

Abstract
Phylogenomic databases provide orthology predictions for species with fully sequenced genomes. Although the goal seems well-defined, the content of these databases differs greatly. Seven ortholog databases (Ensembl Compara, eggNOG, HOGENOM, InParanoid, OMA, OrthoDB, Panther) were compared on the basis of reference trees. For three well-conserved protein families, we observed a generally high specificity of orthology assignments for these databases. We show that differences in the completeness of predicted gene relationships and in the phylogenetic information are, for the great majority, not due to the methods used, but to differences in the underlying database concepts. According to our metrics, none of the databases provides a fully correct and comprehensive protein classification. Our results provide a framework for meaningful and systematic comparisons of phylogenomic databases. In the future, a sustainable set of ‘Gold standard’ phylogenetic trees could provide a robust method for phylogenomic databases to assess their current quality status, measure changes following new database releases and diagnose improvements subsequent to an upgrade of the analysis procedure.
Keywords: conceptual comparison, phylogenomic databases, quality assessment, reference gene trees
INTRODUCTION
Phylogenomic databases provide predictions of evolutionary relationships, mostly for protein-coding genes of species with fully sequenced genomes. Such information is essential for comparative genomics as well as for the study of the divergence of individual gene families. The most common usage is function prediction for yet uncharacterized proteins. This approach is based on the commonly accepted assumption that orthologs—genes derived by speciation—are more likely to share a common function, in contrast to paralogs—genes derived by gene duplication—which are expected to diverge functionally over time [1, 2].
Several studies conducted in recent years have dealt with the comparison and quality assessment of orthology predictions [3–8]. However, direct comparisons regarding the outcomes of these comparative analyses are not possible, as each study was based on a unique set of ortholog databases. The majority of these studies use consistency of functional annotation within ortholog groups as a measure for accurate orthology predictions. The application of such function-based measures is disputable for various reasons: (i) only few proteins have been characterized in depth; (ii) proteins can perform more than one function; (iii) proteins are frequently part of complexes and thus participate in different functions; (iv) paralogs can accomplish the same function; and (v) orthologs can diverge functionally [2]. For these reasons, most authors regret the lack of ‘Gold Standard’ phylogenetic trees for this quality assessment. Yet, the form this standard should take remains elusive.
We present a comparison of databases with an emphasis on how different underlying concepts constrain the results obtained. These differences are illustrated by means of three gene histories, for which reliable reference trees have been reconstructed. Finally, a score-based quantitative comparison is proposed.
Phylogenomic databases under comparison
A large number of valuable ortholog databases are made available to the scientific community. Criteria for the selection of the databases included the taxonomic range and sampling density, the applied methodology and the database concept, especially whether or not the concept is hierarchical. A special interest was furthermore the information provided by ortholog databases to which UniProtKB cross-links. Seven phylogenomic databases are compared in this study, namely Ensembl Compara (http://www.ensembl.org/) [9], eggNOG (http://eggnog.embl.de/) [10], HOGENOM (http://pbil.univ-lyon1.fr/databases/hogenom/) [11], InParanoid (http://inparanoid.sbc.su.se/) [12], OMA (http://omabrowser.org/) [13], OrthoDB (http://cegg.unige.ch/orthodb) [14] and Panther (http://www.pantherdb.org/) [15]. Each of these databases represents a unique specialization (Table 1). Compara, HOGENOM and Panther reconstruct phylogenetic trees while the other databases provide ortholog groups. The number of analyzed proteomes varies between 23 and 1000, and the taxonomic range spans any cellular organism on the one hand or a single phylum on the other. HOGENOM, for instance, is devoted to microbial organisms (bacteria, archaea and unicellular eukaryotes) with completely sequenced genomes, and does not intend to be exhaustive for multicellular organisms. In contrast, Compara focuses on chordate genomes and proteomes, plus a few invertebrates and fungi as outgroups. Panther analyzes about the same number of proteomes as Compara, but these are from selected representatives of all three major kingdoms. A hierarchical protein classification is provided by eggNOG, OrthoDB and, most recently, OMA. eggNOG computes ortholog groups for up to six major taxonomic levels, while OMA does so for all taxonomic nodes. Both databases cover a large number of proteomes from all kingdoms. The most fine-grained hierarchical classification is given by OrthoDB, which seeks to identify all descendants of the common ancestral gene at each speciation node for vertebrates, arthropods, fungi and animal phylogenies. Yet another grouping strategy comprises non-hierarchical clusters of orthologs, including those which are the result of pairwise species comparisons; the most well-known representative is InParanoid. Finally, there are the pure orthologous groups of OMA, which only include genes that are orthologous to each other and do not involve pairs of inparalogs. In addition to providing ortholog groups, eggNOG and OMA use orthology assignments to construct a species tree. Noteworthy, Panther classifies families into subfamilies which are thought to capture groups that are similar in sequence or equivalent in function—but are not designed to define orthologous groups.
