

Abstract
Next-generation sequencing platforms are dramatically reducing the cost of DNA sequencing. With these technologies, bases are inferred from light intensity signals, a process commonly referred to as base-calling. Thus, understanding and improving the quality of sequence data generated using these approaches are of high interest. Recently, a number of papers have characterized the biases associated with base-calling and proposed methodological improvements. In this review, we summarize recent development of base-calling approaches for the Illumina and Roche 454 sequencing platforms.
Keywords: Base-calling, next generation sequencing, deep sequencing, illumina/solexa, roche/454, bustard
INTRODUCTION
Over the last three decades, DNA sequencing has become a workhorse in computational biology, comparative genomics and biology in general. Traditionally, sequencing has been performed using Sanger’s method [1], whose refinement over the years culminated with long reads of up to ~1000 bp at an error rate as low as 10−5error per base [2]. A staggering demand for cheap and fast sequencing technology and substantial funding [3] has lead to the development of numerous new approaches to sequencing. Many of these approaches have been incorporated in commercial products including Roche 454 (Roche 454 Sequencing, http://www.454.com/), Illumina (Illumina Inc, http://www.illumina.com/), SOLiD (Applied Biosystems, https://products.appliedbiosystems.com/), Polonator (Applied Biosystems, https://products.appliedbiosystems.com/), Helicos (Helicos BioScience Corporation, http://www.helicosbio.com/), Pacific Biosciences (Pacific Biosciences, http://www.pacificbiosciences.com/) and Intelligent Bio Systems (Intelligent Bio Systems, http://intelligentbiosystems.com/). These next-generation sequencing technologies improve both speed and cost at the price of a lower accuracy and shorter read lengths compared to Sanger sequencing. Reducing the cost allows the exploration of new problem domains using sequencing such as assessing the variability of genomes [4–7]. Illumina announced a service to sequence a human genome for less than $20 000 (http://investor.illumina.com/phoenix.zhtml?c=121127&p=irol-newsArticle&ID=1434418). Ultimately pushing the price down to $1000 will allow to sequence the genome of an individual as a routine medical test [8].
The next-generation sequencing technologies all rely on a complex interplay of chemistry, hardware and optical sensors. Adding to this complexity is software to analyze the sensor data to predict the individual bases. This last step in the process is referred to as base-calling. While the overall production pipelines are similar across sequencing platforms, they differ in mechanistic details which affect the types of errors made during sequencing. The characterization of errors associated with the different sequencing platforms is of crucial importance to downstream analysis [9]. The accuracy of sequencing can be improved by increasing the coverage, i.e resequencing the same DNA sample multiple times. The data is then aggregated into a consensus sequence with lower error rate [10]. Conversely, more accurate base-callers reduce the coverage required to reach a given accuracy and therefore directly decrease the sequencing costs.
In this review, we focus on recent progress in base-calling algorithms for the Illumina and Roche 454 platforms. Both are well-established next-generation sequencers for which third party programs have been developed as alternative to the vendor base-calling implementation. For a broader overview of next-generation sequencing technology and data processing pipeline, we refer to [11]. In the next section we briefly describe the technology of the Illumina platform with a focus on its biases. We then review several recently published alternative base-callers and compare their performances in terms of accuracy and speed. We then turn to the Roche 454 platform again focusing on the difficulties associated with its technology. We finish this review with a discussion on benefits and drawbacks of the different approaches described and motivations for future developments in this active area of research.
ILLUMINA
The Illumina platform relies on the generation of a single strand DNA library by random fragmentation of a DNA sample. After addition of universal adapters to the templates, the templates are spread in an eight lane flow cell and immobilized on glass [12]. Following in place bridge amplification, this process generates a large number of clusters of identical templates on the glass surface. The sequence of the templates in the clusters is then determined using reversible terminators chemistry [13]. In every sequencing cycle a single fluorescently labeled, 3′-blocked nucleotide is synthesized to each complementary strand. After incorporation, the fluorescent labeling can be detected using imaging technology. Finally, the labels and terminators are chemically removed in order to prepare the complementary strands for the next sequencing cycle. A more detailed description of the process can be found in [14].
