a significant effect of both treatment (F3,99 = 11.46, p < 0.0001) and time (F2,99 = 17.15, p < 0.0001) with a significant interaction of the factors (F6,99 = 2.21, p = 0.048). Increased NREM sleep following general anesthesia with halothane was accompanied by a corresponding significant decrease in wakefulness displaying identical temporal dependency as the NREM changes (Figure 1D, G). Once again, during wakefulness our ANOVA model revealed a significant effect of both the treatment (F3,99 = 12.91, p < 0.0001) and time (F2,99 = 125.95, p < 0.0001) with no significant interaction of the factors (F6,99 = 2.00, p = 0.07).
Mice exposed to general anesthesia subsequently exhibited agent-specific significant changes in their sleep architecture
Following anesthetic exposure, the underlying structure of sleep-wake cycles exhibited significant changes. As shown in Figure 2, anesthetic treatment significantly altered REM bout length (F3,99 = 6.22, p = 0.0007) and number (F3,99 = 5.11, p =0.0025). Significant temporal effects on REM bout length (F2,99 = 4.51, p = 0.0133) and number (F2,99 = 5.14, p = 0.0075) were also evident. Initial significant daytime increases in REM bout length following isoflurane and sevoflurane were offset by decreases in the number of REM bouts; whereas halothane had no initial effect upon daytime REM sleep structure. When the active phase begins at lights out, ZT12, a non-significant increase in REM bout number for isoflurane and sevoflurane or with the significant increase in REM bout number following halothane exposure (Figure 2D) multiplied with non-significant changes in the halothane group REM bout length (Figure 2A) to yield a profoundly significant near doubling of total REM time in the first six-hours of the dark cycle (Figure 1). In the second half of the night, there were no significant changes in REM bout length or its average duration, yet the product of increased bout length and number continued to cause a significant REM rebound only for halothane (Figure 1).
Fig. 2.

Sleep architecture at baseline and following volatile anesthetic exposure. Average length for each bout of (A) REM, (C) NREM, and (E) wakefulness along with the number of bouts for (B) REM, (D) NREM, and (F) wakefulness at baseline (black), and following six hours of sevoflurane (yellow), isoflurane (purple), or halothane (red) in L2, the 2nd portion of the light cycle; D1, the first portion of the dark cycle; and D2, the 2nd portion of the dark cycle. *,**, *** respectively indicate p < 0.05, p < 0.001, or p < 0.001 relative to corresponding baseline totals by Bonferroni post-hoc analysis of two-way ANOVA. NREM signifies non rapid eye movement whereas REM signifies rapid eye movement.
Diversity of the interaction between volatile anesthetics and the neural systems regulating sleep and wakefulness is supported by effects upon NREM sleep architecture. As shown in Figures 2B and 2E, anesthetic treatment significantly altered NREM sleep bout length (F3,99 = 6.77, p = 0.0003) and number (F3,99 = 5.86, p = 0.001). However, time appeared to affect NREM bout number only (F2,99 = 7.20, p = 0.0012) without affecting NREM bout length (F2,99 = 0.36, p=0.70). There were no significant interactions between the bout length and anesthetic treatment factors. Compared with pre-exposure baseline data, six-hour isoflurane exposure significantly shortened the average NREM bout duration but correspondingly increased the number of NREM bouts. Conversely, six-hour halothane exposure led only to an initial significant increase in the length of NREM bouts (Figure 2B, E). There were no significant changes in the architecture of NREM sleep following exposure to sevoflurane.
With respect to baseline wakefulness, anesthetic drug treatment significantly altered both average wake bout duration (F3,99 = 9.47, p < 0.0001) as well as the number of waking bouts (F3,99 = 6.04, p = 0.0008). Effects of time upon the structure of waking were also highly significant for both bout length (F2,99 = 26.72, p < 0.0001) and number of waking bouts (F2,99 = 6.33, p = 0.0026) with a significant interaction of treatment and time occurring only for average bout duration (F6,99 = 3.77, p = 0.0023), but not for number of waking bouts (F6,99 = 0.92, p = 0.48). Exposure to six-hours of volatile anesthetics during the 1st half of the rest phase caused a subsequent significant decrease in the average duration of wake bouts, during the 1st half of the active period, D1. This is consistent with a fragmenting of post-anesthetic arousal state maintenance with a significant degradation in the stability of wake state (Figure 2C). This net decrease in wake bout length was offset by increases in the number of wake bouts during D1 (Figure 2F). In the case of isoflurane, wake bout number was significantly increased during both halves of the dark period.