Table 1:
Comparison of selected phylogenomic databases
| Database | Nb. species and taxonomic range | Nb databases inquired for input data | Homology detection and clustering | Multiple sequence alignment and tree-building | Grouping strategy | Goals include | Updates per year (estimations) |
|---|---|---|---|---|---|---|---|
| Compara (Ensembl 58) | 47 chordates and outgroups | 1 | BlastP Hcluster_sg | M-Coffee, TreeBeSt (NJtree, NJ and ML, species tree) | (i) Phylogenetic trees, (ii) Ortholog groups from species pairs | Gene phylogeny | 6 |
| eggNOG (release 2) | 630 species | 4 | Blast RBH triangular linkage clustering | Muscle, MAFFT and filters PhyML | Hierarchical groups based on up to six taxonomic levels | Comparative genomics, species phylogeny | 1 |
| HOGENOM (release 5) | 964 species | 12 | BlastP2 (low complexity filters) single-linkage clustering ≥50% similarity, ≥80% overlap | Muscle, Gblocks BioNJ, PhyML, FASTTREE and TREEFINDER | Phylogenetic trees | Gene phylogeny | 1 |
| InParanoid (release 7) | 99 eukaryotes and E. coli | 22 | BLAST (compositional adjustment, SEG) ≥50% overlap | Kalign NJ (100 replicates) | Ortholog groups from species pairs | Comparison of species pairs | 1 |
| OMA (May-2010) | 1000 species | 12 | Smith–Waterman with minimum length requirement | – | (i) Pure ortholog groups, (ii) Ortholog groups from species pairs and (iii) Hierarchical groups based on taxonomic nodes | Comparative genomics, phyletic profiles, species phylogeny | 2 |
| OrthoDB (release 3) | 40 vertebrates 23 arthropods 32 fungi | 8 | Smith–Waterman, RBH, triangular linkage clustering | – | Hierarchical groups based on a species phylogeny | Comparative genomics, species phylogeny | 1 |
| Panther (release 7) | 48 species | 13 | BlastP, HSP, single-linkage clusters (SLC) | MAFFT GIGA | (i) Phylogenetic trees and (ii) Ortholog groups from species pairs | Gene phylogeny, Function prediction | 1 |
In summary, the phylogenomic databases investigated here differ substantially in goal, scope, methodology and output. And yet, all their results provide estimations of evolutionary relationships among genes and species. In order to compare them, we must first characterize the information they provide.
METHODS
Sequence information
Representative members of the Popeye domain family, the NOX family NADPH oxidases and the eukaryotic V-type ATP synthase beta subunit subfamily were obtained from the UniProtKB/Swiss-Prot release 57.13 and UniProtKB releases 15.14 and 2010_06 according to their annotation. Further homologs were predicted by similarity searches with BlastP on the Expasy Proteomics Server [16]. The preliminary datasets were complemented by data from UniProtKB/TrEMBL, Ensembl release 57, BeeBase (http://genomes.arc.georgetown.edu/drupal/beebase/) and WormBase (http://www.wormbase.org/). A list of sequence identifiers, species names and database identifiers is given in Supplementary Table S1; sequences are available in the Supplementary Datasets S1–S3.
Reference trees
The methods used are summarized below, and a detailed description of each analysis is provided along with the individual phylogenetic trees in Supplementary Figures S4–S6. The sequence analysis was performed on local computers of the Swiss Institute of Bioinformatics, at phylogeny.fr [17] as well as at the high performance computing center Vital-IT (http://www.vital-it.ch/). The sequences of the three data sets were aligned using MUSCLE [19]. Sequences with gaps within conserved regions were removed and short isoforms were replaced by appropriate ones if available. Gene models were corrected if possible, or otherwise excluded. A multiple sequence alignment (MSA) was constructed with ProbCons [19], and data models were built through gap removal, Gblocks (stringent and less stringent parameter settings), or manual selection of conserved regions. Phylogenies were inferred using maximum likelihood (ML), Bayesian Markov-Chain Monte Carlo (MCMC) and neighbor joining (NJ). ML-trees were calculated with PhyML [20] using the amino acid replacement models Jones, Taylor and Thornton (JTT) [21] or Whelan and Goldman (WAG) [22], accounting for rate heterogeneity across sites using an eight-category discrete gamma distribution and estimating the shape parameter, and in some analyses the number of invariant sites from the data. Branch support values were calculated with the approximate Likelihood Ratio Test (aLRT) based on a Shimodaira–Hasegawa-like or Chi2-based procedure [23]. Bayesian analyses were performed using MrBayes 3.1.2 [24]. Two independent runs of four chains and one million generations were run using fixed models that performed best when applying PhyML. To test the consistency and robustness of tree topologies, consensus trees were generated from 1000 bootstrap replicates using the BioNJ algorithm [25] and the JTT model of amino acid substitution. Finally, a consensus tree was constructed considering all the analysis results and species trees as used by TreeBeST (http://treesoft.sourceforge.net) for chordates, and reconstructed by OrthoDB for arthropods and fungi. The user-defined tree was tested against the ML tree and alternative models using TREE-PUZZLE [26]. It is noted that even though sequence names used in the reference trees are in the format of UniProtKB/Swiss-Prot entry names, the identifiers are mostly not valid UniProtKB entry names.