The Illumina platform suffers from numerous biases due to imperfect chemistry and sensors (Figure 1). During template preparation mixed clusters occur whenever multiple templates are colocated [15]. Such clusters need to be excluded from downstream analysis. While sequencing, a strand which has failed to incorporate a base in a given cycle will continue to lag behind. This is referred to as phasing. On the other hand, if multiple bases are synthesized in a single cycle, this is called pre-phasing. Phasing, pre-phasing and the decay of signal intensity from one cycle to another, again due to imperfect chemistry, result in an increase of base-calling errors towards the end of reads. Furthermore, in early chemistries (e.g. FC-104-100x), an accumulation of Thymine (T) due to incomplete cleavage of the T-dye has been reported [15]. Yet other biases are due to the limitations of the optical detection. The emission frequency spectra corresponding to the four dyes partly overlap. As a result, the intensity quadruples detected at each cycles show some positive correlation. This effect, commonly referred to as cross-talk, has been found to be cycle dependent [16]. Finally, due to optical effects, the intensity is uneven across each tile, with lower intensity toward the edges [17].
Figure 1:

Illustration of the commonly modeled biases in base-callers for the Illumina platform. ϕ: Phasing can be observed as leading (gray arrow) and lagging (black arrows) signal increase before and after each intensity peak. This is illustrated by the averaged intensities of the cytosine channel when sequencing GCAGTAGTGTTGGTT CTGTAGTGGAATGTGCGGTTGTTGAGAATTCAGTA. Cross-talk correction and normalization have been applied and the first cycle has been omitted. δ: Signal decay is illustrated by the intensity signal of sequencing the micro satellite sequence ACACAC … Shown are the averaged intensities of cytosine (red) and adenine (blue) after crosstalk correction and normalization. Again, the first cycle is not shown. µ: Mixed clusters occur whenever more than one template collocate on the tile. ω: The image shows local averages of the fluorescence intensities across the area of a tile. Due to optical effects, stronger intensities are measured toward the center of the image. Σ: The intensity quadruples of the four bases are not orthogonal. Shown is the projection of measured intensities of the first sequencing cycle of the phiX174 data onto the axes corresponding to A and C. τ: In past chemistries the T-fluophore was not washed away efficiently and hence accumulated with growing number of cycles. The illustration shows the intensity values for one tile of a 51-cycle PhiX 174 RF1 run after correction by Bustard. Shown is the 95th percentile for the signal intensities in each channel and cycle. Figure credits: ϕ, δ from [16]; ω, Σ from [17]; τ from [18].
BASE-CALLING
The Illumina sequencing platform is shipped with GApipeline, which implements image analysis (Firecrest), base-calling (Bustard) and alignment to reference sequences. Bustard applies a cycle independent correction for cross-talk, followed by the correction of phasing and pre-phasing. After these corrections have been applied the base with the highest intensity is chosen. For quality control, a sample of the bacteriophage ϕX174 genome is usually included in one of the eight lanes of the flow cell. A more detailed description of the base-calling algorithm implemented in Bustard can be found in [19].
Within the last 2 years, numerous papers have been published which improve upon the native base-calling implementation. The first among them which was Alta-Cyclic. Alta-Cyclic uses a parametric model for dephasing and then corrects for cross-talk using a cycle dependent cross-talk matrix. Support Vector Machines (SVM) are used to determine the base based on the four intensity values. To account for signal decay and cycle dependent cross-talk Alta-Cyclic uses a different SVM for every cycle. In order to optimize the SVMs and phasing parameters supervised learning is used. Alta-Cyclic performs a grid search to find phasing parameters for which the SVMs can optimally predict the bases in a reference sequence, which requires training the SVMs at every grid point. The model is optimized for every run of the Illumina platform independently. This procedure is not only computationally expensive but it also requires resequencing part of a reference genome in order to generate enough training data. The ϕX174 control lane can be used for training.
Another approach is implemented in a package called Rolexa [17]. Like Bustard, Rolexa first applies a cycle dependent linear transform accounting for cross-talk between the different bases, before using a binomial distribution for dephasing. Finally, it can be observed that due to optical effects clusters near the center of each tile appear brighter than those near the edges. Rolexa corrects for these optical effects by fitting a two dimensional Lowess model to the intensities of each tile. After applying these three corrections Rolexa uses a clustering algorithm based on Gaussian mixtures for base-calling. From this a measure of uncertainty can be computed which is used to call the most likely bases as well as reporting IUPAC codes. IUPAC codes are used to encode ambiguities in the base-calling process through additional letters. For instance, S stands for either C or G. However, since all other implementations report Phred scores [20] [the log probabilities of an error: 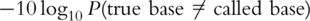 ] rather than IUPAC codes, this approach is difficult to compare. An advantage of Rolexa is that it does not depend on supervised learning, thereby eliminating the need to resequence known templates for training and thereby increasing overall yield.