Latency to enter REM sleep was significantly reduced during the active period following exposure to sevoflurane, isoflurane, or halothane
Anesthetic treatment significantly changed sleep latency (F1,63 = 3.58, p = 0.0185). Moreover, there was a significant interaction between the time and treatment factors (F3,63 = 13.62, p < 0.0001). In comparison to the pre-anesthetic exposure baseline, mice exhibited a significantly shorter latency to entering REM sleep, consistent with an increased homeostatic pressure to enter REM sleep. REM sleep latency in D1 decreased more than three-fold from over three hours to under one hour for all three anesthetic agents and was highly significant by post-hoc Bonferroni corrected analysis as shown in Figure 3. Immediately following the anesthetic exposure through the end of the light cycle, all mice exhibited an increased latency to enter REM sleep, which reached significance only for isoflurane. An analysis of the residuals indicated that variance increased with latency. Nonetheless, the results are nearly identical (F3,63 = 13.37, p <0.0001) when a variance stabilizing square root transformation is applied to the response.
Fig 3.

Sleep latency at baseline and following volatile anesthetic exposure. Time until the first bout of REM and NREM sleep following emergence in L2, the 2nd portion of the light cycle and D1, the first portion of the dark cycle. *,*** respectively indicate p < 0.05 or p < 0.001 relative to baseline by Bonferroni post-hoc analysis of two-way ANOVA. NREM signifies non rapid eye movement whereas REM signifies rapid eye movement.
Latency to enter NREM sleep was significantly reduced during the active period only following exposure to halothane
NREM sleep latency was significantly changed by time (F1,63 = 7.32, p = 0.009). Moreover, there was a significant interaction between time and treatment (F3,63 = 3.09, p = 0.03). Post-hoc Bonferroni corrected testing revealed a significant reduction in NREM sleep latency only following halothane exposure (Figure 3).
Homeostatic pressure to enter deep state of NREM sleep was not increased following exposure to sevoflurane, isoflurane, or halothane
Unlike general anesthesia, sleep is homeostatically regulated. Following sleep deprivation, there is an increased propensity for the deprived subject to enter the deep, restorative phases of slow wave NREM sleep, which is characterized by highly synchronized, large amplitude waveforms observed on the electroencephalogram with a frequency less than 4Hz in the delta range. This increased homeostatic propensity or drive to sleep manifests by an increased amount of slow wave sleep and is measured by performing a Fourier transformation on the electroencephalogram and computing the power in the frequency bins below 4Hz (delta power).48 If states of general anesthesia elicited by volatile anesthetic exposure physiologically deprived subjects of NREM sleep, then relative to baseline, a significant increase in NREM delta power should be induced following anesthetic exposure. However, if general anesthesia fulfils the requirements of NREM sleep then no increases in delta power would be expected. Following six-hours of volatile anesthetic exposure given at the onset of the rest phase, we failed to uncover any ensuing significant increase in delta power (Figure 4). Instead, in comparison to baseline, NREM sleep delta power was actually reduced following volatile anesthetic exposure. Two-way mixed model ANOVA revealed a significant effect of anesthetic treatment on delta power (F3,99 = 9.50, p < 0.0001). Meanwhile, there was no significant effect of time (F2,99 = 0.57, p = 0.56) and no significant interaction between time and treatment (F6,99 = 1.85, p = 0.097) upon delta power in NREM sleep following volatile anesthetic exposure. Nevertheless, we observe that the largest reduction in delta power occurred at the beginning of the active phase where previous isoflurane exposure significantly reduced homeostatic drive to sleep. Detailed spectral decomposition of the electroencephalographic power in all NREM sleep epochs as a function of time (Figure 5) also demonstrated that relative to baseline, there was reduced power in slow (< 4Hz) frequency bins that was most pronounced during D1 and D2.
Fig 4.
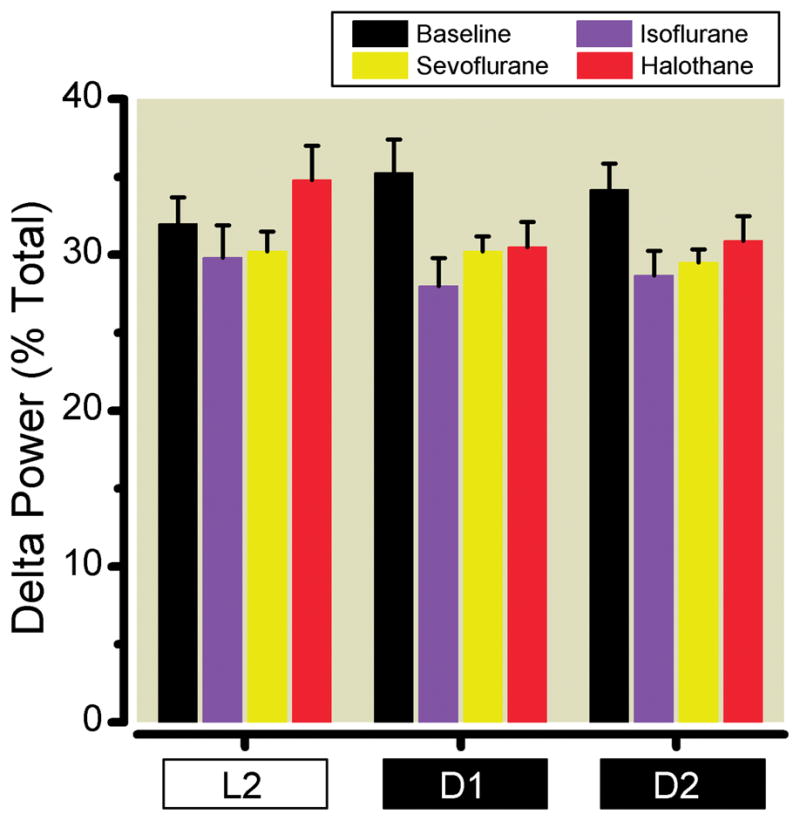
Delta power (1.0–4.0 Hz) at baseline (black), and following six hours of sevoflurane (yellow), isoflurane (purple), or halothane (red) in L2, the 2nd portion of the light cycle; D1, the first portion of the dark cycle; and D2, the 2nd portion of the dark cycle.