Mapping of data from ortholog databases to the reference gene trees
Data was mapped to ortholog groups of the following databases: Compara (Ensembl 56), eggNOG (2.0), HOGENOM (05), InParanoid (7.0), OMA (October 2009), OrthoDB (3); Panther (7.0 beta). Whenever possible, Swiss-Prot cross-references to the phylogenomic databases were used to identify the relevant ortholog groups. Sequence mapping can be hindered for two reasons: (i) UniProtKB frequently uses special taxonomic identifiers for bacterial strains, if the complete genome has been sequenced; and (ii) over 4% of the sequence data is updated during the annotation process, which can prevent the mapping of sequences. We verified such cases manually in order to maximize data matching, taking into account available identifiers for genes, transcripts, and proteins.
For the examples shown here, the comparison between the reference tree and the databases was performed using the browser or the data sets. Here, the purpose was to obtain all relevant gene identifiers and to identify false positive hits for the selected species, which in some cases could not be obtained automatically based simply on the gene identifiers. Sequences of possible false positive hits were analyzed using MSA and tree reconstruction approaches. Consequently, a few more orthologs were identified and added to the reference dataset and tree. Blast services from phylogenomic databases were employed to search for genes that could not be mapped according to gene identifiers. For InParanoid, we obtained the relevant gene identifiers from the InParanoid browser, and extracted the corresponding ortholog information from the database.
Gene relationships
For the quantitative analysis, we determined the number of predictions for three types of pairwise gene relationships: orthology, orthology/paralogy and ‘extended’ gene relationships. The latter take into account the number of gene duplications since the last common node of a gene pair. In this manner, a higher resolution for the topological correctness at internal nodes was obtained. We define the ‘extended’ relationships (x, y)-orthology and (x, y)-paralogy, where x and y specify how many duplications took place on the evolutionary path from the point where the two genes in question began diverging. For instance, a pair of orthologs with a single lineage-specific duplication resulting in genes A1, A2 and B are (1,0)-orthologs. Note that this concept is slightly different from the commonly used n:m orthology concept (where n, m is typically ‘1’ or ‘many’): n and m refer to the number of respective co-orthologs, while x and y in the extended gene relationships refer to the number of duplications on the respective paths of the relevant gene pairs since their common ancestry.
Metric and quantitative analysis
Terms used in the context of scoring are defined as follows: ‘True positives’ are predicted gene relationships that coincide with those of the reference model. ‘False negatives’ are gene relationships failed to be predicted according to the reference model and the species list of a given phylogenomic database. The lack of predicted gene relationships can arise from a number of causes. For example, the gene may not be part of the input dataset, the gene model may be incorrect, the gene product may be an isoform, or an ortholog being wrongly predicted as paralog. As the content of databases is benchmarked here rather than the orthology prediction methods, we do not differentiate between these causes. The selection of up-to-date and complete input data is seen as one of the important tasks of phylogenomic databases. False positives are predicted gene relationships which do not correspond to those inferred from the reference tree and which are either outparalogs or not homologs at all. ‘True negatives’ are gene relationships, which are correctly predicted not to be the type of gene relationship in question. ‘Expected OTUs’ (Operational Taxonomic Units) are all relevant genes according to the reference tree and the species list of a database. ‘Mapped OTUs’ are all relevant genes according to the reference tree and the species list of a database. ‘Supplementary gene list’ specifies genes that have not been used in the analysis of the reference tree, e.g. due to incomplete or erroneous gene models. This list is thought to be helpful when automating the benchmarking procedure. Currently, these genes are not considered when calculating scores. However, it is conceivable to annotate some gene relationships based on gene synteny or analysis of small datasets of closely related genes.
A list of all possible gene relationships with annotated orthology/paralogy was created for each reference tree, which was then used as a template to construct database-specific lists by removing genes from non-relevant species. The expected number of orthologous and paralogous relationships was obtained from these lists. The number of true positives, false positives and false negatives was determined from the database results. For pure orthologous groups, only the number of true positive and false positive ortholog predictions could be determined, as no paralogs are specified in this concept. For pairwise groups, the status of each orthologous and paralogous prediction was determined from the groups. For hierarchical groups, we calculated precision and sensitivity for the most specific groups and for trees that were reconstructed according to the hierarchy. Reconciled trees were benchmarked to the hierarchical reference groups.
