

Abstract
A single recurrent mutation in the regulatory subdomain of a bone morphogenetic protein type I receptor kinase has been linked to heterotopic ossification in classic fibrodysplasia ossificans progressiva (FOP). As a result of a substitution at 1 residue by only 1 other side chain (Arg206His) in just 1 of the 4 type I BMP receptors (ALK2/ACVR1), soft connective tissues progressively metamorphose through an endochondral process into cartilage that is replaced by bone. The substitution of arginine for histidine, also a basic residue yet with the singular property of ionization/protonation over the physiological pH range, led to the hypothesis of an aberrant, pH-sensitive switch mechanism for the ligand-independent activation of BMP signaling through the mutant receptor kinase in patients presenting with classic FOP. To test a potential aspect of the putative pH-dependent mechanism, i.e. loss of autoinhibition of the kinase mediated by the inhibitory protein FKBP12, in vitrointeraction analyses with purified wild-type and R206H ALK2 kinase and FKBP12 proteins were performed. Interactions between the kinases and inhibitory proteins were analyzed qualitatively and quantitatively by native gel electrophoresis and HPLC size exclusion chromatography and with an optical biosensor (Octet; ForteBio). Binding of inhibitory protein by the R206H mutant was diminished 3-fold relative to the wild type kinase at a physiological pH, yet below this value (<∼7.5) pronounced nonspecific interactions, particularly with the mutant, prevented comparative evaluations. In conclusion, substitution with histidine leads to partial loss of inhibition of the mutant type I receptor through diminished binding of FKBP12, which may act as a gradient reader in morphogenetic contexts.
Key Words: BMP/TGF-β signaling, FKBP12, Ser/Thr kinase receptors, Skeletal dysplasias, Fibrodysplasia ossificans progressiva
Introduction
Fibrodysplasia ossificans progressiva (FOP) is the most disabling condition of ectopic skeletogenesis known to mankind. Beginning in childhood, soft tissues such as skeletal muscle, tendon, ligament, and fascia are transformed through an endochondral process into ribbons, sheets, and plates of heterotopic bone that progressively lock joints in place and block movement. As shown by a large body of evidence collected over a decade, the heterotopic ossification is associated with dysregulation of bone morphogenetic protein (BMP) signaling [Shafritz et al., 1996; Ahn et al., 2003; de la Pena et al., 2005; Fiori et al., 2006]. Consistent with those findings, an identical heterozygous mutation in ACVR1 (ALK2), a type I BMP receptor, was identified in all classically affected individuals [Shore et al., 2006]. Through structure-based homology modeling [Huse et al., 1999], the highly conserved R206H substitution lying in the glycine-serine regulatory subdomain was hypothesized to lead to ligand-independent activation of the receptor in FOP due to the introduction of an aberrant pH-sensitive switch [Groppe et al., 2007]. In addition to the classic syndrome caused by the R206H substitution, a small fraction of patients present atypical and variant forms of FOP linked to mutations at this and 6 other sites in the regulatory subdomain and the active site of the type I receptor kinase [Kaplan et al., 2009].
To examine the effect of the single recurrent R206H mutation linked to classic FOP, in vitrointeraction analyses with purified wild-type and R206H ALK2 kinase and FKBP12 proteins were performed. The results of 3 independent methods showed that binding of the inhibitory protein by the R206H mutant was not severely affected but was rather diminished just several fold relative to wildtype kinase. Below a physiological pH, pronounced nonspecific interactions, particularly with the mutant, prevented exploration of the aberrant pH-sensitive switch hypothesis. Nonetheless, the independent in vitro analyses with purified components clearly demonstrate that substitution with histidine only mildly perturbs binding of the FKBP12 inhibitory protein, which may act as a gradient reader in morphogenetic or other contexts such as the congenital developmental defect and heterotopic ossification of FOP.