Fig. 5.

Average spectral power during non rapid eye movement sleep at baseline (black), and following six hours of sevoflurane (yellow), isoflurane (purple), or halothane (red) over (A) L2, the 2nd portion of the light cycle (B) D1, the 1st portion of the dark cycle and (C) D2, the 2nd portion of the dark cycle.
Discussion
Herein we demonstrate that the state of general anesthesia does not fully substitute for natural sleep. In contrast to propofol, a six-hour general anesthetic initiated and maintained by the volatile anesthetics sevoflurane, isoflurane, or halothane does indeed incur a REM sleep deficit, while only halothane accrues an additional NREM sleep debt. Each volatile anesthetic fragmented subsequent wakefulness during the animal’s ensuing active period. This was most severe for halothane, which also significantly reduced the total duration of wakefulness.
The response to sleep depriving stimuli depends upon the circadian phase in which it is administered.49 Our anesthetic delivery was specifically chosen to coincide with the onset of the light cycle—the rodent rest phase. We reasoned that should the anesthetic state function in analogous manner to a “sleep deprivation” by precluding subjects from activating the neural circuits necessary for NREM and/or REM sleep, then the ensuing propensity to sleep, also known as the homeostatic pressure, would be maximized by delivering the anesthetic at the end of the waking period.
One readily detected manifestation of sleep deprivation is the ensuing rebound when deprived subjects are presented with the opportunity for recovery sleep.50 For NREM sleep, this recovery may manifest as increased total NREM sleep duration; increased intensity of NREM sleep, indicated by an increase in NREM delta power; increased consolidation of NREM sleep bouts; and decreased latency to enter NREM sleep. Conversely, for REM sleep there is no known means to increase REM intensity or quality within a single bout, hence it has been proposed that REM sleep deficits can only be compensated by a shorter latency to the first REM bout and by increased total REM sleep time.50
As with a six-hour sleep deprivation produced by gentle handling or wheel running,51,52 a six-hour volatile anesthetic exposure also caused a delayed REM rebound that was expressed during the first half of the mouse’s active phase for sevoflurane and isoflurane and for the entire active phase for halothane. Consistent with the increased REM pressure during the mouse’s active phase, we demonstrate a significantly shorter latency to enter REM sleep. The magnitude of REM sleep rebound in the first half of the active phase caused by exposure to isoflurane, sevoflurane, and halothane was 184%, 207%, and 228% respectively of the baseline level of REM sleep, was nearly identical to the 220% seen following six-hours of automated sleep deprivation by wheel running in mice,52 and was comparable to the rebound following 24 hours of total sleep deprivation in rats.53
Whereas the REM sleep rebound appeared largely comparable to that expected in rodents following traditional sleep deprivation, NREM sleep following sevoflurane or isoflurane exposure did not increase in total amount, in average NREM sleep bout duration, or in NREM sleep intensity as defined by increased delta power. While there was a decreased latency to enter NREM sleep in the active phase following isoflurane or sevoflurane exposure, these changes failed to reach significance. Hence by all criteria, a six-hour exposure to isoflurane and sevoflurane did not induce a NREM rebound. Halothane, which is known to exhibit distinct cortical and subcortical effects from the halogenated ether anesthetics isoflurane and sevoflurane at equipotent doses,40,54–57 showed a distinct profile with evidence for rebound NREM sleep. Following halothane exposure, mice significantly increased the total time spent in NREM sleep both immediately and into the second half of their active phase. This increase was accompanied by an initial increase in NREM sleep bout length in accordance with published results in mice following six-hours of sleep deprivation by gentle handling,58 and with decreased latency to enter NREM sleep. Similar to other anesthetics,18,25,27,59 but unlike sleep deprivation,48 NREM sleep following halothane exposure was not associated with increased delta power.