Extended orthology/paralogy relationships were obtained directly from the reconciled trees. For hierarchical groups, the unresolved trees were reconstructed according to Figure 1. Specified gene relationships were evaluated on the assumption that branches with non-overlapping taxonomic ranges (i.e. all different species) are orthologs and branches with overlapping taxonomic ranges (i.e. some common species) are paralogs. Unclear gene relationships at multifurcating nodes were set to ‘undefined’. For pairwise groups, the information was considered specified if there was no more than one gene duplication since the last common ancestor for each lineage; in all other cases, the gene relationships were considered ‘undefined’. In case of OMA pairwise groups, the branch of the reference gene was not considered. Extended gene relationships are not calculated from pure orthologous groups that conceptually contain no information on gene duplications. For plain trees, Robinson–Foulds distances were calculated.
Figure 1:

Concepts of selected phylogenomic databases. Rows (from top to bottom) indicate the different database concepts, the structure of ortholog groups, the completeness of predicted gene relationships and the implied tree structures. Latter visualizes the captured phylogenetic information.
Precision and sensitivity were calculated for the three types of gene relationships. The Positive Predictive Value (PPV) is calculated as: 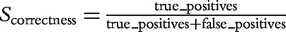 . This score reflects the correctness of the predicted gene relationships, regardless of the size of an ortholog group, the number of family members or the existence of hierarchical levels. The True Positive Rate (TPR) complements the PPV by taking into account the number of false negative hits. TPR is calculated as
. This score reflects the correctness of the predicted gene relationships, regardless of the size of an ortholog group, the number of family members or the existence of hierarchical levels. The True Positive Rate (TPR) complements the PPV by taking into account the number of false negative hits. TPR is calculated as 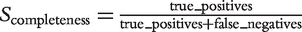 .
.
All scores were normalized between 0 and 1, with higher values indicating a better fit to the reference tree. The distinction between the quality descriptors PPV and TPR is relevant in systems with a sensitivity-specificity trade-off, as it was observed in earlier benchmarking studies. Consequently, these ratios have not been combined into a single quality score.
RESULTS AND DISCUSSION
Conceptual comparison of phylogenomic databases
There are five main conceptual frameworks which emerge from the databases we compared: (i) pure orthologous groups; (ii) orthologs of species pairs; (iii) hierarchical ortholog groups; (iv) reconciled trees; and (v) trees with no annotation. In Figure 1, the different grouping strategies are presented in the form of annotated trees with resolved or unresolved nodes according to the information they capture.
‘Pure orthologous groups’ consist of genes that all share orthologous relationships [16]. Hence, only a part of all possible orthologous—but no paralogous—gene relationships are captured. Accordingly, the phylogenetic information of such groups corresponds to unresolved trees with all nodes presenting speciation events. In general, pure orthologous groups are suitable when precision is of higher importance than sensitivity. OMA has chosen this gene classification as a basis for the reconstruction of the species tree.
‘Ortholog clusters of species pairs’ include orthologs or co-orthologs from only two species per cluster, resulting in a large number of small groups. Expressed in terms of a tree, the root node is always a speciation event and all other internal nodes present gene duplications, which multifurcate if more than two gene copies exist in a species. In principle, this approach can comprise all orthologous gene pairs, but it captures only species-specific in paralogs; paralogs between different species are not captured at all. Orthology assignment for species pairs is the well-established strategy of InParanoid, but also provided by Compara, Panther and OMA. The concept for OMA ‘pairwise’ varies in that orthologous genes are given for each reference gene. Thus, lineage-specific gene duplications are provided for only one of the two branches.
‘Hierarchical ortholog groups’, which are defined for particular taxonomic levels, consist of sequences that descend from a single ancestor in the taxonomic range in question. eggNOG provides hierarchical groups with respect to major taxonomic levels, while OrthoDB provides hierarchical groups with respect to any split in their species tree. In the latter, higher resolution between the more closely related species can be achieved. If all nodes are resolved, the hierarchical groups collectively imply the gene tree topology. Even if not explicitly indicated, a split of groups along a lineage indicates a gene duplication event. Evolutionary events are not defined at internal nodes except for the root nodes of eggNOG and OrthoDB, which are expected to be speciation events. Gene relationships can be inferred based on the assumption that groups with one gene per species are 1:1 orthologs, and groups with overlapping taxonomic ranges are paralogs [28]. Consequently, for gene families with multiple gene duplications, gene relationships are often only specified for the genes of more closely related species. In this case, all gene relationships within and between groups can be specified, even when speciation nodes are unresolved. Panther provides two hierarchical levels, namely families and mutually exclusive groups of subfamilies. As subfamilies are not intended to reflect gene phylogeny, this concept is not considered in our study.
The most fine-grained classification is the gene tree. Unlabeled gene trees possess no orthology assignment per se and need further interpretation for the prediction of gene relationships. ‘Reconciled gene trees’ include details on evolutionary events, generally assigning speciation or gene duplication at each internal node. Hence, all possible orthologous and paralogous gene relationships can be directly derived from a resolved tree. Annotated gene trees are constructed by Compara, HOVERGEN and Panther; plain gene trees by HOGENOM. eggNOG and InParanoid also provide gene trees but they are not used to infer orthology.