Materials and Methods
Protein Preparations
The cytoplasmic kinase domains of wild-type and R206H ALK2 were expressed in Sf21 insect cells after infection with high-titer recombinant baculovirus stocks produced with the Invitrogen Bac-to-Bac system (DH10Bac bacmid and pFastBacHT vectors). The N-terminally tagged (6xHis-TEV) kinases were isolated from the insect cell lysates by Ni2+-NTA (Qiagen) affinity chromatography. After binding in batch, the recombinant proteins were eluted, cleaved with TurboTEV protease, and purified to near homogeneity by a subsequent reverse affinity step. Recombinant 6xHis-TEV-FKBP12 was expressed with the pET system (BL21DE3 and modified pET15b), isolated from Escherichia coli lysates by affinity chromatography, cleaved with TEV, and purified by reverse affinity. The charge variant for native gel assays was purified by a single affinity step and not subjected to proteolysis.
Native Polyacrylamide Gel Electrophoresis
Complex formation was analyzed by native polyacrylamide gel electrophoresis (PAGE) as previously described [Groppe et al., 2008]. Briefly, protein samples were electrophoresed through 4% stacking/12% resolving gels with native Tris-glycine gel and running buffers (Invitrogen-Novex recipes) and visualized by Coomassie staining.
Size Exclusion Chromatography
Protein samples were injected onto a Tosoh TSK-GEL G3000PWxl (polymer-coated bead) column equilibrated with 25 mM Na HEPES (pH 8.0), 50 mM arginine-glutamate, and 150 mM NaCl on a Dionex UltiMate 3000 high-performance liquid chromatographer (HPLC). Three wavelength elution data (218, 280, and 310 nm) were collected and analyzed using the Chromeleon software package.
Optical Biosensor (Octet) Biolayer Interferometry
Protein-protein interactions were quantitated with an Octet QK (FortéBio, Menlo Park, Calif., USA) by biolayer interferometry (BLI). FKBP12 protein was PEG-biotinylated with NHS-PEG4biotin (Thermo-Pierce), buffer exchanged on PD-10 desalting columns, and immobilized on streptavidin-coated fiber optic tips (FortéBio). Proteins were diluted into optimized binding buffer (25 mM Na HEPES (pH 8.0), 50 mM arginine-glutamate, and 150 mM NaCl).
Results
Qualitative Analyses by Native PAGE
In a recent protein structure-function study, analysis of complex formation by native polyacrylamide gel electrophoresis provided a qualitative comparison of wild-type and mutant TGF-β ligand-receptor interactions in vitro [Groppe et al., 2008]. Because the rate and direction of migration depends on the inherent charge of a protein at the pH of the buffer system, resulting in retrograde migration of FKBP12, a charge variant was engineered through modification of a flexible N-terminal affinity tag (6xHis-TEV). In the kinase-FKBP12 native gel assay (fig. 1), the wild-type kinase became saturated with FKBP12 at a 1:1 molar ratio; however, excess inhibitory protein failed to produce significantly more mutant complex by mass action, indicative of weaker interaction, by an estimate of several fold.
Fig. 1.
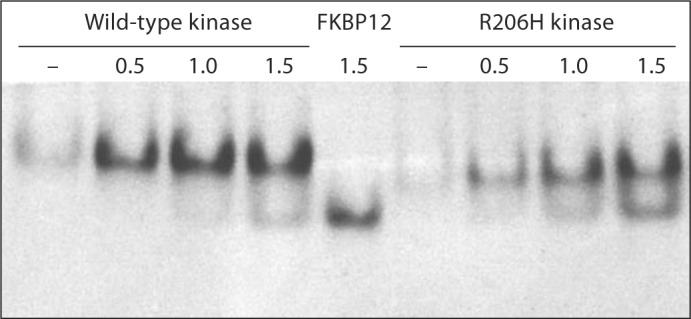
Native PAGE analysis of FKBP12 protein interactions with wild-type and R206H ALK2 kinases. Center lane: FKBP12 only. Dashes: Kinase only. Values refer to molar equivalents of FKBP12 relative to a fixed mass of kinase. Note the faster migration of the less positive R206H mutant at the nonphysiological pH of the gel buffer (∼8.3).