Cumulatively our findings suggest that sevoflurane and isoflurane may satisfy the homeostatic drive for NREM sleep. Specifically, in the case of isoflurane and sevoflurane NREM sleep pressure may be discharged by the state of general anesthesia.18,25,59 However, like halothane and unlike propofol, sevoflurane and isoflurane do not satisfy the homeostatic drive for REM sleep. This conclusion is supported by additional studies in rodents in which sleep deprived rats anesthetized with isoflurane or sevoflurane were unable to dissipate preexisting REM sleep deficits during the volatile anesthetic.18,25
Results obtained herein suggest that the brain is still able to track time elapsed under general anesthesia.60 Our results with three distinct volatile anesthetics suggest that REM and NREM homeostatic tracking mechanisms could be distinct. Were the homeostatic markers of NREM and REM sleep dependent only upon synaptic activity, an increased probability of synaptic failure in the presence of general anesthetics could erase information stored by the homeostat. Alternatively, the homeostatic encoding of accrued sleep debts could be stored purely by a neurochemical signal that amasses with neuronal activity, but exists in an activity-independent, stable form. In this latter case, dysfunction in synaptic neurotransmission by volatile anesthetics would not be sufficient to reset the homeostat. Such a possibility would lead to a permissive state in which the homeostat might be frozen in place, but still able to track preexisting debts, or unaffected by the presence of the anesthetics and able to run as if no drug were present. Data by Mashour and colleagues18,25 demonstrate the latter possibility. Our study, in which no sleep debts were accrued prior to anesthetic exposure, allows further insight into homeostatic functioning under anesthesia. Finding a distinct interaction of anesthetic drugs with neural circuits regulating arousal is consistent with agent-specific distinct interactions with the homeostat. The absence of a NREM rebound following sevoflurane or isoflurane exposure implies either a timekeeping failure or that the anesthetic state has partially substituted for NREM sleep. Halothane anesthesia would appear fundamentally different by not satisfying homeostatic need for NREM or REM sleep. As additional knowledge is gained about the interactions of distinct anesthetics with the REM and NREM homeostats, it might be possible to use general anesthetics to decode the anatomic loci and intrinsic regulation underlying sleep need.
Study Limitations
Several methodological issues merit consideration when examining our results. To minimize experimental variability, mice were exposed in parallel to identical gas flow conditions while euthermia was preserved. Groups of 4 or 6 mice were treated simultaneously. This design prevented true randomization of anesthetic exposure, potentially biasing our results based by the order of volatile anesthetic presentation. This flaw may be mitigated as short-term tolerance to volatile anesthetics does not occur, which makes cross-tolerance also doubtful.47,61 We did not acquire respiratory data and cannot formally exclude intermittent hypoxia or hypoventilation during anesthetic exposures. We attempted to normalize exposures using equipotent anesthetic concentrations that were 1.2–1.3 times the ED50 for loss of righting in mice.45 Sevoflurane and halothane equipotency was also confirmed by a second anesthetic endpoint—indistinguishable levels of burst suppression. However, with isoflurane we observed a significantly higher incidence of burst suppression. As REM rebound occurred following exposure to all volatile anesthetics and NREM rebound occurred only following halothane exposure, we believe that the depth of anesthesia as defined by burst suppression or potentially by relatively greater isoflurane levels cannot fully explain our findings. Of note, shorter anesthetic exposures may not be sufficient to accrue potential sleep debts.62
Conclusion and Implications
In summary, we demonstrate that a six-hour exposure to hypnotic doses of volatile anesthetics incurs a REM sleep deficit in mice, which is followed by a significant REM rebound hours after the cessation of the anesthetic. Such results are consistent with the notion that the volatile anesthetic anesthetized brain is prevented from accessing the neuronal circuits driving REM sleep while the REM homeostat continues to register the accruing deficit. Although isoflurane and sevoflurane appear to fulfill NREM sleep need, halothane does not. While extrapolating results from a murine model to humans is fraught with difficulties, earlier human studies also demonstrate similar adaptations in sleep following anesthesia. However, these studies either permitted unmonitored daytime napping and measured only night time sleep electroencephalographically in healthy volunteers37 or showed a significant REM sleep rebound, but were conducted in the setting of actual surgery and potentially confounded by pain and polypharmacy.35,36 Nevertheless, our results in healthy adult mice support the idea of REM rebound following volatile anesthetic exposure and potentially herald important clinical morbidity. REM sleep loss can cause hyperalgesia.63 Moreover, delayed REM rebound following volatile anesthetic exposures may place patients at risk for episodes of hypoxemia64 and is especially concerning in patients with obstructive sleep apnea in whom hypoventilation, carbon dioxide retention, oxygen desaturations and bradyarrhythmias often accompany REM sleep.65 REM rebound has also been linked to hemodynamic instability, myocardial infarction, stroke, and postoperative delirium.64 Together with other preclinical animal studies18,25,27,28,59 our data suggest that important agent-specific differences may exist in the ways that individual general anesthetic drugs affect sleep. Hence opportunities exist to determine whether the potential disruption of sleep by distinct anesthetics actually heralds heightened morbidity in vulnerable human populations; and whether an agent with a more favorable post-anesthetic sleep profile, like propofol,27,28 might make a better choice to reduce potential perioperative complications associated with REM sleep rebounds.