Thus, concepts differ in the extent to which they capture phylogenetic information, which may therefore only be partial, even for perfect results. In fact, only two of the discussed strategies—reconciled trees and hierarchical ortholog groups—have the potential to characterize all orthology/paralogy relationships of a homologous group. This aspect will be analyzed in more detail in the next section.
Qualitative and quantitative evaluation
Reference trees
High confidence gene trees are made available through scientific publications. But even for many apparently well-characterized gene families, the best estimation of the gene tree often includes ambiguous key nodes, which renders them difficult to utilize as reference trees. What is more, for the great majority, the data are no longer up-to-date, or trees include genes from not yet fully sequenced genomes, and which are therefore not present in phylogenomic databases. Because of this, we constructed reference trees for three gene families: the Popeye-domain containing family, the NOX ‘ancestral-type’ subfamily of NADPH oxidases (NOX1-4) and the V-type ATPase beta subunit. These gene families have been selected according to the following characteristics: (i) they contain one or more lineage-specific gene duplications, which we assume is a challenge for orthology prediction; (ii) they possess relatively simple gene phylogenies with no major changes in the domain architecture and no horizontal gene transfer (in contrast to more complex gene histories, simple ones are generally expected to be correctly resolved by orthology prediction methods); (iii) their sequences contain strong phylogenetic signal, which is important for the construction of the reference tree. It should be noted that each family was chosen prior to the database comparison.
Minimal requirements for reference trees derived from phylogenetic analysis were defined as follows:
The reference tree can be a consensus tree derived from multiple analyses, differing for instance, in the type of input data, the species composition, or the analytic methods applied. A reference tree could even be a dendrogram, as only the tree topology is of importance for the prediction of gene relationships.
All duplication nodes of a reference tree should be significantly supported by at least one state-of-the-art method, but not necessarily within a single gene tree. Therefore multiple analyses are performed for sub-datasets, until relevant nodes are resolved. Topological differences of close speciation nodes, found between the gene tree and the expected species tree, can be the result of incomplete lineage sorting [28]. Topological incongruence is acceptable if there is no evidence of either hidden paralogy or of horizontal gene transfer and, beyond that, if the constrained topology is not rejected by statistical tests. Orthology can also be supported by gene synteny.
It is desirable to analyze all relevant gene copies predicted in the genomes of the selected species. Exceptions are sequences that might hinder the analysis, e.g. sequences derived from incomplete or erroneous gene models or events of gene conversion [29, 30].
The robustness of tree topologies can be further tested by adding sequences of species other than those selected for the reference tree.
Finally, all available information and findings should be considered in total to confirm findings and, likewise, uncover inconsistencies.
Despite all efforts to improve phylogenetic inference, a reference tree still remains a tentative model of past history. Update and maintenance are essential when novel related sequences, improved tree-building approaches, or new knowledge on gene and genome evolution become available. In particular, the identification of new gene duplications within subtrees of apparent 1:1 orthologs will help to discriminate between true orthologs and pseudo-orthologs [31]. Additionally, a larger choice for the selection of suitable outgroups can improve the analysis. Supplementary Text S1–S5 in ‘Supplementary Data’ provides reference gene trees in extended Newick format (NHX) with annotated gene duplications, lists of gene pairs with annotated gene relationships and a list of genes that were excluded from the analysis, e.g. fragments.
Data mapping
In principle, there are two strategies for the mapping of sequence data between a reference tree and the corresponding ortholog group from a phylogenomic database: mapping by sequence identity or mapping by gene identifiers. The advantage of the first solution is the standardization of parameters, which is of importance when benchmarking orthology prediction methods. This approach, however, fails to match a considerable number of genes due to differing sequence versions, gene models, natural variants and isoforms which are used for the orthology classification by the phylogenomic databases. Even minor differences in sequence can influence the outcome of an analysis. In contrast to previous studies, we have chosen to address here the typical user question regarding the existence of orthologs in two or more species. This question can best be answered via the mapping of gene identifiers, as mapping based on sequence identity will miss many correct orthology assignments. In order to provide a visual of the results, one of the reference trees is depicted along with the mapped ortholog predictions in Figure 2. All three reference trees are shown in Supplementary Figures S1–S3, together with relevant Supplementary information.