Semiquantitative Analyses by HPLC Size Exclusion Chromatography
Analysis of the Noggin-BMP7 [Groppe et al., 2002], BMP [Greenwald et al., 2003] and TGF-β ligand-receptor [Groppe et al., 2008 ] complexes by HPLC size exclusion chromatography (SEC) allowed for the rapid characterization and comparison of protein preparations with respect to binding affinity; hence, it was a desirable approach for semiquantitative analyses of FKBP12 interactions with the wild-type and R206H mutant kinases. After identification of an optimized buffer that suppressed nonspecific interactions between the column matrix and the kinases, in particular the mutant, the inhibitory protein and kinases were analyzed alone and as mixtures at 1:1 or 1:2 molar equivalents (fig. 2). In keeping with the qualitative results of the native gel assays, even 2 molar equivalents of FKBP12 (middle chromatogram) were insufficient for the formation of a stable complex with the mutant kinase in comparison to the wild type, which was saturated with one, judging from residual FKBP12 protein and elution times relative to free kinases (complexes were eluted 0.37 min earlier with wild type but only 0.08 min with the mutant). SDS-PAGE analysis of the pooled peak fractions from each separation showed that the mutant complex was appreciably less stable (seemingly more than the several-fold decrease estimated by native gel analysis); however, the results of SEC may exaggerate the differences in affinities due to separation of the components during elution.
Fig. 2.
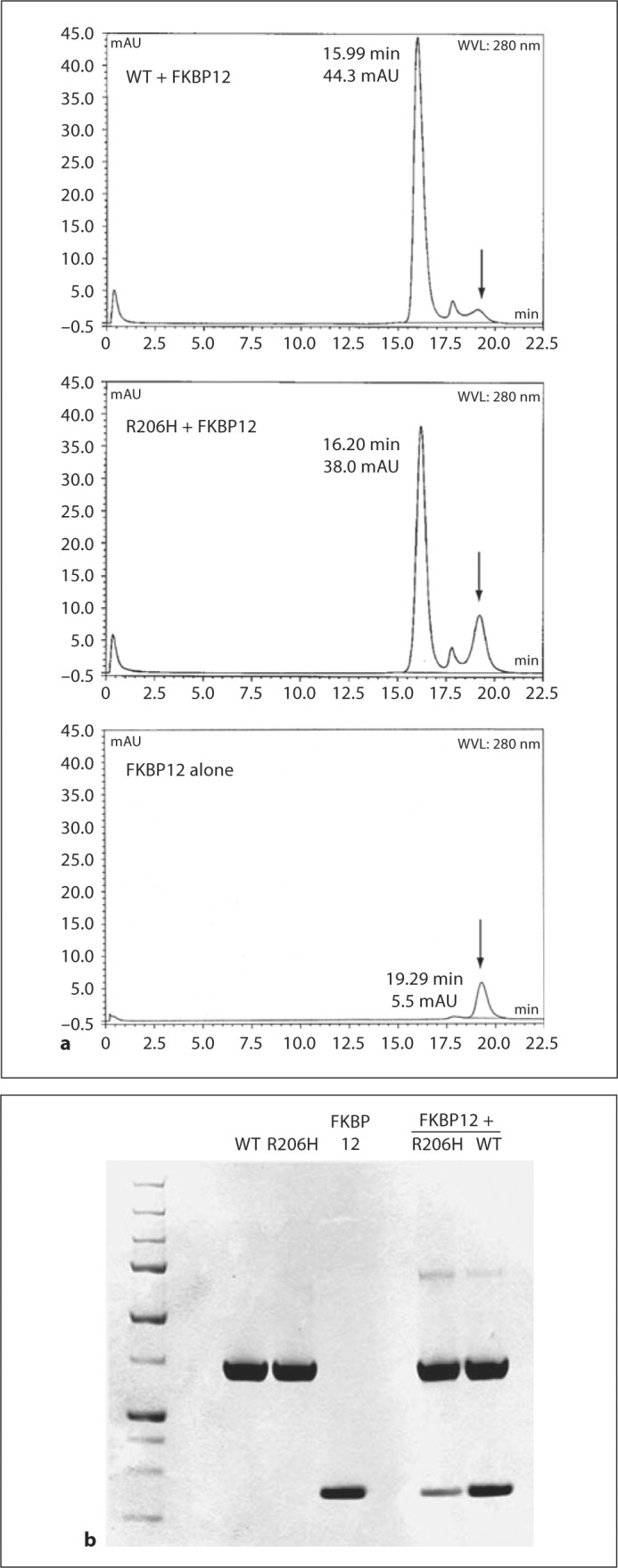
HPLC SEC analyses of FKBP12-kinase interactions. a Chromatograms (mAU, 280 nm) of kinase proteins mixed with FKBP12 at 1:1 (WT) and 1:2 (R206H) molar equivalents and of FKBP12 alone (1 equivalent). Arrows mark the position of FKBP12 elution. b The 3 left lanes of lower SDS-PAGE analysis are reference samples, and the 2 right lanes are peak fractions of the kinases mixed with FKBP12 at 1:1 (WT) and 1:2 (R206H) equivalents (see above).