FinalBox Summary.
What we already know about this topic
“Administration of propofol to rodents satisfies the need for both rapid eye movement (REM) and non-REM sleep.”
What this article tells us that is new
In contrast to published actions of propofol, for which no REM sleep rebound occurred, REM sleep rebound occurs following exposure to volatile anesthetics in rodents, suggesting that volatile anesthetics do not fully substitute for natural sleep.
Acknowledgments
Support: This work was supported by research grant numbers GM077357, GM088156, and the American Recovery and Reinvestment Act (ARRA) funds through grant GM077357 from the National Institutes of Health, Bethesda, Maryland, from the Mary Elizabeth Groff Foundation, Radnor PA, the Foundation for Anesthesia Education and Research, Rochester, MN, and by the Department of Anesthesiology and Critical Care, University of Pennsylvania, Philadelphia, PA
The authors wish to thank Sigrid Veasey, M.D. (Associate Professor, Department of Medicine, University of Pennsylvania) and Polina Fenik, M.S. (Research Specialist, Center for Sleep and Circadian Neurobiology, University of Pennsylvania) for their assistance with electroencephalographic recordings in mice.
Footnotes
Attribution: Department of Anesthesiology and Critical Care, University of Pennsylvania, Philadelphia, PA
Available at http://grants.nih.gov/grants/olaw/references/PHSPolicyLabAnimals.pdf Accessed last on May 10, 2011
References
- 1.Halsey MJ, Prys Roberts C, Strunin L. Receptors and transmembrane signaling: Cellular and molecular aspects of anaesthesia. British Journal of Anaesthesia. 1993;71:1. [Google Scholar]
- 2.Lydic R, Biebuyck JF. Sleep neurobiology: Relevance for mechanistic studies of anaesthesia. Br J Anaesth. 1994;72:506–8. doi: 10.1093/bja/72.5.506. [DOI] [PubMed] [Google Scholar]
- 3.Karan SB, Perlis M, Ward D. Anesthesia and sleep medicine: An opportunity to be mutually informative? Seminars in Anesthesia, Perioperative Medicine and Pain. 2007;26:42–8. [Google Scholar]
- 4.Ferrarelli F, Massimini M, Sarasso S, Casali A, Riedner BA, Angelini G, Tononi G, Pearce RA. Breakdown in cortical effective connectivity during midazolam-induced loss of consciousness. Proc Natl Acad Sci U S A. 2010;107:2681–6. doi: 10.1073/pnas.0913008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massimini M, Ferrarelli F, Huber R, Esser SK, Singh H, Tononi G. Breakdown of cortical effective connectivity during sleep. Science. 2005;309:2228–32. doi: 10.1126/science.1117256. [DOI] [PubMed] [Google Scholar]
- 6.Murphy M, Bruno M, Riedner BA, Boveroux P, Noirhomme Q, Landsness E, Brichant J-F, Phillips C, Massimini M, Laureys S, Tononi G, Boly M. Propofol anesthesia and sleep: A high-density EEG study. Sleep. 2011;34:283–91A. doi: 10.1093/sleep/34.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imas OA, Ropella KM, Ward BD, Wood JD, Hudetz AG. Volatile anesthetics disrupt frontal-posterior recurrent information transfer at gamma frequencies in rat. Neurosci Lett. 2005;387:145–50. doi: 10.1016/j.neulet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Wagner T, Axmacher N, Lehnertz K, Elger CE, Fell J. Sleep-dependent directional coupling between human neocortex and hippocampus. Cortex. 2010;46:256–63. doi: 10.1016/j.cortex.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Lee U, Kim S, Noh GJ, Choi BM, Hwang E, Mashour GA. The directionality and functional organization of frontoparietal connectivity during consciousness and anesthesia in humans. Conscious Cogn. 2009;18:1069–78. doi: 10.1016/j.concog.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Horovitz SG, Braun AR, Carr WS, Picchioni D, Balkin TJ, Fukunaga M, Duyn JH. Decoupling of the brain’s default mode network during deep sleep. Proc Natl Acad Sci U S A. 2009;106:11376–81. doi: 10.1073/pnas.0901435106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boveroux P, Vanhaudenhuyse A, Bruno MA, Noirhomme Q, Lauwick S, Luxen A, Degueldre C, Plenevaux A, Schnakers C, Phillips C, Brichant JF, Bonhomme V, Maquet P, Greicius MD, Laureys S, Boly M. Breakdown of within- and between-network resting state functional magnetic resonance imaging connectivity during propofol-induced loss of consciousness. Anesthesiology. 2010;113:1038–53. doi: 10.1097/ALN.0b013e3181f697f5. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlin NL, Eikermann M. This is no humbug: Anesthetic agent-induced unconsciousness and sleep are visibly different. Anesthesiology. 2010;113:1007–9. doi: 10.1097/ALN.0b013e3181f69825. [DOI] [PubMed] [Google Scholar]