Figure 2:

Reference tree for the V-type ATPase β-subunit subfamily and corresponding ortholog predictions from seven phylogenomic databases. The different grouping strategies are clearly reflected: OMA, InParanoid and the unlabeled trees of HOGENOM occur as mutually exclusive groups, while all other databases possess hierarchical grouping strategies. Most orthology predictions coincide with those of the reference tree, but none of the phylogenomic databases is in full agreement with all of them: OMA groups are split into more groups than necessary, which results in less predicted gene relationships; InParanoid predicts the B2 subunit of Ornithorhynchus anatinus to be an ortholog of the human B1 subunit and lacks some of the arthropod orthologs; OrthoDB assigns corresponding 1:1 orthologs only for closely related species such as primates or rodents; eggNOG gives contradictory information on the B2 subunit of Xenopus tropicalis; the tree topology of Panther suggests lineage-specific duplications for the paralogs of X. tropicalis, Caenorhabditis elegans and C. briggsae; the tree of Compara includes an additional duplication event within the vertebrate B2 clade; HOGENOM differs from the reference tree only by the inversion of a speciation node (data not shwn) and lacks one of the expected orthologs in the data set. Missing orthologs are also observed for OMA, InParanoid and Panther. Explanation: the left block (headed ‘Ortholog hierarchies’) indicates the ortholog classification derived from the reference tree, with the largest homolog group given in the first column; different levels of orthologous hierarchies are shown as patterned cells in the right-handed columns. Corresponding groups defined by the phylogenomic databases are patterned accordingly, if relevant to the benchmarked ortholog classification. Triangle: gene duplication event. White cell: gene of species that are not covered by the database. Plain gray cell: gene assigned to an unexpected ortholog group. Descending diagonal: expected gene that was missing in an ortholog group. Ascending diagonal: false positive prediction. Black horizontal bar: groups of the same hierarchical level within the same column. For OrthoDB the black bar also separates the three taxonomic sections of the database (VeRTebrate, ARThropods, FUNgi). For more details, see Supplementary Figure S3.
Gene relationships to be evaluated
As the conceptual discussion above has demonstrated, the various grouping strategies provide unequal degrees of phylogenetic information. To be able to compare the databases quantitatively, we chose to reduce their predictions to three categories of pairwise gene relationships: (i) orthology, (ii) orthology and paralogy and (iii) ‘extended’ orthology/paralogy. The last type represents an attempt to capture more phylogenetic information than provided by simple orthology/paralogy. This is accomplished by taking into account the number of gene duplications for each lineage since the last common node for the compared genes (see Methods section).
Measures
We have developed simple and intuitive measures in order to answer two typical user questions:
‘Are the predicted relationships correct?’ This question deals with the number of false positive hits, and we calculate for this purpose the positive predictive value from the number of true positives and false positives.
‘Are the predicted relationships complete?’ This question concerns the fraction of false negative hits, which can be expressed via the true positive rate.
Both scores are calculated according to the three aforementioned gene relationships assigned or implied by each database. Unlabeled trees capture considerable phylogenetic signals, but provide no information regarding the three types of gene relationships considered here. As an alternative evaluation approach for unlabeled trees, we quantified ‘correctness’ in terms of the agreement between the topologies of plain trees and reference trees (pruned to a common set of leaves) in terms of the Robinson–Foulds distance [32]. All scores were normalized between 0 and 1, whereby values of 1 correspond to a perfect match with the reference tree. Neither the number of species nor the taxonomic range has any impact on the scoring.
Score-based quantitative analysis
Scores calculated for each database and each gene family are presented in Table 2. The authors stress that values are based on only three gene families (3783 gene relationships) and that the results can, therefore, only be indicative. Most databases achieve high score values for correct orthology predictions. Maximum values for specificity are obtained by OMA groups, OrthoDB groups and Compara trees, which are each based on a different concept. Tree topologies from Panther differ slightly from the reference trees, resulting in the assignment of wrong gene relationships in the reconciled tree. For databases with hierarchical group concepts, scores were calculated from groups and by considering the hierarchical topology. In the first case, gene relationships were mostly specified at high resolution, while gene relationships at other levels were considered undefined—hence the high precision and the low sensitivity. When gene relationships are derived from reconstructed trees, many more gene relationships can be evaluated. The precision scores of the two measures differ most for OrthoDB, as groups seem to be split according to speciation prior to duplication. In this manner, high precision is achieved between closely related species. But the hierarchy at early vertebrate radiation nodes is not consistent with the gene phylogeny, e.g. teleostei and tetrapods in the POP and NOX families. Similar trends are observed when considering the precision of both, orthologous and paralogous gene relationships (Table 2).