Quantitative Analyses by BLI Optical Biosensor
In order to obtain a third independent and more quantitative determination of the relative affinity of the wild-type and R206H kinases, an optical biosensor instrument was employed. Previously, surface plasmon resonance measurements allowed for the quantitation and comparison of wild-type and mutant TGF-β ligand-receptor interactions in vitro [Groppe et al., 2008]. Here, a comparable optical biosensor instrument (Octet; ForteBio) based on BLI was employed (fig. 3). Typically, 5- to 10-fold higher concentrations of the mutant kinase were required to yield a comparable effect (shift of light, nm), in keeping with the lower affinity for the inhibitory FKBP12 protein observed using the native gel and HPLC SEC methods. At the higher concentrations of mutant protein required for a reliable signal, and despite the optimized buffer, nonspecific background was strongly diminished although not eliminated; hence, the measurement of binding and the calculation of affinity may be exaggerated. Nevertheless, about a 3-fold decrease in affinity for the mutant was determined; this is more consistent with the native gel than the SEC/PAGE analyses. Due to significant nonspecific binding, particularly with the more metastable mutant, assays performed below a physiological pH produced an enhanced signal that did not saturate (plateau), precluding investigation of the aberrant pH-sensitive switch hypothesis.
Fig. 3.

Octet optical biosensor analyses of wild-type and R206H kinase interactions with the FKBP12 inhibitory protein. a, b Processed response data (λ shift; nm) of association (FKBP12-immobilized tips in kinase samples; 1,200 s) and dissociation (FKBP12-immobilized tips moved to buffer; 1,200 s) phases. Note the slower apparent on-rate of the mutant. c, d Steady-state analysis fits of maximal effects measured over 2 ranges of concentration (0.025–0.45 μM, WT; 0.25–1.75 μM, R206H) for determination of the equilibrium dissociation constants (KD) of the 2 interactions.
Discussion
The results of 3 independent in vitrointeraction analyses with purified wild-type and R206H ALK2 kinase and FKBP12 proteins showed that binding of the inhibitory protein by the mutant was not severely affected but was rather diminished only several fold relative to the wild-type kinase. Consistent with these in vitro findings, cell-based assays with co-immunoprecipitations following transient transfections provided estimates of an approximately 2-fold lower affinity [Shen et al., 2009; Song et al., 2010]. Intriguingly, in contrast to the modest effect of the mutation on FKBP12 binding, transcriptional reporter construct assays with cultured cells showed that the mutant receptor was not inhibited by the negative regulator [van Dinther et al., 2010].
Interaction of the FKBP12 inhibitory protein with the type I receptors of the TGF-β superfamily has been hypothesized to act as a gradient reader [Wang and Donahoe, 2004], playing a role in the morphogenetic activities of the signal ligands (TGF-β, BMP, GDF, activin, and nodal). Diminished FKBP12 binding could perturb the read-out of the gradient, increasing the signaling output and leading to developmental defects or homeostatic aberrations such as the congenital developmental defect and heterotopic ossification of FOP. In the context of the gradient reader mechanism, a modest 3-fold decrease in affinity for FKBP12 could account for the dysregulation of BMP signaling by the R206H ALK2 receptor kinase linked to classic FOP. Because phosphorylation of the regulatory subdomain of the type I kinase abrogates binding by the inhibitory protein [Huse et al., 2001, 2002] the effect of diminished interaction may be amplified, increasing over time or in certain subcellular environments.