- 13.Alkire MT, Haier RJ, Fallon JH. Toward a unified theory of narcosis: Brain imaging evidence for a thalamocortical switch as the neurophysiologic basis of anesthetic-induced unconsciousness. Conscious Cogn. 2000;9:370–86. doi: 10.1006/ccog.1999.0423. [DOI] [PubMed] [Google Scholar]
- 14.Fiset P, Paus T, Daloze T, Plourde G, Meuret P, Bonhomme V, Hajj-Ali N, Backman SB, Evans AC. Brain mechanisms of propofol-induced loss of consciousness in humans: A positron emission tomographic study. J Neurosci. 1999;19:5506–13. doi: 10.1523/JNEUROSCI.19-13-05506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veselis RA, Reinsel RA, Beattie BJ, Mawlawi OR, Feshchenko VA, DiResta GR, Larson SM, Blasberg RG. Midazolam changes cerebral blood flow in discrete brain regions: An H2(15)O positron emission tomography study. Anesthesiology. 1997;87:1106–17. doi: 10.1097/00000542-199711000-00015. [DOI] [PubMed] [Google Scholar]
- 16.Nofzinger EA, Buysse DJ, Miewald JM, Meltzer CC, Price JC, Sembrat RC, Ombao H, Reynolds CF, Monk TH, Hall M, Kupfer DJ, Moore RY. Human regional cerebral glucose metabolism during non-rapid eye movement sleep in relation to waking. Brain. 2002;125:1105–15. doi: 10.1093/brain/awf103. [DOI] [PubMed] [Google Scholar]
- 17.Tung A, Szafran MJ, Bluhm B, Mendelson WB. Sleep deprivation potentiates the onset and duration of loss of righting reflex induced by propofol and isoflurane. Anesthesiology. 2002;97:906–11. doi: 10.1097/00000542-200210000-00024. [DOI] [PubMed] [Google Scholar]
- 18.Pal D, Lipinski WJ, Walker AJ, Turner AM, Mashour GA. State-specific effects of sevoflurane anesthesia on sleep homeostasis: Selective recovery of slow wave but no rapid eye movement sleep. Anesthesiology. 2011;114:302–10. doi: 10.1097/ALN.0b013e318204e064. [DOI] [PubMed] [Google Scholar]
- 19.Kaputlu I, Sadan G, Ozdem S. Exogenous adenosine potentiates hypnosis induced by intravenous anaesthetics. Anaesthesia. 1998;53:496–500. doi: 10.1046/j.1365-2044.1998.00330.x. [DOI] [PubMed] [Google Scholar]
- 20.Tanase D, Baghdoyan HA, Lydic R. Dialysis delivery of an adenosine A1 receptor agonist to the pontine reticular formation decreases acetylcholine release and increases anesthesia recovery time. Anesthesiology. 2003;98:912–20. doi: 10.1097/00000542-200304000-00018. [DOI] [PubMed] [Google Scholar]
- 21.Tung A, Mendelson WB. Anesthesia and sleep. Sleep Med Rev. 2004;8:213–25. doi: 10.1016/j.smrv.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Lydic R, Baghdoyan HA. Sleep, anesthesiology, and the neurobiology of arousal state control. Anesthesiology. 2005;103:1268–95. doi: 10.1097/00000542-200512000-00024. [DOI] [PubMed] [Google Scholar]
- 23.Alkire MT, Hudetz AG, Tononi G. Consciousness and anesthesia. Science. 2008;322:876–80. doi: 10.1126/science.1149213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franks NP. General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–86. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 25.Mashour GA, Lipinski WJ, Matlen LB, Walker AJ, Turner AM, Schoen W, Lee U, Poe GR. Isoflurane anesthesia does not satisfy the homeostatic need for rapid eye movement sleep. Anesth Analg. 2010;110:1283–9. doi: 10.1213/ANE.0b013e3181d3e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Achermann P. The two-process model of sleep regulation revisited. Aviat Space Environ Med. 2004;75:A37–43. [PubMed] [Google Scholar]
- 27.Tung A, Bergmann BM, Herrera S, Cao D, Mendelson WB. Recovery from sleep deprivation occurs during propofol anesthesia. Anesthesiology. 2004;100:1419–26. doi: 10.1097/00000542-200406000-00014. [DOI] [PubMed] [Google Scholar]
- 28.Tung A, Lynch JP, Mendelson WB. Prolonged sedation with propofol in the rat does not result in sleep deprivation. Anesth Analg. 2001;92:1232–6. doi: 10.1097/00000539-200105000-00028. [DOI] [PubMed] [Google Scholar]