Table 2:
Benchmarking results based on three reference trees
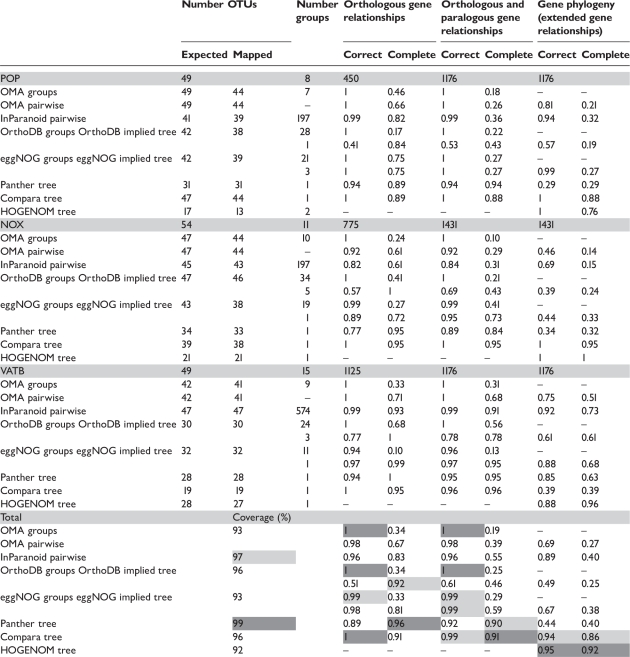 |
The analyzed databases are OMA pure orthologous groups and pairwise groups, InParanoid, OrthoDB, eggNOG, Panther trees and HOGENOM. Databases with a hierarchical grouping concept are scored in two ways, based on the ortholog groups and based on the implied trees. For HOGENOM, the calculation is based on Robinson Foulds distances. Columns: ‘Expected OTUs’: number of genes expected to be present in an ortholog group according to the species list of the phylogenomic database. ‘Mapped OTUs’: number of genes of the reference tree that are mapped to the ortholog groups; ‘Number groups’: number of groups relevant to the reference tree. Scores are calculated for the three types of gene relationships: orthology, orthology/paralogy and ‘extended’ gene relationships. The weighted average is shown bottom left of the table. For each column, the best achieved values are shaded dark gray, the second-best light gray. ‘Coverage’ indicates the weighted average of mapped genes, in percent. For each family, the number of genes and the number of relevant gene relationships are indicated within the gray header row.
There is a strong variability in the recall of predicted gene relationships. The concept of pure orthologous groups does not allow a comprehensive coverage: the sensitivity score drops with an increase in the number of OMA groups for a family. As no paralogous gene relationships are predicted, the sensitivity score for all gene relationships—orthologs and paralogs—decreases along with the number of paralogs in a family. For pairwise group concepts, the recall is significantly higher for orthologous relationships than for all gene relationships, an observation that can, at least in part, be explained by the database concept. The sensitivity scores for databases with reconciled trees are among the highest, but less than 1.0 for differing reasons. False negatives identified for Compara can primarily be ascribed to the underlying sequence input data. Panther complements the Ensembl proteomes with UniProtKB [33] sequences and profits from a nearly complete input dataset for the three gene families; false negatives are mostly a result of tree topologies that differ from that of the reference tree.
The precision score for ‘gene phylogeny’ is generally lower than the one calculated for all gene relationships, but it can also increase when many gene relationships are no longer defined, in which case the sensitivity score decreases (Table 2, right column). Non-hierarchical methodologies can only compete in this measure if a family includes no more than one duplication per lineage. Databases with hierarchical grouping strategies have the potential to perform well, but the scores indicate inconsistencies, because the measure applied here takes into account internal node topologies that differ from those of the reference tree. This can be observed for the NOX family, where precision scores decrease significantly for OrthoDB and eggNOG. The highest overall scores for precision and sensitivity are achieved by tree-based methods, namely Compara and HOGENOM. Panther trees show lower score values due to various inconsistencies in tree topologies. It should be noted that our set of well-conserved proteins might be biased in favor of tree-based strategies.
In summary, our scoring schemes are consistent with both the quality of predicted gene relationships and the concepts underlying the databases. For the three examples, we observe a generally high specificity of orthology predictions regarding the phylogenomic databases examined in this study. However, the results largely differ with respect to sensitivity and gene phylogeny.
CONCLUSIONS
For three rather simple gene histories, none of the phylogenomic databases was in perfect agreement with the reference trees. Preliminary results suggest that there is generally high precision in orthology predictions. Most of the variation in sensitivity of orthology predictions can be explained by conceptual differences and incomplete datasets. Gene phylogenies can qualitatively and quantitatively be best resolved by databases utilizing a tree concept.
Reference trees constitute a robust benchmark for measuring precision and sensitivity of phylogenetic information provided by databases. Towards the end of this study, we shared the results with groups that provide phylogenomic databases. In response, the information was used by the authors of the Panther database to identify and fix a bug in their tree reconstruction software. The reference trees were also used to verify the correctness of the latest OrthoDB release after an update of the analysis procedure. This positive feedback drives home the point that, in the future, the maintenance of ‘gold standard’ phylogenetic trees represents a highly desirable and profitable undertaking.
SUPPLEMENTARY DATA
Supplementary data are available online at http://bib.oxfordjournals.org/.
Key Points.
Phylogenomic databases differ substantially in concept. Reconciled trees represent the most informative grouping strategy.
None of the phylogenomic databases agrees perfectly with our reference gene trees.
Orthology predictions, as provided by the databases, are generally correct—at least for simple gene trees.
Most of the variation in sensitivity of orthology predictions can be explained by conceptual differences and incomplete data sets.
Reference gene trees provide a robust way for the quality assessment of orthology predictions.