Acknowledgements
This study was supported by grant number 5R03AR056838 from the NIAMS/NIH and an extramural award from The Center for Research in FOP and Related Disorders at the University of Pennsylvania School of Medicine.
Glossary
Abbreviations used in this paper
| ALK | activin-like kinase |
| BLI | biolayer interferometry |
| BMP | bone morphogenetic protein |
| FKBP12 | FK506 binding protein 12 kDa |
| FOP | fibrodysplasia ossificans progressiva |
| HPLC | high-performance liquid chromatography |
| PAGE | polyacrylamide gel electrophoresis |
| SEC | size exclusion chromatography |
References
- Ahn J., Serrano de la Pena L., Shore E.M., Kaplan F.S. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Joint Surg Am. 2003;85:667–674. doi: 10.2106/00004623-200304000-00013. [DOI] [PubMed] [Google Scholar]
- de la Pena L.S., Billings P.C., Fiori J.L., Ahn J., Kaplan F.S., Shore E.M. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- Fiori J.L., Billings P.C., de la Pena L.S., Kaplan F.S., Shore E.M. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- Greenwald J., Groppe J., Gray P., Wiater E., Kwiatkowski W., Vale W., Choe S. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell. 2003;11:605–617. doi: 10.1016/s1097-2765(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Groppe J., Greenwald J., Wiater E., Rodriguez-Leon J., Economides A.N., Kwiatkowski W., Affolter M., Vale W.W., Belmonte J.C., Choe S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- Groppe J., Hinck C.S., Samavarchi-Tehrani P., Zubieta C., Schuermann J.P., Taylor A.B., Schwarz P.M., Wrana J.L., Hinck A.P. Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell. 2008;29:157–168. doi: 10.1016/j.molcel.2007.11.039. [DOI] [PubMed] [Google Scholar]
- Groppe J.C., Shore E.M., Kaplan F.S. Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Relat Res. 2007;462:87–92. doi: 10.1097/BLO.0b013e318126c049. [DOI] [PubMed] [Google Scholar]
- Huse M., Chen Y.G., Massagué J., Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- Huse M., Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Huse M., Muir T.W., Xu L., Chen Y.G., Kuriyan J., Massagué J. The TGFβ receptor activation process: an inhibitor-to substrate-binding switch. Mol Cell. 2001;8:671–682. doi: 10.1016/s1097-2765(01)00332-x. [DOI] [PubMed] [Google Scholar]
- Kaplan F.S., Xu M., Seemann P., Connor J.M., Glaser D.L., Carroll L., Delai P., Fastnacht-Urban E., Forman S.J., Gillessen-Kaesbach G., Hoover-Fong J., Köster B., Pauli R.M., Reardon W., Zaidi S.-A., Zasloff M., Morhart R., Mundlos S., Groppe J., Shore E.M. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009;30:379–390. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafritz A.B., Shore E.M., Gannon F.H., Zasloff M.A., Taub R., Muenke M., Kaplan F.S. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- Shen Q., Little S.C., Xu M., Haupt J., Ast C., Katagiri T., Mundlos S., Seemann P., Kaplan F.S., Mullins M.C., Shore E.M. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest. 2009;119:3462–3472. doi: 10.1172/JCI37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore E.M., Xu M., Feldman G.J., Fenstermacher D.A., Brown M.A., Kaplan F.S. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Song G.A., Kim H.J., Woo K.M., Baek J.H., Kim G.S., Choi J.Y., Ryoo H.M. Molecular consequences of the ACVR1 (R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542–22553. doi: 10.1074/jbc.M109.094557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinther M., Visser N., de Gorter D.J., Doorn J., Goumans M.J., de Boer J., ten Dijke P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J Bone Miner Res. 2010;25:1208–1215. doi: 10.1359/jbmr.091110. [DOI] [PubMed] [Google Scholar]
- Wang T., Donahoe P.K. The immunophilin FKBP12: a molecular guardian of the TGF-β family type I receptors. Front Biosci. 2004;9:619–631. doi: 10.2741/1095. [DOI] [PubMed] [Google Scholar]
- Weiser R., Wrana J.L., Massagué J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]