] rather than IUPAC codes, this approach is difficult to compare. An advantage of Rolexa is that it does not depend on supervised learning, thereby eliminating the need to resequence known templates for training and thereby increasing overall yield.
BayesCall [19] and Seraphim [21] implement more complex, fully parametric models. In addition to cross-talk, phasing and pre-phasing, they also explicitly model the signal decay. Furthermore Seraphim accounts for differences in the PCR amplification step for each read [21] and BayesCall adds parameters that model other residual effects which are propagated from one cycle to the next [19]. For BayesCall [19] the complete model is cycle dependent which dramatically increases the number of parameters. The parameters are estimated using an expectation maximization procedure. As in the clustering approach used in Rolexa [17], expectation maximization does not rely on supervised learning and therefore eliminates the need for training data. In both papers the base with the maximum posterior probability is called. Since the probabilities of the other bases can be readily computed, it is straightforward to report meaningful quality metrics. A faster version of BayesCall is naiveBayesCall [22]. naiveBayesCall makes use of the same model as BayesCall and also uses the same algorithm for parameter estimation. During base-calling approximate algorithms improve speed by orders of magnitude while only slightly sacrificing accuracy [22].
A completely different approach was taken in Ibis [18]. Rather than modeling every potential source of errors, multi class SVMs are applied directly to the raw intensity signal. Using simulation, it was inferred that under a simple model of phasing, pre-phasing and T accumulation, most information is contained in the intensities of the previous, current and next cycle. Hence the SVMs use the intensity values of the current cycle, its predecessor and successor as input. In order to train the cycle dependent SVMs a known sequence has to be included or when resequencing a genome it is also possible to use the reference genome for training.
The intensity data needs to be extracted from the raw images before any of the above can be used. This image processing step is usually performed using Bustard's Firecrest module. BING [23] and Swift [15] are alternative implementations of the complete data processing pipeline. Both image processing algorithms differ in many mechanistic details from Bustard. With BING, one has the option of base-calling each pixel in the image tile independently rather than first identifying clusters of the same templates. During base-calling, both implementations rely on serial corrections, similar to Bustard, and do not implement elaborate statistical procedures. As with Bustard, Swift gives access to the post-image processing data and hence could also be used in conjunction with one of the base-callers described above.
A summary of all implementations and the respective statistical methodologies is shown in Table 1. On a practical note, all base-callers reviewed here support the longer reads introduced with Illumina’s Genome Analyzer II.
Table 1:
A summary of the available applications used for base-calling on the Illumina platform
| Name | Statistical approach | Biases explicitly corrected | Training data required | Quality score | Practical notes | References |
|---|---|---|---|---|---|---|
| Bustard | Parametric Model | Σ, ϕ, δ | No | Phred | Not freely retrievable | |
| Alta-Cyclic | Mixed Parametric and SVM | Σ, ϕ, δ | Yes | Phred | No longer maintained; requires a Sun Grid Engine cluster environment | [16] |
| Rolexa | Parametric Model | Σ, ϕ, ω | No | IUPAC | No longer maintained | [17] |
| Swift | Parametric Model | Σ, ϕ, µ | No | Phred | No longer maintained | [15] |
| BayesCall/ naiveBayesCall | Parametric Model | Σ, ϕ, δ | No | Phred | [19, 22] | |
| Seraphim | Parametric Model | Σ, ϕ, δ | No | Phred | We did not succeed installing it | [21] |
| Ibis | Fully empirical SVM | (n/a) | Yes | Phred | [18] | |
| BING | Parametric Model | Σ, ϕ | No | None | Not freely retrievable; requires own image processing as input | [23] |
We give a short description of the statistical approach used by each application. Next, the biases explicitly modeled and corrected by the application are reported (see Figure 1 for details). Alta-cyclic and Ibis rely on supervised learning and require training data. Finally uncertainty measurements or sequencing quality is either reported as Phred scores or using IUPAC codes. For details, please refer to the main text.