FUNDING
The Swiss Federal Government through the Federal Office of Education and Science (B.B.), by the Etat de Vaud and the Swiss National Science Foundation (M.R.R.). Funding for open access charge: Swiss Institute of Bioinformatics.
Acknowledgements
The authors wish to thank all databases for providing their resources to the scientific communities. A special thanks for helpful discussions and feedback to Adrian M. Altenhoff, Peer Bork, Laurent Duret, Evgenia V. Kriventseva and Robert M. Waterhouse, Paul Thomas, Albert J. Vilella, Arnold Kuzniar and Alan Bridge, Vivienne Baillie Gerritsen and Dwight Klett for correcting the manuscript and three anonymous reviewers for helpful comments. Conceived by I.X., designed by B.B. with contributions from C.D., analyzed and interpreted by B.B., C.D., M.R.R. and I.X., written by B.B. with contributions from all authors. All authors read and approved the manuscript.
Biographies
Brigitte Boeckmann is a researcher in the Swiss-Prot group at SIB Geneva. Her main interests focus on the evolution of proteins, proteomes and function.
Marc Robinson-Rechavi is associate professor in bioinformatics at the Department of Ecology and Evolution in the University of Lausanne, and Group Leader at the Swiss Institute of Bioinformatics. His main interest is in the evolution of animal genomes in the context of organismal function and development.
Ioannis Xenarios is a professor in computational biology at the University of Lausanne. He is also director of the Swiss-Prot group and Vital-IT group at the Swiss Institute of Bioinformatics.
Christophe Dessimoz is senior post-doc and lecturer in the CBRG group, computer science, ETH Zurich. His interest lies in understanding the forces that shape genes, genomes and species by using computational and statistical methods.
References
- 1.Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 2.Studer RA, Robinson-Rechavi M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009;25:210–6. doi: 10.1016/j.tig.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Alexeyenko A, Lindberg J, Pérez-Bercoff A, et al. Overview and comparison of ortholog databases. Drug Discov Today: Technol. 2006;3:137–43. doi: 10.1016/j.ddtec.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Chen F, Mackey AJ, Vermunt JK, et al. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS One. 2007;2:e383. doi: 10.1371/journal.pone.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hulsen T, Huynen MA, de Vlieg J, et al. Benchmarking ortholog identification methods using functional genomics data. Genome Biol. 2006;7:R31. doi: 10.1186/gb-2006-7-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol. 2009;5:e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vilella AJ, Severin J, Ureta-Vidal A, et al. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–35. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuzniar A, van Ham RC, Pongor S, et al. The quest for orthologs: finding the corresponding gene across genomes. Trends Genet. 2008;24:539–51. doi: 10.1016/j.tig.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Flicek P, Aken BL, Ballester B, et al. Ensembl's 10th year. Nucleic Acids Res. 2010;38:D557–62. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller J, Szklarczyk D, Julien P, et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010;38:D190–5. doi: 10.1093/nar/gkp951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penel S, Arigon AM, Dufayard JF, et al. Databases of homologous gene families for comparative genomics. BMC Bioinformatics. 2009;10(Suppl 6):S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostlund G, Schmitt T, Forslund K, et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38:D196–203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altenhoff AM, Schneider A, Gonnet GH, et al. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–94. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waterhouse RM, Zdobnov EM, Tegenfeldt F, et al. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2011;39:D283–8. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mi H, Dong Q, Muruganujan A, et al. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38:D204–10. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasteiger E, Gattiker A, Hoogland C, et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–8. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dereeper A, Guignon V, Blanc G, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–69. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Do CB, Mahabhashyam MS, Brudno M, et al. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–40. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guindon S, Dufayard JF, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 21.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–82. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 22.Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–9. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- 23.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–52. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 24.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 25.Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–95. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt HA, von Haeseler A. Maximum-likelihood analysis using TREE-PUZZLE. Curr Protoc Bioinformatics. 2007 doi: 10.1002/0471250953.bi0606s17. Chapter 6:Unit 6 6. [DOI] [PubMed] [Google Scholar]
- 27.van der Heijden RT, Snel B, van Noort V, et al. Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinformatics. 2007;8:83. doi: 10.1186/1471-2105-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galtier N, Daubin V. Dealing with incongruence in phylogenomic analyses. Philos Trans R Soc Lond B Biol Sci. 2008;363:4023–9. doi: 10.1098/rstb.2008.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JM, Cooper DN, Chuzhanova N, et al. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 2007;8:762–75. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- 30.Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- 31.Dessimoz C, Boeckmann B, Roth AC, et al. Detecting non-orthology in the COGs database and other approaches grouping orthologs using genome-specific best hits. Nucleic Acids Res. 2006;34:3309–16. doi: 10.1093/nar/gkl433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson DF, Foulds LR. Comparison of phylogenetic trees. Math Biosci. 1981;53:131–47. [Google Scholar]
- 33.The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–48. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]