- 29.Campbell JL, Gu Q, Guo D, Nash HA. Genetic effects in Drosophila on the potency of diverse general anesthetics: A distinctive pattern of altered sensitivity. J Neurogenet. 2009;23:412–21. doi: 10.3109/01677060903177800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antkowiak B. Different actions of general anesthetics on the firing patterns of neocortical neurons mediated by the GABA(A) receptor. Anesthesiology. 1999;91:500–11. doi: 10.1097/00000542-199908000-00025. [DOI] [PubMed] [Google Scholar]
- 31.Hans P, Dewandre PY, Brichant JF, Bonhomme V. Comparative effects of ketamine on Bispectral Index and spectral entropy of the electroencephalogram under sevoflurane anaesthesia. Br J Anaesth. 2005;94:336–40. doi: 10.1093/bja/aei047. [DOI] [PubMed] [Google Scholar]
- 32.Kubota T, Anzawa N, Hirota K, Yoshida H, Kushikata T, Matsuki A. Effects of ketamine and pentobarbital on noradrenaline release from the medial prefrontal cortex in rats. Can J Anaesth. 1999;46:388–92. doi: 10.1007/BF03013235. [DOI] [PubMed] [Google Scholar]
- 33.Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M. The sedative component of anesthesia is mediated by GABA(A) receptors in an endogenous sleep pathway. Nat Neurosci. 2002;5:979–84. doi: 10.1038/nn913. [DOI] [PubMed] [Google Scholar]
- 34.Van Dort CJ, Baghdoyan HA, Lydic R. Neurochemical modulators of sleep and anesthetic states. Int Anesthesiol Clin. 2008;46:75–104. doi: 10.1097/AIA.0b013e318181a8ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knill RL, Skinner MI, Novick T, Vandenberghe HM, Moote CA. The night of intense REM sleep after anesthesia and surgery increases urinary catecholamines. Can J Anaesth. 1990;37:S12. [PubMed] [Google Scholar]
- 36.Knill RL, Moote CA, Skinner MI, Rose EA. Anesthesia with abdominal surgery leads to intense REM sleep during the first postoperative week. Anesthesiology. 1990;73:52–61. doi: 10.1097/00000542-199007000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Moote CA, Knill RL. Isoflurane anesthesia causes a transient alteration in nocturnal sleep. Anesthesiology. 1988;69:327–31. doi: 10.1097/00000542-198809000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Yanagida H, Yamamura H. The study of natural sleep after general anaesthesia in the cat. Br J Anaesth. 1972;44:1229–33. doi: 10.1093/bja/44.12.1229. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi S, Kushikata T, Matsuki A. Effects of isoflurane and ketamine on sleep in rabbits. Psychiatry Clin Neurosci. 2001;55:239–40. doi: 10.1046/j.1440-1819.2001.00840.x. [DOI] [PubMed] [Google Scholar]
- 40.Kelz MB, Sun Y, Chen J, Cheng Meng Q, Moore JT, Veasey SC, Dixon S, Thornton M, Funato H, Yanagisawa M. An essential role for orexins in emergence from general anesthesia. Proc Natl Acad Sci U S A. 2008;105:1309–14. doi: 10.1073/pnas.0707146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veasey SC, Valladares O, Fenik P, Kapfhamer D, Sanford L, Benington J, Bucan M. An automated system for recording and analysis of sleep in mice. Sleep. 2000;23:1025–40. [PubMed] [Google Scholar]
- 42.Leach NT, Sun Y, Michaud S, Zheng Y, Ligon KL, Ligon AH, Sander T, Korf BR, Lu W, Harris DJ, Gusella JF, Maas RL, Quade BJ, Cole AJ, Kelz MB, Morton CC. Disruption of Diacylglycerol Kinase Delta (DGKD) Associated with Seizures in Humans and Mice. Am J Hum Genet. 2007;80:792–9. doi: 10.1086/513019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gross BA, Walsh CM, Turakhia AA, Booth V, Mashour GA, Poe GR. Open-source logic-based automated sleep scoring software using electrophysiological recordings in rats. J Neurosci Methods. 2009;184:10–8. doi: 10.1016/j.jneumeth.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ouyang M, Hellman K, Abel T, Thomas SA. Adrenergic signaling plays a critical role in the maintenance of waking and in the regulation of REM sleep. J Neurophysiol. 2004;92:2071–82. doi: 10.1152/jn.00226.2004. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, Chen J, Pruckmayr G, Baumgardner JE, Eckmann DM, Eckenhoff RG, Kelz MB. High throughput modular chambers for rapid evaluation of anesthetic sensitivity. BMC Anesthesiology. 2006;6:13. doi: 10.1186/1471-2253-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friedman EB, Sun Y, Moore JT, Hung HT, Meng QC, Perera P, Joiner WJ, Thomas SA, Eckenhoff RG, Sehgal A, Kelz MB. A Conserved Behavioral State Barrier Impedes Transitions between Anesthetic-Induced Unconsciousness and Wakefulness: Evidence for Neural Inertia. PLoS One. 2010;5:e11903. doi: 10.1371/journal.pone.0011903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chalon J, Tang CK, Roberts C, Walpert L, Hoffman C, Ramanathan S, Turndorf H. Murine auto- and cross-tolerance to volatile anaesthetics. Can Anaesth Soc J. 1983;30:230–4. doi: 10.1007/BF03013800. [DOI] [PubMed] [Google Scholar]