PERFORMANCE COMPARISON
The rapid and at times simultaneous emergence of new base-calling approaches makes it difficult to assess their relative performance. Though comparative studies reported by authors of individual packages must be interpreted with caution, they can provide us with some insights. Kircher et al. [18] reported that Ibis outperforms Alta-Cyclic and Rolexa which in turn are more accurate than Bustard. However, note that Rolexa was forced to not make use of IUPAC codes in this comparison. In the report of Kao et al. [19], BayesCall was shown to outperform Alta-Cyclic. In terms of the Phred quality scores, both Ibis and BayesCall have been shown to report more accurate scores than Alta-Cyclic, which itself improves upon Bustard [18, 19]. With respect to the running time, Kircher et al. [18] reported the following timings. Bustard was clearly the fastest implementation tested, requiring 50 min on a single processor for parameter estimation and base-calling of the complete control lane of a 51 cycle data set. Ibis required 3 times, Rolexa 21 times and Alta-Cyclic 73 times more computational resources than Bustard. Alta-Cyclic was run on a cluster, reducing the effective time for base-calling. For BayesCall and Seraphim, no direct comparison is available. From the timings reported in the respective publication, it appears that BayesCall requires roughly 20 h for parameter estimation and 6 h to call 1 million bases for a 76 cycle data set. Thus, without parallel computing, it takes several days to process a single lane. However as discussed above, a significantly faster version of BayesCall, called naiveBayesCall [22], was recently published. As for Seraphim, the reported time for base-calling and mapping reads on the control lane is under 2 h on a 15 node cluster, including parameter estimation.
We sought to compare all base callers reviewed here on the same data set and hardware. However, this proved very difficult, as many of the packages are either not freely retrievable, no longer maintained or fraught with practical problems (Table 1). Despite considerable efforts, we did not succeed in obtaining, installing or running Bing, Seraphim and Swift. The other base callers could be assessed using a data set of 286 847 reads of length 51 from the phiX174 control lane, obtained using V1 chemistry (Figure 2A). With the exception of Rolexa, all base callers show a clear improvement upon Bustard. Ibis performs best, closely followed by naiveBayesCall and Alta-cyclic.
Figure 2:

Comparison of Base-Callers for the Illumina Platform. (A) Error rate of base callers for Illumina Platform (FC-104-10xx). The test data consists of 286 847 reads (length 51, chemistry FC-104-10xx) from the phiX174 control lane, provided by Martin Kirchner, who also provided the results for Bustard 1.95 and Alta-cyclic. The method used here is identical to that of [18]. (B) Time required on a 2 GHz AMD Opteron with eight cores for training (blue) and base-calling (green). For Ibis, training was performed on a set of 1.15 million reads disjoint from the test set. For (naive)BayesCall, an unsupervised learning method, parameter estimation was performed on the test data set itself (the base caller randomly selects 250 reads for this purpose). For Rolexa, for which there was no clear separation between a parameter estimation phase and a base-calling phase, all time was attributed to base-calling. Note that training only needs to be performed once, but base-calling on a full lane involves about 40 times more reads than in our test set. (C) Phred score accuracy. Deviation from the 45-degree line indicate either underestimation (curve above the line) or overestimation (curve below the line) of the quality of the base called. We only report data for quality scores with at least 20 000 bases.
Regarding computational cost, we measured separately training/parameter estimation time and base-calling time for the four packages that we could run on our benchmark linux computer: Ibis, BayesCall, naiveBayesCall and Rolexa (Figure 2B). For training time, Ibis was an order of magnitude faster than (naive)BayesCall, while Rolexa did not require any distinct parameter estimation phase at all. But in practice, most time is spent calling bases. With this respect, Ibis is by far the fastest of the four packages. The efficiency improvements afforded by naiveBayesCall over its predecessor are very significant, and make it usable in practice. Remarkably, the two most accurate base callers also happen to be the fastest ones.
The quality score reported differs among the software packages: Phred scores are reported by Ibis; an Illumina specific encoding is used by Bustard and AltaCyclic, and the error probability is returned by BayesCall and naiveBayesCall. In order to compare these different measures we converted all of them to Phred scores. We then compare the reported Phred scores with the Phred scores computed from the observed error rate of bases with the respective Phred score (Figure 2C). In this comparison, Bustard significantly deviates from the optimal line, AltaCyclic shows overestimation of the Quality for high quality base-calls, and BayesCall and naiveBayesCall consistently overestimate the quality of their calls, except for very low quality base-calls. We note that this effect is less pronounced for naiveBayesCall. Due to the smoothness of the curve obtained from (naive)BayesCall, it might be possible to find a simple and effective correction for the respective quality scores. Overall, the base caller closest to the optimal line is Ibis.