- 48.Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–21. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vyazovskiy VV, Achermann P, Tobler I. Sleep homeostasis in the rat in the light and dark period. Brain Res Bull. 2007;74:37–44. doi: 10.1016/j.brainresbull.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 50.Borbely AA, Tobler I, Hanagasioglu M. Effect of sleep deprivation on sleep and EEG power spectra in the rat. Behav Brain Res. 1984;14:171–82. doi: 10.1016/0166-4328(84)90186-4. [DOI] [PubMed] [Google Scholar]
- 51.Morrow JD, Opp MR. Sleep-wake behavior and responses of interleukin-6-deficient mice to sleep deprivation. Brain Behav Immun. 2005;19:28–39. doi: 10.1016/j.bbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Fenzl T, Romanowski CP, Flachskamm C, Honsberg K, Boll E, Hoehne A, Kimura M. Fully automated sleep deprivation in mice as a tool in sleep research. J Neurosci Methods. 2007;166:229–35. doi: 10.1016/j.jneumeth.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Schwierin B, Borbely AA, Tobler I. Prolonged effects of 24-h total sleep deprivation on sleep and sleep EEG in the rat. Neurosci Lett. 1999;261:61–4. doi: 10.1016/s0304-3940(98)01006-4. [DOI] [PubMed] [Google Scholar]
- 54.Hudetz AG. Effect of volatile anesthetics on interhemispheric EEG cross-approximate entropy in the rat. Brain Res. 2002;954:123–31. doi: 10.1016/s0006-8993(02)03358-9. [DOI] [PubMed] [Google Scholar]
- 55.Antunes LM, Golledge HD, Roughan JV, Flecknell PA. Comparison of electroencephalogram activity and auditory evoked responses during isoflurane and halothane anaesthesia in the rat. Vet Anaesth Analg. 2003;30:15–23. doi: 10.1046/j.1467-2995.2003.00085.x. [DOI] [PubMed] [Google Scholar]
- 56.Orth M, Bravo E, Barter L, Carstens E, Antognini JF. The differential effects of halothane and isoflurane on electroencephalographic responses to electrical microstimulation of the reticular formation. Anesth Analg. 2006;102:1709–14. doi: 10.1213/01.ane.0000205752.00303.94. [DOI] [PubMed] [Google Scholar]
- 57.Gompf HS, Chen J, Sun Y, Yanagisawa M, Aston-Jones G, Kelz MB. Halothane-induced Hypnosis is not Accompanied by Inactivation of Orexinergic Output in Rodents. Anesthesiology. 2009;111:1001–9. doi: 10.1097/ALN.0b013e3181b764b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stenberg D, Litonius E, Halldner L, Johansson B, Fredholm BB, Porkka-Heiskanen T. Sleep and its homeostatic regulation in mice lacking the adenosine A1 receptor. J Sleep Res. 2003;12:283–90. doi: 10.1046/j.0962-1105.2003.00367.x. [DOI] [PubMed] [Google Scholar]
- 59.Nelson AB, Faraguna U, Tononi G, Cirelli C. Effects of anesthesia on the response to sleep deprivation. Sleep. 2010;33:1659–67. doi: 10.1093/sleep/33.12.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daley JT, Kelz MB. Time in general anesthesia: Depriving the homeostat? Sleep. 2010;33:1583–4. doi: 10.1093/sleep/33.12.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milutinovic PS, Zhao J, Sonner JM. Tolerance to isoflurane does not occur in developing Xenopus laevis tadpoles. Anesth Analg. 2009;108:176–80. doi: 10.1213/ane.0b013e31818ca33e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jang HS, Jung JY, Jang KH, Lee MG. Effects of isoflurane anesthesia on post-anesthetic sleep-wake architectures in rats. Korean J Physiol Pharmacol. 2010;14:291–7. doi: 10.4196/kjpp.2010.14.5.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roehrs T, Hyde M, Blaisdell B, Greenwald M, Roth T. Sleep loss and REM sleep loss are hyperalgesic. Sleep. 2006;29:145–51. doi: 10.1093/sleep/29.2.145. [DOI] [PubMed] [Google Scholar]
- 64.Kaw R, Michota F, Jaffer A, Ghamande S, Auckley D, Golish J. Unrecognized sleep apnea in the surgical patient: Implications for the perioperative setting. Chest. 2006;129:198–205. doi: 10.1378/chest.129.1.198. [DOI] [PubMed] [Google Scholar]
- 65.Chung SA, Yuan H, Chung F. A systemic review of obstructive sleep apnea and its implications for anesthesiologists. Anesth Analg. 2008;107:1543–63. doi: 10.1213/ane.0b013e318187c83a. [DOI] [PubMed] [Google Scholar]