For the practical use of the base-callers, their performance on more recent chemistry is of high relevance. We assessed Ibis and naiveBayesCall, which have the lowest error rates on V1 (FC-104-100x) chemistry, on a V4 (FC-103-300x) chemistry data set with 217 904 reads of length 81. For the V4 chemistry, we obtained an error rate of 1.02% for naiveBayesCall, while Ibis achieves an error rate as low as 0.97%. The absolute error rate is markedly lower than with the older chemistry, a remarkable achievement given that the read length is ~60% longer. In line with the results obtained on V1 chemistry, Ibis also outperforms naiveBayesCall in terms of the reported quality scores on the V4 chemistry (data not shown).
BASE-CALLING FOR ROCHE 454 LIFE SCIENCES
The Roche 454 platform starts by constructing an adaptor flanked single strand DNA sequence library. The sequence fragments are bound to beads and amplified on the beads by emulsion PCR in order to increase the downstream signal intensity. Ideally, during this process a single template is attached to each bead leading to uniform clusters on each bead. The beads are then deposited onto an array of picoliterscale wells [24] such that each well contains a single bead. After these preparatory steps, the actual sequencing begins using the pyrosequencing method [25]. In every sequencing cycle, a single species of nucleotides is introduced. In wells where the nucleotides are incorporated, this results in the release of pyrophosphate which eventually leads to a burst of light. The light is detected using a CCD sensor and software detects wells containing template DNA. This step includes image analysis and base-calling. For a more detailed description we refer to the original paper [24].
A number of sources of errors have been described [9]. Firstly, there is a risk of mixed clusters, caused by the binding of different DNA fragments to a single bead. In such a case, it will be impossible to detect a clear signal and the acquired data from the wells containing such beads has to be excluded. Secondly, in every cycle there is a slight chance of incomplete synthesis of the complementary DNA strand which leads to phasing. Similarly if the reagent of a previous cycle was not perfectly removed, it is possible that multiple different bases are incorporated, resulting in pre-phasing [24]. The main source of errors is, however, due to thresholding. Thresholds are needed to determine whether a base was incorporated or not. The thresholds necessary to determine the lenghts of homopolymers are even more delicate. Homopolymers are consecutive runs of the same base. Since all bases of a homopolymer are included in a single cycle, the length of the homopolymer has to be inferred from the signal intensity. Incorrect prediction of homopolymer lengths leads to insertions and deletions which are by far the most frequent errors associated with the pyrosequencing technology [26].
In the original 454 paper, wells containing templates are identified by detecting the key sequence ‘TCAG’ at the start of the sequence [24]. The number of incorporated bases is determined from the intensity of the emitted light. It is shown that the intensity is linear with the lengths of the homopolymer, thus allowing for easy classification. A prior on the homopolymer lengths of 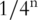 is used. In order to compensate for an incomplete extension rate of 0.1–0.3% and a carry forward rate (pre-phasing) of 1–2% a detailed physical model is proposed. If, frequent ambiguous intensity levels are detected for a given read, that read is filtered out as a low quality read. This allows to exclude wells containing multiple templates. Finally, a Phred like quality score [20] is assigned to every called base. This quality score corresponds to the log-probability that the base was not an overcall, that is, the predicted homopolymer length was not too long.
is used. In order to compensate for an incomplete extension rate of 0.1–0.3% and a carry forward rate (pre-phasing) of 1–2% a detailed physical model is proposed. If, frequent ambiguous intensity levels are detected for a given read, that read is filtered out as a low quality read. This allows to exclude wells containing multiple templates. Finally, a Phred like quality score [20] is assigned to every called base. This quality score corresponds to the log-probability that the base was not an overcall, that is, the predicted homopolymer length was not too long.
In Pyrobayes, Quinlan et al. [26] proposed to improve the above procedure by adapting an empirical prior on the homopolymer lengths and by using a classifier based on an empirical measure of the signal intensity. This challenges the validity of a simple linear classifier. As they illustrated in their report, using this more empirical approach does not reduce the total error rate. However, Pyrobayes clearly outperforms the native base-caller in substitution error rate and in the accuracy of the Phred quality scores. Thus, they argued that Pyrobayes is superior in the context of single nucleotide polymorphism (SNP) prediction.
DISCUSSION AND OUTLOOK
The advent of next-generation sequencing platforms during the past few years has lead to a recent burst in base-calling software. We have reviewed base-calling methods for Illumina and Roche 454, the two leading platforms, with most of the efforts concentrated on the former.
The various base-callers differ in the statistical methodologies used to infer the correct base and in the way they report uncertainty. At this point, it remains to be seen which approach will ultimately achieve highest accuracy: a mechanistic model such as in BayesCall or Seraphim, a strictly empirical approach such as in Ibis, or some intermediate solution such as in Alta-Cyclic. Currently, the two most accurate base-callers, Ibis and naiveBayesCall, have diametrically different methodological approaches and yet achieve close accuracy. As we suggested above, models that avoid supervised learning have the advantage of a potentially increased yield in the case of de novo sequencing since they do not require resequencing of a known reference sequence for training. Furthermore, the parameters of mechanistic models have a clear interpretation and can give valuable insights to sources of noise in the underlying technology. For instance, estimates of the pre-phasing and phasing rate can be obtained [19]. This information could drive future improvements of the technology. On the other hand, the SVM used by Ibis [18] are advantageous when adapting the applications to future releases of the Illumina platform or an entirely different platform because only very few assumptions about the type of biases are made. These assumptions are more likely to hold true for different technologies than the numerous assumptions made by mechanistic models.
When reporting uncertainty for bases called, most base-calling implementations relies on Phred scores rather than IUPAC encoding used by Rolexa [17]. In principle, reporting the probabilities of all four bases would provide downstream analyses with the complete information derived by the base-calling algorithms. Whether summarizing this information using Phred scores or using IUPAC codes is preferable cannot be decided independently from the subsequent analysis tools. However, Phred scores, as opposed to IUPAC codes, are more widely used and hence there is a wealth of tools which can handle them [21].
The approaches also differ significantly in computational resources required, ranging from Bustard, which is reported to be the fastest [18], to Alta-Cyclic and BayesCall, which requires orders of magnitudes more computational resources. On the other hand Ibis requires only about three times more resources than Bustard while also being very competitive in terms of accuracy. In this case, the gained accuracy may well justify the increased computational costs.
It is anticipated that in the future next-generation sequencing technologies will continue to improve rapidly. By improving accuracy, read length and quality score, base-callers have the potential to reduce costs, increase yield and simplify downstream analysis. Designing and updating near optimal base-callers not only for Illumina and Roche 454 but also for other next-generation platforms will continue to be an important research task. A first third party base-caller for the SOLiD system [27] has recently been developed and, as for Illumina and Roche 454, significant improvements are reported. Further research in this area can contribute toward closing the gap between the time required for sequence data generation and analysis [28].
Key Points.
Base-calling, the inference of DNA sequences from physical signals, is a crucial step of the sequencing process.
Improving the accuracy of base-calling decreases coverage requirements and costs, and is therefore of high interest.
For both Illumina and Roche 454, the leading next-generation sequencing platforms, several alternatives to the vendor base-caller have been recently proposed, which correct various types of systematic errors.
Some base-callers explicitly model the biases, while other rely on reference sets to train general purpose classifiers; as we discuss in the main text, both approaches have their pros and cons.
Acknowledgements
We thank Martin Kirchner who provided us with the two data sets we used in our comparison, as well as results from AltaCyclic and Bustard1.9.5. We also acknowledge helpful correspondence with several other authors of base callers. In particular, we thank Jacques Rougemont for comments on the article, and for providing us with feedback on Rolexa. We had helpful correspondence with Tom Skelly on Swift, Hector Corrada Bravo on Seraphim, Jeffrey Kriseman on BING and Yaniv Erlich on AltaCyclic. Finally we would like to thank four anonymous reviewers for their comments on the article. We thank Martin Bishop and Alison Bentley for their editorial support. This article started as assignment for the graduate course ‘Reviews in Computational Biology’ (263-5151-00L) at ETH Zurich.
Biographies
Christian Ledergerber is a PhD student in the CBRG group, computer science, ETH Zurich. Previously, he spent a year in the Pfister Lab on computer vision at Harvard University. He is interested in computational biology, statistics, theoretical computer science and climbing.
Christophe Dessimoz is a post-doc and lecturer in the CBRG group, computer science, ETH Zurich. He strives to understand the forces that shape genes, genomes and species, using computational and statistical methods.
References
- 1.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Procd Natl Acad Sci USA. 1977;74(12):5463–7. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 3.National Institute of Health. THE $1000 GENOME. http://grants.nih.gov/grants/guide/rfa-files/RFA-HG-04-003.html, http://www.genome.gov/10000368 (28 November 2010, date last accessed) [Google Scholar]
- 4.International HapMap Consortium. The international HapMap project. Nature. 2003;426(6968):789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 5.Nayanah S. 1000 genomes project. Nat Biotech. 2008;26(3):256. doi: 10.1038/nbt0308-256b. [DOI] [PubMed] [Google Scholar]
- 6.Begun DJ, Holloway AK, Stevens K, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biology. 2007;5(11):e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigel D, Mott R. The 1001 genomes project for arabidopsis thaliana. Genome Biol. 2009;10(5):107. doi: 10.1186/gb-2009-10-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan EY. Advances in sequencing technology. Mutat Res. 2005;573(1–2):13–40. doi: 10.1016/j.mrfmmm.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Huse SM, Huber JA, Morrison HG, et al. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8(7):R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9(9):868–77. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta S, Datta S, Kim S, et al. Statistical analyses of next generation sequence data: a partial overview. J Proteomics Bioinform. 2010;3:183–90. doi: 10.4172/jpb.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedurco M, Romieu A, Williams S, et al. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 2006;34(3):e22. doi: 10.1093/nar/gnj023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turcatti G, Romieu A, Fedurco M, et al. A new class of cleavable fluorescent nucleotides: synthesis and optimization as reversible terminators for DNA sequencing by synthesis. Nucleic Acids Res. 2008;36(4):e25. doi: 10.1093/nar/gkn021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whiteford N, Skelly T, Curtis C, et al. Swift: primary data analysis for the Illumina Solexa sequencing platform. Bioinformatics. 2009;25(17):2194–9. doi: 10.1093/bioinformatics/btp383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erlich Y, Mitra PP, Delabastide M, et al. Alta-Cyclic: a self-optimizing base caller for next-generation sequencing. Nature Methods. 2008;5(8):679–82. doi: 10.1038/nmeth.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rougemont J, Amzallag A, Iseli C, et al. Probabilistic base calling of solexa sequencing data. BMC Bioinformatics. 2008;9:431. doi: 10.1186/1471-2105-9-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 2009;10(8):R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao W-C, Stevens K, Song YS. BayesCall: a model-based base-calling algorithm for high-throughput short-read sequencing. Genome Res. 2009;19(10):1884–95. doi: 10.1101/gr.095299.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. error probabilities. Genome Res. 1998;8(3):186–94. [PubMed] [Google Scholar]
- 21.Bravo HC, Irizarry RA. Model-based quality assessment and base-calling for second-generation sequencing data. Biometrics. 2009;66(3):665–74. doi: 10.1111/j.1541-0420.2009.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao W-C, Song YS. Research in Computational Molecular Biology, volume 6044/2010 of Lecture Notes in Computer Science. Springer Berlin/Heidelberg: naiveBayesCall: an efficient model-based base-calling algorithm for high-throughput sequencing; pp. 233–47. May 2010. [Google Scholar]
- 23.Kriseman J, Busick C, Szelinger S, et al. Bing: biomedical informatics pipeline for next generation sequencing. J Biomed Inform. 2009;43(3):428–43. doi: 10.1016/j.jbi.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11(1):3–11. doi: 10.1101/gr.11.1.3. [DOI] [PubMed] [Google Scholar]
- 26.Quinlan AR, Stewart DA, Strömberg MP, et al. Pyrobayes: an improved base caller for SNP discovery in pyrosequences. Nature Methods. 2008;5(2):179–81. doi: 10.1038/nmeth.1172. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, Irizarry RA, Bravo HC. Intensity normalization improves color calling in SOLiD sequencing. Nature Methods. 2010;7(5):336–7. doi: 10.1038/nmeth0510-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mcpherson JD. Next-generation gap. Nature Methods. 2009;6(11 Suppl.):S2–5. doi: 10.1038/nmeth.f.268. [DOI] [PubMed] [Google Scholar]